

Abstract
Cholangiocytes are the target cells in cholestatic models of ductal hyperplasia including bile duct ligation (BDL). We have shown that: (i) cholangiocytes express VEGFR-2 and VEGFR-3; (ii) VEGF-A and VEGF-C stimulate cholangiocyte proliferation via an autocrine mechanism; and (iii) chronic administration of VEGF-A prevents cholangiocyte damage induced by hepatic artery ligation. Caffeic acid phenethyl ester (CAPE) induces growth inhibition in different cells. Taurocholic acid (TC) protects cholangiocytes against injury induced by parasympathetic or sympathetic denervation. The aims of this study were to determine if: (i) CAPE induces bile duct damage; and (ii) TC prevents CAPE-induced bile duct damage by increasing cholangiocyte VEGF expression. Methods: Normal and BDL rats (immediately after surgery) were fed 1% TC or control diet in the absence/presence of daily IP injections of CAPE (10 mg/Kg BW). One week later, we evaluated: (i) cholangiocyte apoptosis, proliferation and ductal mass in liver sections; (ii) functional activity by measuring secretin-stimulated bile and bicarbonate secretion; and (iii) VEGF-A/C and VEGFR-2/R-3 expression in liver sections. In vitro, BDL cholangiocytes were exposed to CAPE (40 μM) in the absence/presence of TC (40 μM) with and without pretreatment with VEGF receptor inhibitors before evaluating cholangiocyte apoptosis and proliferation. Results: Chronic CAPE administration to BDL rats increased cholangiocyte apoptosis and decreased ductal mass. This effect was associated with reduced expression of VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3. In vivo, TC feeding partly prevented CAPE-induced changes in cholangiocyte apoptosis and growth and loss of ductal secretion. The protective effect of TC was associated with enhanced VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3. In vitro, TC partially prevented CAPE-induced increases in apoptosis and decreases in cholangiocyte proliferation. These changes were reversed by pretreatment with VEGF-receptor inhibitors. Conclusion: Manipulation of cholangiocyte VEGF expression by bile acids may be important in preventing the impairment of cholangiocyte proliferation by exogenous agents.
Keywords: intrahepatic biliary epithelium, biliary hyperplasia, bile acids, apoptosis, VEGF
Introduction
The intrahepatic biliary tree consists of a complex, ramified network of interconnecting tubular conduits of different diameters and function, lined by cholangiocytes (1, 2). Constitutively, normal cholangiocytes have low replicative activity (3, 4), but they proliferate or undergo apoptosis in cholangiopathies that are characterized by an abnormal balance between cholangiocyte proliferation and death (5). Cholangiocytes are the target cells in a number of animal models of cholestasis including bile duct ligation (BDL), bile acid feeding or administration of carbon tetrachloride (CCl4), models that are characterized by changes in cholangiocyte growth and/or apoptosis (6–10). In rats with BDL, cholangiocyte hyperplasia is associated with enhancement of basal and secretin-stimulated ductal secretion (3, 10). On the other hand, reduced biliary growth and enhanced cholangiocyte apoptosis is coupled with decreased secretin-stimulated cholangiocyte secretory activity (8, 9). These features are typical of the terminal stages of different cholangiopathies including primary biliary cirrhosis (11).
Caffeic acid phenethyl ester (CAPE) is the active component of propolis, a honeybee hive product. CAPE exhibits a broad spectrum of biological properties including antiproliferative (12) and anti-metastatic activities (13). For example, CAPE is able to suppress the growth of HepG2 (a human hepatocellular carcinoma cell line) xenografts in nude mice (14). No information exists on the role of CAPE on the regulation of cholangiocyte proliferation in cholestasis.
Bile acids modulate cholangiocyte pathobiology by entering these cells via a specific transporter (Na+-dependent bile acid transporter, ASBT) localized at the apical pole of cholangiocytes (15, 16). Entry of bile acids into these cells by the ASBT induces changes in cholangiocyte secretion, proliferation and apoptosis (6, 17–19). Taurocholic acid (TC) plays an important role in the regulation of cholangiocyte function (19–21). For example, TC feeding to normal rats stimulates cholangiocyte proliferation and secretin-stimulated choleresis (22). Furthermore, depletion of endogenous bile acid pools in BDL rats reduces cholangiocyte proliferation and secretin-stimulated ductal secretion and ASBT expression and bile acid transport activity compared to controls (23). TC prevents the induction of cholangiocyte death by apoptosis, and the loss of proliferative and functional responses of cholangiocytes in response to toxins (e.g., CCl4) and cholinergic or adrenergic denervation (19–21).
Vascular endothelial growth factor (VEGF) is a mitogen for vascular endothelial cells and regulates vascular pathophysiology (24, 25). While VEGF-A binds to vascular endothelial growth factor receptor type 2 (VEGFR-2), VEGF-C exerts its function by either interaction with VEGFR-2 or VEGFR-3 (24, 26). VEGF is secreted by several epithelia, where it modulates cell growth by autocrine and paracrine mechanisms (27, 28). We have previously demonstrated that VEGF-A and VEGF-C are secreted by cholangiocytes and play an important role in modulating cholangiocyte proliferation and apoptosis in response to cholestasis (26) and hepatic artery ligation (29).
The aims of this study were to assess: (i) the chronic in vivo and acute in vitro effects of CAPE on cholangiocyte apoptosis, proliferation and ductal secretion in cholestatic BDL rats; (ii) if CAPE effects on cholangiocyte apoptosis and growth are associated with changes in VEGF-A/C and VEGF receptor expression; (iii) if TC partially prevents the effects of CAPE on cholangiocyte function by affecting cholangiocyte VEGF expression; and (iv) if in vitro, inhibition of VEGF receptors can block the protective effects of TC on CAPE-induced bile duct damage.
Materials and Methods
Materials
Reagents were purchased from Sigma Chemical (St. Louis, MO) unless otherwise indicated. Rat chow, containing 1% TC or AIN-76 (bile acid control diet), was prepared from Dyets Inc. (Bethlehem, PA). The substrate for γ-glutamyl transpeptidase (γ-GT), N-(-L-glutamyl)-4-methoxy-2-naphthylamide, was purchased from Polysciences (Warrington, PA).
Animal Models
Male Fischer 344 rats (150−175 gm) were purchased from Charles River (Wilmington, MA) and kept in a temperature-controlled environment (20−22°C) with a 12:12 hour light-dark cycle. Animals had free access to water and chow. The experimental groups used in the studies are shown in Table 1. Specifically, we used rats that, immediately after BDL (for preparation of liver blocks or isolation of cholangiocytes) (3, 30) or BDI (for bile collection during intravenous infusion of Krebs Ringer Henseleit, KRH or secretin), were fed AIN-76 (bile acid control (BAC) diet) or 1% TC (an approximate dose of 275 μmol/day) (6) in the absence or presence of daily IP injections of 0.1% DMSO or CAPE (10 mg/kg body weight) (31) in 1:1 DMSO:NaCl for 1 week. Since we have previously shown (6, 19) that daily IP injections of DMSO to BDL rats fed bile acid control diet or TC do not affect the functional activity of cholangiocytes, we did not include these groups of animals in the study. We also evaluated the effect of CAPE on apoptosis and proliferation of normal rats. Before each experimental procedure, the animals were anaesthetized with sodium pentobarbital (50 mg/kg body weight) according to the regulations of the panel on euthanasia of the American Veterinarian Medical Association. All experiments were conducted under the guidelines of the local IACUC.
Table 1.
Animal Modelsa
| Animals | Treatment |
|---|---|
| BDL + bile acid control feeding + DMSO | Immediately after surgery, rats were fed bile acid control diet for 1 week |
| BDL + TC feeding + DMSO | Immediately after surgery, rats were fed TC for 1 week |
| BDL + bile acid control feeding + CAPE | Immediately after surgery, rats were fed bile acid control diet in the presence of daily injections of CAPE for 1 week |
| BDL + TC feeding + CAPE | Immediately after surgery, rats were fed TC in the presence of daily injections of CAPE for 1 week |
BDL, bile duct ligation; CAPE, caffeic acid phenethyl ester; DMSO, dimethyl sulfoxide; TC, taurocholic acid.
Evaluation of Portal Inflammation, Lobular Damage, Necrosis and Cholangiocyte Apoptosis
Paraffin embedded liver sections (4 μm, 3 sections analyzed per group) from the selected groups of animals were stained with hematoxylin and eosin (H&E) before determining lobular damage, necrosis and the degree of portal inflammation (32). Ten randomly selected portal areas and surrounding lobular areas were evaluated using light microscopy of H&E-stained sections. Liver sections were examined in a coded fashion with the microscope ECLIPSE E600 (Nikon Eclipse, Tokyo, Japan) (using BX-51 light microscopy Olympus, Tokyo, Japan). Two hundred cells per slide were counted in a coded fashion in ten non-overlapping fields.
Cholangiocyte apoptosis was evaluated by the quantitative terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) kit (ApopTag; Chemicon International, Inc.) (9, 33) according to the vendor's instructions in paraffin-embedded liver sections (4 μm, 3 sections analyzed per group). The number of apoptotic cholangiocytes in liver sections was evaluated in a coded fashion using a BX-51 light microscopy (Olympus, Tokyo, Japan). Two hundred cells per slide were counted in ten non-overlapping fields. We also evaluated the expression of the apoptotic marker Bax (34) by immunohistochemistry in liver sections from all the studied groups (see below).
Immunohistochemistry
Liver pieces were fixed in 10% buffered formalin for 2 hours and paraffin embedded at 55°C. Liver sections (4 μm, 3 sections analyzed per group) were mounted on glass slides coated with acetone amino-propyltriehoxylan (2%) solution. After deparaffination, endogenous peroxidase activity was blocked by a 20-minute incubation in methanolic hydrogen peroxide (2.5%). Sections were hydrated in graded ethanol and rinsed in 1× phosphate-buffered saline (1× PBS, pH 7.4). Endogenous biotin activity was blocked by using the Biotin Blocking System (DakoCytomation code X0590, Glostrup, Denmark) and the samples were rinsed in 1× PBS. Sections were incubated overnight at 4°C with the following antibodies from Santa Cruz Biotechnology, Inc.: (i) CK-19 (G-14, code sc-33120, dilution 1:100); (ii) PCNA (PC10, code sc-56, dilution: 1:200); (iii) VEGF-A (code sc-152, dilution: 1:100); VEGF-C (code sc-9047, dilution: 1:100), VEGFR-2 (A-3, code sc-6251, dilution: 1:100) and VEGFR-3 (C-20, code sc-321, dilution: 1:100). Samples were rinsed with 1× PBS for 5 minutes, incubated for 20 minutes at room temperature with secondary biotinylated antibody (Dako-Cytomation LSAB Plus System-HRP, code K0690, Glostrup, Denmark), then with Dako ABC for 20 minutes (DakoCytomation LSAB Plus System-HRP, code K0690, Glostrup, Denmark) and developed with 3−3′-diaminobenzidine (DakoCytomation Liquid DAB Plus Substrate Chromogen System, code K3468, Glostrup, Denmark). For all immunoreactions, negative controls (non-immune serum with the omission of the primary antibody) were also included. Immunohistochemistry observations were taken by BX-51 light microscopy (Olympus, Tokyo, Japan) with a Videocam (Spot Insight; Diagnostic Instruments, Inc., Sterling Heights, MI) and processed with an Image Analysis System (IAS; Delta Sistemi, Rome, Italy).
The intrahepatic bile duct mass was measured as the area occupied by the number of CK-19-positive bile ducts/total area × 100 in liver sections (4 μm, 3 sections analyzed per group) from the selected group of animals. The numbers of PCNA, VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3 positive cholangiocytes were assessed in sections (4 μm, 6 sections analyzed per group) for each group. Positive cells were counted in six non-overlapping fields (magnification ×20) for each slide and the data expressed as percentage of positive cells. Light microscopy and immunohistochemical observation was performed by three independent pathologists in a blinded manner.
Effect of CAPE on Basal and Secretin-Stimulated Bile and Bicarbonate Excretion
To demonstrate the effects of TC feeding on CAPE-induced functional damage of bile ducts, we measured basal and secretin-stimulated choleresis (a functional marker of cholangiocyte proliferation) (9, 10, 35) in the selected groups of animals. Briefly, following anesthesia with sodium pentobarbital, the selected BDI rats were surgically prepared for bile collection as described (10). When steady-state bile flow was reached, the selected BDI animal was infused with secretin (100 nM) for 30 minutes and by a final infusion of KRH for 30 minutes. Bile was collected every 10 minutes in pre-weighed tubes and immediately stored at −70°C before determination of bicarbonate concentration. Bile bicarbonate concentration (measured as total CO2) was determined by an ABL™ 520 Blood Gas System (Radiometer Medical A/S, Copenhagen, Denmark).
Purification of Cholangiocytes
In vitro experiments were performed in virtually pure (98% by γ-GT histochemistry) (36) BDL cholangiocytes isolated by immunomagnetic separation using a monoclonal antibody (IgM, kindly provided by Dr. R. Faris, Brown University, Providence, RI) against an unidentified antigen expressed by all intrahepatic cholangiocytes (37). Cell number and viability (greater than 97%) were assessed by standard Trypan blue exclusion.
In Vitro Effects of CAPE on Apoptosis and Proliferation in Purified BDL Cholangiocytes
BDL cholangiocytes were incubated at 37°C with 0.2% BSA (basal) or CAPE (40 μM) (38) for 5 hours in the absence or presence of TC (40 μM) (22), with or without pretreatment with a VEGFR-2 (SU5416: VEGF Receptor 2 Kinase Inhibitor III, Calbiochem, Gibbstown, NJ) (39) or VEGFR-3 (MAZ51: VEGF Receptor 3 Kinase Inhibitor, Calbiochem, Gibbstown, NJ) (40) inhibitor. Subsequently, we evaluated expression for PCNA and the pro-apoptotic protein Bax (34) by immunoblots (17) in protein (10 μg) from whole cell lysates. After stripping, the amount of protein loaded was normalized by immunoblots for β-actin (17). The intensity of the bands was determined by scanning video densitometry using the phospho-imager, Storm 860, (GE Healthcare, Piscataway, NJ) and the ImageQuant TL software version 2003.02 (GE Healthcare, Little Chalfont, Buckinghamshire, England).
Statistical Analysis
All data are expressed as mean ± SEM. The differences between groups were analyzed by Student's t test when two groups were analyzed, or analysis of variance (ANOVA) if more than two groups were analyzed. A P value of less than 0.05 was used to indicate statistically significant differences.
Results
Evaluation of Portal Inflammation, Necrosis, Lobular Damage and Cholangiocyte Apoptosis
There was no significant difference in lobular damage, necrosis and the degree of portal inflammation in the experimental groups found in Table 1. Figure 1 shows a representative section from each group of animals examined and no visible changes are noted. Administration of CAPE to BDL rats significantly increased the number of TUNEL-positive cholangiocytes compared to BDL controls and BDL rats fed TC (Fig. 2A and Table 2). TC feeding partly prevented CAPE-induced increases in cholangiocyte apoptosis (Fig. 2A and Table 2). The number of apoptotic cholangiocytes (Fig. 2A–B and Table 2) was significantly decreased in BDL rats fed TC and injected with CAPE compared to BDL rats treated with CAPE alone; however, this number was not as low as BDL rats treated with DMSO (controls) in the absence or presence of the TC rich diet. Consistent with previous studies (19), TC feeding did not alter cholangiocyte apoptosis compared to BDL controls (Fig. 2A and Table 2). We found similar changes in Bax expression (Fig. 2B). There was no positive staining for Bax in the BDL control or BDL + TC groups. However, after chronic administration with CAPE, Bax expression greatly increases and is consequently decreased in animals treated with CAPE and fed a TC enriched diet. These data suggest that TC partially prevents CAPE-induced cell death. CAPE did not induce damage of cholangiocytes in normal rats (not shown).
Figure 1.
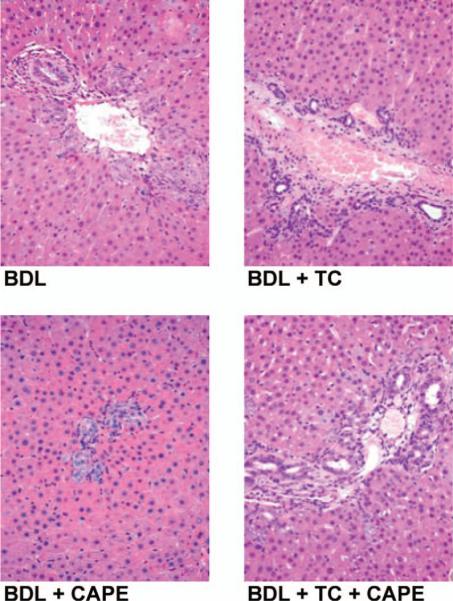
Evaluation of lobular damage, necrosis and portal inflammation by light microscopy in liver sections (4 μm thick, stained for H&E) from rats that, immediately after BDL, were fed AIN-76 (BAC) or 1% TC in the presence of daily IP injections of 0.1% DMSO or CAPE for 1 week. There was no significant difference in lobular damage, necrosis and degree of portal inflammation in the selected experimental groups. Original magnification, ×125. BAC, bile acid control; BDL, bile duct ligation; CAPE, caffeic acid phenethyl ester; DMSO, dimethyl sulfoxide; H&E, hematoxylin and eosin; TC, taurocholic acid. A color version of this figure is available in the online journal.
Figure 2.
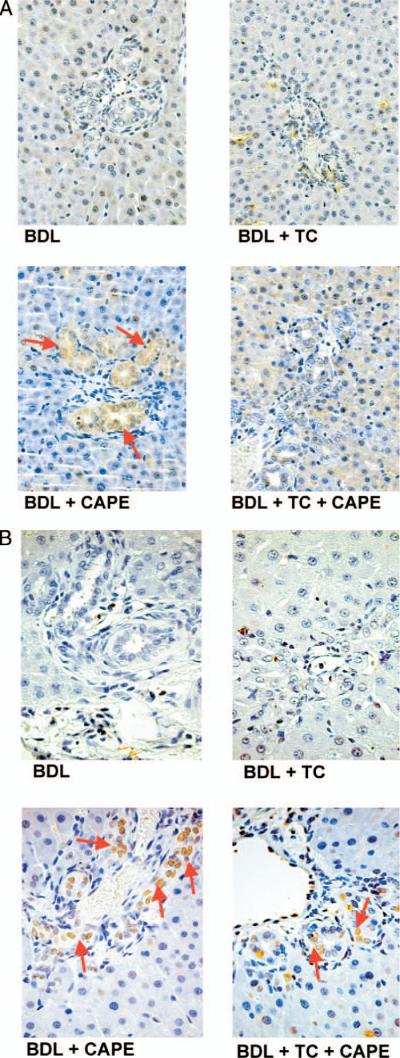
(A) Apoptosis was measured by TUNEL staining in rats that, immediately after BDL, were fed AIN-76 (BAC) or 1% TC in the presence of daily IP injections of 0.1% DMSO or CAPE for 1 week (for quantitative data see Table 2). (B) Semiquantitative immunohistochemistry for Bax in rats that, immediately after BDL, were fed AIN-76 or 1% TC in the presence of daily IP injections of 0.1% DMSO or CAPE for 1 week. Original magnification, ×250. BAC, bile acid control; BDL, bile duct ligation; CAPE, caffeic acid phenethyl ester; DMSO, dimethyl sulfoxide; TC, taurocholic acid; TUNEL, terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling. A color version of this figure is available in the online journal.
Table 2.
Evaluation of Apoptosis (TUNEL), Cholangiocyte Proliferation (PCNA) and Bile Duct Mass (CK-19) in Liver Sections from Rats that Immediately After BDL Were Fed Bile Acid Control Diet or 1% TC in the Presence of Daily IP Injections of 0.1% DMSO or CAPE for 1 Weeka
| Parameters | BDL + bile acid control feeding + DMSO | BDL + TC feeding + DMSO | BDL + bile acid control feeding + CAPE | BDL + TC feeding + CAPE |
|---|---|---|---|---|
| Cholangiocyte apoptosis | 5.00 ± 0.58 | 5.80 ± 1.08 | 63.40 ± 2.01* | 26.60 ± 2.04# |
| PCNA-positive cholangiocytes | 46.20 ± 1.43 | 48.80 ± 1.69 | 20.60 ± 1.50* | 40.20 ± 1.71# |
| CK-19-positive cholangiocytes | 5.04 ± 0.26 | 5.43 ± 0.24 | 1.48 ± 0.22* | 4.39 ± 0.25& |
BDL, bile duct ligation; CAPE, caffeic acid phenethyl ester; DMSO, dimethyl sulfoxide; TC, taurocholic acid.
P < 0.001 vs. all other groups.
P < 0.05 vs. BDL rats fed control diet or TC.
P < 0.05 vs. BDL rats fed TC.
Evaluation of Cholangiocyte Proliferation
Coupled with increased apoptosis (Fig. 2A–B and Table 2), chronic administration of CAPE to BDL rats decreased the number of PCNA- and CK-19-positive cholangiocytes in liver sections compared with BDL rats fed a bile acid control diet in combination with DMSO injections (controls) (Figs. 3 and 4 and Table 2). BDL rats fed a TC rich diet and injected with DMSO exhibited similar levels of apoptotic-, PCNA-, and CK-19-positive cholangiocytes (Figs. 3 and 4 and Table 2) compared to control rats. Similarly, in rats fed a TC rich diet in combination with CAPE treatment, the levels of the number of positive PCNA- and positive CK-19-cholangiocytes were similar to controls (Figs. 3 and 4 and Table 2) demonstrating a partial protective effect of TC against CAPE treatment. CAPE did not change the number of PCNA-positive cholangiocytes and intrahepatic ductal mass of normal rats (not shown).
Figure 3.
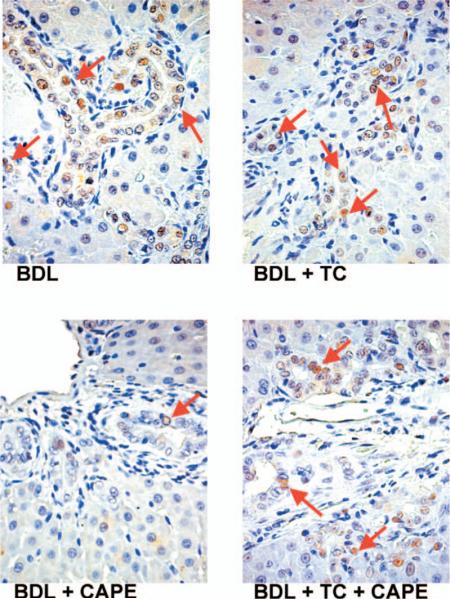
Immunohistochemistry for the number of PCNA-positive cholangiocytes in rats that, immediately after BDL, were fed AIN-76 (BAC) or 1% TC in the presence of daily IP injections of 0.1% DMSO or CAPE for 1 week (for quantitative data see Table 2). Original magnification, ×250. BAC, bile acid control; BDL, bile duct ligation; CAPE, caffeic acid phenethyl ester; DMSO, dimethyl sulfoxide; PCNA, proliferating cell nuclear antigen; TC, taurocholic acid. A color version of this figure is available in the online journal.
Figure 4.
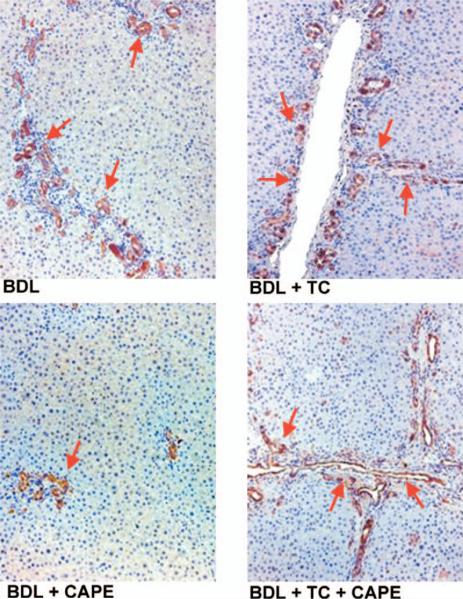
Immunohistochemistry for the number of CK-19-positive cholangiocytes in rats that, immediately after BDL, were fed AIN-76 (BAC) or 1% TC in the presence of daily IP injections of 0.1% DMSO or CAPE for 1 week (for quantitative data see Table 2). Original magnification, ×125. BAC, bile acid control; BDL, bile duct ligation; CAPE, caffeic acid phenethyl ester; CK-19, cytokeratin-19; DMSO, dimethyl sulfoxide; TC, taurocholic acid. A color version of this figure is available in the online journal.
Measurement of Ductal Bile Secretion
Basal and secretin-stimulated bile flow and secretion of control BDI rats were similar to that of previous studies (Fig. 5) (10). Consistent with the notion that CAPE induces functional damage of bile ducts, secretin failed to increase bile and bicarbonate secretion in BDI rats treated with CAPE (Fig. 5). Similar to other studies showing that TC increases biliary growth (6), we found that TC feeding to BDI rats significantly increased both basal and secretin-stimulated bile and bicarbonate secretion compared to BDI control rats (Fig. 5). Keeping with the concept that TC feeding ameliorates the functional damage of bile ducts by CAPE, in BDI rats treated with CAPE + TC there was enhanced basal and secretin-stimulated bile flow and bicarbonate secretion compared to control BDI rats (Fig. 5).
Figure 5.
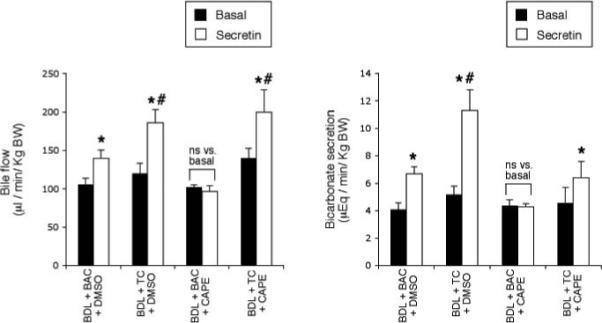
Effect of secretin on bile flow and bicarbonate secretion in rats that, immediately after BDL, were fed AIN-76 (BAC) or 1% TC in the presence of daily IP injections of 0.1% DMSO or CAPE for 1 week. * P < 0.05 vs. its corresponding value of basal bile flow and bicarbonate secretion. # P < 0.05 vs. secretin-stimulated bile flow and bicarbonate of BDL controls. BAC, bile acid control; BDL, bile duct ligation; CAPE, caffeic acid phenethyl ester; DMSO, dimethyl sulfoxide; TC, taurocholic acid. Data are mean ± SE of 6 experiments.
In Vivo Evaluation of VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3 Expression
Immunohistochemistry in liver sections showed that the expression of VEGF-A and VEGF-C (Fig. 6A) and the receptors VEGFR-2 and VEGFR-3 (Fig. 6B) was lower in cholangiocytes from BDL rats treated with CAPE compared with BDL controls and rats fed a TC enriched diet (Fig. 6A–B). TC feeding prevented the CAPE-induced decrease in VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3 expression that was similar to that of BDL control rats (Fig. 6A–B). Quantitative data for cholangiocyte VEGF protein and receptor expression is found in Figure 6C.
Figure 6.


Evaluation of VEGF-A (A), VEGF-C (A), VEGFR-2 (B), and VEGFR-3 (B) in liver sections from rats that, immediately after BDL, were fed AIN-76 (BAC) or 1% TC in the presence of daily IP injections of 0.1% DMSO or CAPE for 1 week. Original magnification, ×125. Quantitative data for the evaluation of Figures 6A and 6B are seen in Figure 6C. BAC, bile acid control; BDL, bile duct ligation; CAPE, caffeic acid phenethyl ester; DMSO, dimethyl sulfoxide; TC, taurocholic acid; VEGF, vascular endothelial growth factor. A color version of this figure is available in the online journal.
In Vitro Effects of CAPE on Cholangiocyte Apoptosis and Proliferation
Similar to our in vivo studies, in BDL cholangiocytes treated with CAPE, we found increased Bax protein expression (Fig. 7A) and decreased PCNA protein expression (Fig. 7B) compared to cholangiocytes treated with BSA. In vitro stimulation with TC alone did not change the expression of Bax (Fig. 7A), but significantly increased PCNA protein expression (Fig. 7B). CAPE-induced changes in Bax and PCNA protein expression were partly prevented by pre-treatment with TC (Fig. 7A–B). The effects of TC on CAPE-induced changes in Bax and PCNA protein expression were partially blocked by pre-incubation with VEGFR-2 or VEGFR-3 inhibitors (Fig. 7A–B). VEGFR-2 or VEGFR-3 inhibitors alone did not change either Bax or PCNA protein expression (not shown).
Figure 7.

Measurement of apoptosis (A) by Bax immunoblots and proliferation (B) by PCNA immunoblots in purified BDL cholangiocytes treated in vitro with 0.2% BSA (basal) or CAPE (40 μM) for 5 hours in the presence or absence of TC (40 μM) with or without pre-incubation with a VEGFR-2 or VEGFR-3 inhibitor. In vitro, CAPE increased (A) Bax and decreased (B) PCNA protein expression compared to cholangiocytes treated with 0.2% BSA. In vitro stimulation with TC alone did not change the expression of Bax, but significantly increased PCNA protein expression. CAPE-induced changes in Bax and PCNA protein expression were prevented by pre-treatment with TC. The effects of TC on CAPE-induced changes in Bax and PCNA protein expression were blocked by pre-incubation with VEGFR-2 or VEGFR-3 inhibitors. BDL, bile duct ligation; CAPE, caffeic acid phenethyl ester; PCNA, proliferating cell nuclear antigen; TC, taurocholic acid; VEGF, vascular endothelial growth factor. Data are mean ± SEM of 3 experiments. * P < 0.05 vs. the corresponding basal value.
Discussion
Our study demonstrates that TC, both in vivo and in vitro, partially prevents CAPE-induced cholangiocyte functional damage by changes in cholangiocyte VEGF expression. Specifically, we have shown that: (i) chronic treatment of BDL rats with CAPE increases apoptosis, and reduces cholangiocyte proliferation and secretin-stimulated ductal secretion; (ii) CAPE-induced changes in cholangiocyte apoptosis and proliferation are associated with reduced expression of VEGF-A, VEGF-C, VEGFR-2, and VEGFR-3; (iii) TC feeding partly prevents CAPE-induced changes in cholangiocyte apoptosis and proliferation, and maintains VEGF-A, VEGF-C, VEGFR-2, and VEGFR-3 expression; (iv) TC feeding reverses the CAPE-induced functional damage of bile ducts; and (v) in purified BDL cholangiocytes, TC moderately prevents CAPE-induced effects on cholangiocyte apoptosis and proliferation by sustaining the VEGF system.
Cholangiocyte proliferation is a typical hallmark of cholangiopathies that induce chronic damage of the biliary tree with dysregulation of the balance between the survival and death of cholangiocytes (9, 33, 41). Cholangiocyte proliferation in the course of cholangiopathies compensates for the loss of injured ducts (9, 10, 33); in fact, proliferating cholangiocytes show higher secretory activity (4, 10, 42), which is fundamental for maintaining a normal ductal secretion despite the loss of damaged bile ducts (9, 33). The cholestatic BDL rat model, which mimics the pathological conditions observed in human cholangiopathies (3, 10), is characterized by enhanced cholangiocyte proliferation (3, 10) and is widely used for evaluating the intracellular mechanisms of cholangiocyte proliferation/loss.
In the rat liver, CAPE treatment prevents CCl4-induced damage (43), inhibits proliferation and collagen synthesis of stellate cells at lower concentrations, and induces stellate cell apoptosis at high concentrations (12). Similar to BDL vagotomized rats (33), CAPE induced an increase in cholangiocyte cell death that was coupled with decreased cholangiocyte proliferation after chronic and acute stimulation. CAPE-induced cholangiocyte damage was achieved without changes in portal inflammation, hepatic damage and necrosis. These data suggest that CAPE could be used therapeutically to treat pathological conditions of abnormal cholangiocyte proliferation such as those observed in autosomal dominant polycystic kidney disease (ADPKD) (44). The data also suggest that CAPE may be an important therapeutic approach for decreasing ductular reaction in chronic liver diseases as a strategy to delay biliary fibrosis given the link between ductular reaction and biliary fibrosis (45).
Many studies have shown that cholangiocyte biology is influenced by bile acids including TC, ursodeoxycholate (UDCA) and tauroursodeoxycholate (TUDCA) (6, 18, 22). TC is not only a potent mitogenic stimulus (6), but is required for the proliferative response by cholangiocytes to cholestasis, as demonstrated by the fact that depletion of endogenous pools of TC reduces cholangiocyte growth and secretin-stimulated ductal secretion (23). We have previously demonstrated that TC administration is protective against cholangiocyte apoptosis induced by acute administration of CCl4 (6), and sympathetic (20) or adrenergic denervation (19).
Cholangiocyte proliferation is also regulated by neuropeptides, hormones and growth factors, including VEGF (17, 29). Cholangiocytes secrete VEGF-A and VEGF-C and express VEGFR-2 (Flk-1) and VEGFR-3 (Flt-4), all of which are amplified in BDL cholangiocytes (26, 29). Therefore, VEGF participates in the adaptive proliferative and apoptotic response of cholangiocytes to cholestasis by autocrine mechanisms (26, 29). For example, Gaudio, et al. found that administration of anti-VEGF-A or anti-VEGF-C antibodies (which neutralizes VEGF and reduces circulating levels of VEGF) decreases BDL-induced hyperplasia, thus confirming the important role of VEGF in the balance between cholangiocyte proliferation and loss (26, 29). Important studies have revealed that VEGF regulates cholangiocyte growth, including a study that has shown that cholangiocytes are able to produce a VEGF gradient demonstrated by an increasing amount of VEGF expression as bile ducts develop (46).
The effects of bile acids on VEGF expression have been shown mainly in cancerous cell types where bile acids affect angiogenesis thus altering cell growth, such as in human colon cancer in which deoxycholic acid significantly enhances the angiogenic potential of the tumor (47) or the promoting role of chenodeoxycholic acid in the development of human esophageal cancer supported by a higher production of VEGF, via the COX-2 pathway (48). VEGF has been shown to play a role in the cholangiocyte proliferative response (26, 29); however, the effects of TC on VEGF expression have not been shown in the context of the regulation of cholangiocyte hyperplasia. Here, we propose that the in vivo and in vitro TC protective effects on CAPE-induced damage of the biliary epithelium are due to changes in cholangiocyte VEGF expression.
In our final set of experiments, we set out to determine if the observed in vivo effects of CAPE and TC + CAPE are specific to cholangiocytes and if acute treatment would elicit similar results. Similar (although not as dramatic) to in vivo results, we found that acute CAPE stimulation in vitro decreased proliferation coupled with increased apoptosis that is blocked by inhibition of VEGF receptor activation. Gaudio, et al. have previously shown that there is an autocrine regulation of cholangiocyte VEGF expression (26, 29). The findings in our study lend support to the concept that autocrine VEGF regulation may be modulated by bile acids in cholangiocytes and that our findings in vivo can be duplicated in vitro.
Other studies support the concept that the activation of the VEGF system represents a protective mechanism during experimental liver damage (29). Our data support these findings, in particular the concept that bile acids, like TC, and VEGF may play an important role to maintain the balance between loss and growth in cholangiopathies. In conclusion, our findings may provide novel perspectives in the regulation of cholangiocyte loss in chronic cholestatic liver diseases via manipulation of cholangiocyte VEGF expression.
Acknowledgments
We sincerely thank Dr. Gianfranco Alpini for his assistance and expert advice in the development of this manuscript. Also, we thank Ms. Julie Venter for her assistance with the animal studies.
The study was supported by a grant award from Scott & White to Heather Francis, the Dr. Nicholas C. Hightower Centennial Chair of Gastroenterology from Scott & White, a grant award from Health and Labour Sciences Research Grants for the Research on Measures for Intractable Diseases (from the Ministry of Health, Labour and Welfare of Japan) and from a Grant-in-Aid for Scientific Research C (19590744) from JSPS to Dr. Ueno, by University funds to Dr. Onori and PRIN 2007 and Federate Athenaeum funds from University of Rome “La Sapienza” to Prof. Gaudio and Prof. Alvaro, by MIUR (PRIN) grants: #2007, prot. HPT7BA_003 to Prof. Alvaro and an NIH K01 grant award (DK078532) to Dr. DeMorrow.
References
- 1.Benedetti A, Bassotti C, Rapino K, Marucci L, Jezequel AM. A morphometric study of the epithelium lining the rat intrahepatic biliary tree. J Hepatol. 1996;24:335–342. doi: 10.1016/s0168-8278(96)80014-6. [DOI] [PubMed] [Google Scholar]
- 2.Alpini G, Roberts S, Kuntz SM, Ueno Y, Gubba S, Podila PV, LeSage G, LaRusso NF. Morphological, molecular, and functional heterogeneity of cholangiocytes from normal rat liver. Gastroenterology. 1996;110:1636–1643. doi: 10.1053/gast.1996.v110.pm8613073. [DOI] [PubMed] [Google Scholar]
- 3.Alpini G, Glaser S, Ueno Y, Pham L, Podila PV, Caligiuri A, LeSage G, LaRusso NF. Heterogeneity of the proliferative capacity of rat cholangiocytes after bile duct ligation. Am J Physiol Gastrointest Liver Physiol. 1998;274:G767–775. doi: 10.1152/ajpgi.1998.274.4.G767. [DOI] [PubMed] [Google Scholar]
- 4.LeSage G, Glaser S, Gubba S, Robertson WE, Phinizy JL, Lasater J, Rodgers RE, Alpini G. Regrowth of the rat biliary tree after 70% partial hepatectomy is coupled to increased secretin-induced ductal secretion. Gastroenterology. 1996;111:1633–1644. doi: 10.1016/s0016-5085(96)70027-6. [DOI] [PubMed] [Google Scholar]
- 5.Alpini G, Prall RT, LaRusso NF. The pathobiology of biliary epithelia. In: Arias IM, Boyer JL, Chisari FV, Fausto N, Jakoby W, Schachter D, Shafritz DA, editors. The Liver; Biology & Pathobiology. 4E. Lippincott Williams & Wilkins; Philadelphia, PA: 2001. pp. 421–435. [Google Scholar]
- 6.Alpini G, Glaser S, Ueno Y, Rodgers R, Phinizy JL, Francis H, Baiocchi L, Holcomb LA, Caligiuri A, LeSage G. Bile acid feeding induces cholangiocyte proliferation and secretion: evidence for bile acid-regulated ductal secretion. Gastroenterology. 1999;116:179–186. doi: 10.1016/s0016-5085(99)70242-8. [DOI] [PubMed] [Google Scholar]
- 7.Marzioni M, Francis H, Benedetti A, Ueno Y, Fava G, Venter J, Reichenbach R, Mancino MG, Summers R, Alpini G, Glaser S. Ca2+-dependent cytoprotective effects of ursodeoxycholic and tauroursodeoxycholic acid on the biliary epithelium in a rat model of cholestasis and loss of bile ducts. Am J Pathol. 2006;168:398–409. doi: 10.2353/ajpath.2006.050126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LeSage G, Benedetti A, Glaser S, Marucci L, Tretjak Z, Caligiuri A, Rodgers R, Phinizy JL, Baiocchi L, Francis H, Lasater J, Ugili L, Alpini G. Acute carbon tetrachloride feeding selectively damages large, but not small, cholangiocytes from normal rat liver. Hepatology. 1999;29:307–319. doi: 10.1002/hep.510290242. [DOI] [PubMed] [Google Scholar]
- 9.LeSage G, Glaser S, Marucci L, Benedetti A, Phinizy JL, Rodgers R, Caligiuri A, Papa E, Tretjak Z, Jezequel AM, Holcomb LA, Alpini G. Acute carbon tetrachloride feeding induces damage of large but not small cholangiocytes from BDL rat liver. Am J Physiol Gastrointest Liver Physiol. 1999;276:G1289–G1301. doi: 10.1152/ajpgi.1999.276.5.G1289. [DOI] [PubMed] [Google Scholar]
- 10.Alpini G, Lenzi R, Sarkozi L, Tavoloni N. Biliary physiology in rats with bile ductular cell hyperplasia. Evidence for a secretory function of proliferated bile ductules. J Clin Invest. 1988;81:569–578. doi: 10.1172/JCI113355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvaro D, Invernizzi P, Onori P, Franchitto A, De Santis A, Crosignani A, Sferra R, Ginanni-Corradini S, Mancino MG, Maggioni M, Attili AF, Podda M, Gaudio E. Estrogen receptors in cholangiocytes and the progression of primary biliary cirrhosis. J Hepatol. 2004;41:905–912. doi: 10.1016/j.jhep.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 12.Zhao WX, Zhao J, Liang CL, Zhao B, Pang RQ, Pan XH. Effect of caffeic acid phenethyl ester on proliferation and apoptosis of hepatic stellate cells in vitro. World J Gastroenterol. 2003;9:1278–1281. doi: 10.3748/wjg.v9.i6.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagaoka T, Banskota AH, Tezuka Y, Harimaya Y, Koizumi K, Saiki I, Kadota S. Inhibitory effects of caffeic acid phenethyl ester analogues on experimental lung metastasis of murine colon 26-L5 carcinoma cells. Biol Pharm Bull. 2003;26:638–641. doi: 10.1248/bpb.26.638. [DOI] [PubMed] [Google Scholar]
- 14.Chung TW, Moon SK, Chang YC, Ko JH, Lee YC, Cho G, Kim SH, Kim JG, Kim CH. Novel and therapeutic effect of caffeic acid and caffeic acid phenyl ester on hepatocarcinoma cells: complete regression of hepatoma growth and metastasis by dual mechanism. FASEB J. 2004;18:1670–1681. doi: 10.1096/fj.04-2126com. [DOI] [PubMed] [Google Scholar]
- 15.Alpini G, Glaser S, Rodgers R, Phinizy JL, Robertson WE, Lasater J, Caligiuri A, Tretjak Z, LeSage G. Functional expression of the apical Na+-dependent bile acid transporter in large but not small rat cholangiocytes. Gastroenterology. 1997;113:1734–1740. doi: 10.1053/gast.1997.v113.pm9352879. [DOI] [PubMed] [Google Scholar]
- 16.Lazaridis KN, Pham L, Tietz P, Marinelli RA, deGroen PC, Levine S, Dawson PA, LaRusso NF. Rat cholangiocytes absorb bile acids at their apical domain via the ileal sodium-dependent bile acid transporter. J Clin Invest. 1997;100:2714–2721. doi: 10.1172/JCI119816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alpini G, Ueno Y, Glaser S, Marzioni M, Phinizy JL, Francis H, LeSage G. Bile acid feeding increased proliferative activity and apical bile acid transporter expression in both small and large rat cholangiocytes. Hepatology. 2001;34:868–876. doi: 10.1053/jhep.2001.28884. [DOI] [PubMed] [Google Scholar]
- 18.Alpini G, Baiocchi L, Glaser S, Ueno Y, Marzioni M, Francis H, Phinizy JL, Angelico M, LeSage G. Ursodeoxycholate and tauroursodeoxycholate inhibit cholangiocyte growth and secretion of BDL rats through activation of PKC alpha. Hepatology. 2002;35:1041–1052. doi: 10.1053/jhep.2002.32712. [DOI] [PubMed] [Google Scholar]
- 19.Marucci L, Alpini G, Glaser S, Alvaro D, Benedetti A, Francis H, Phinizy JL, Marzioni M, Mauldin J, Venter J, Baumann B, Ugili L, LeSage G. Taurocholate feeding prevents CCl4-induced damage of large cholangiocytes through PI3-kinase-dependent mechanism. Am J Physiol Gastrointest Liver Physiol. 2003;284:G290–G301. doi: 10.1152/ajpgi.00245.2002. [DOI] [PubMed] [Google Scholar]
- 20.Marzioni M, Ueno Y, Glaser S, Francis H, Benedetti A, Alvaro D, Venter J, Fava G, Alpini G. Cytoprotective effects of taurocholic acid feeding on the biliary tree after adrenergic denervation of the liver. Liver Int. 2007;27:558–568. doi: 10.1111/j.1478-3231.2007.01443.x. [DOI] [PubMed] [Google Scholar]
- 21.Marzioni M, LeSage G, Glaser S, Patel T, Marienfeld C, Ueno Y, Francis H, Alvaro D, Tadlock L, Benedetti A, Marucci L, Baiocchi L, Phinizy JL, Alpini G. Taurocholate prevents the loss of intrahepatic bile ducts due to vagotomy in bile duct-ligated rats. Am J Physiol Gastrointest Liver Physiol. 2003;284:G837–G852. doi: 10.1152/ajpgi.00398.2002. [DOI] [PubMed] [Google Scholar]
- 22.Alpini G, Glaser S, Robertson W, Phinizy JL, Rodgers RE, Caligiuri A, LeSage G. Bile acids stimulate proliferative and secretory events in large but not small cholangiocytes. Am J Physiol Gastrointest Liver Physiol. 1997;273:G518–G529. doi: 10.1152/ajpgi.1997.273.2.G518. [DOI] [PubMed] [Google Scholar]
- 23.Alpini G, Glaser S, Alvaro D, Ueno Y, Marzioni M, Francis H, Baiocchi L, Stati T, Barbaro B, Phinizy JL, Mauldin J, LeSage G. Bile acid depletion and repletion regulate cholangiocyte growth and secretion by a phosphatidylinositol 3-kinase-dependent pathway in rats. Gastroenterology. 2002;123:1226–1237. doi: 10.1053/gast.2002.36055. [DOI] [PubMed] [Google Scholar]
- 24.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 25.Larrivee B, Karsan A. Signaling pathways induced by vascular endothelial growth factor (review). Int J Mol Med. 2000;5:447–456. doi: 10.3892/ijmm.5.5.447. [DOI] [PubMed] [Google Scholar]
- 26.Gaudio E, Barbaro B, Alvaro D, Glaser S, Francis H, Ueno Y, Meininger CJ, Franchitto A, Onori P, Marzioni M, Taffetani S, Fava G, Stoica G, Venter J, Reichenbach R, DeMorrow S, Summers R, Alpini G. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology. 2006;130:1270–1282. doi: 10.1053/j.gastro.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 27.Schoeffner DJ, Matheny SL, Akahane T, Factor V, Berry A, Merlino G, Thorgeirsson UP. VEGF contributes to mammary tumor growth in transgenic mice through paracrine and autocrine mechanisms. Lab Invest. 2005;85:608–623. doi: 10.1038/labinvest.3700258. [DOI] [PubMed] [Google Scholar]
- 28.Sartelet H, Decaussin M, Devouassoux G, Nawrocki-Raby B, Brichon PY, Brambilla C, Brambilla E. Expression of vascular endothelial growth factor (VEGF) and its receptors (VEGF-R1 [Flt-1] and VEGF-R2 [KDR/Flk-1]) in tumorlets and in neuroendocrine cell hyperplasia of the lung. Hum Pathol. 2004;35:1210–1217. doi: 10.1016/j.humpath.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 29.Gaudio E, Barbaro B, Alvaro D, Glaser S, Francis H, Franchitto A, Onori P, Ueno Y, Marzioni M, Fava G, Venter J, Reichenbach R, Summers R, Alpini G. Administration of r-VEGF-A prevents hepatic artery ligation-induced bile duct damage in bile duct ligated rats. Am J Physiol Gastrointest Liver Physiol. 2006;291:G307–G317. doi: 10.1152/ajpgi.00507.2005. [DOI] [PubMed] [Google Scholar]
- 30.Alpini G, Ulrich C, Roberts S, Phillips JO, Ueno Y, Podila PV, Colegio O, LeSage G, Miller LJ, LaRusso NF. Molecular and functional heterogeneity of cholangiocytes from rat liver after bile duct ligation. Am J Physiol Gastrointest Liver Physiol. 1997;272:G289–G297. doi: 10.1152/ajpgi.1997.272.2.G289. [DOI] [PubMed] [Google Scholar]
- 31.Park JH, Lee JK, Kim HS, Chung ST, Eom JH, Kim KA, Chung SJ, Paik SY, Oh HY. Immunomodulatory effect of caffeic acid phenethyl ester in Balb/c mice. Int Immunopharmacol. 2004;4:429–436. doi: 10.1016/j.intimp.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 32.Taffetani S, Glaser S, Francis H, DeMorrow S, Ueno Y, Alvaro D, Marucci L, Marzioni M, Fava G, Venter J, Vaculin S, Vaculin B, Lam IP, Lee VH, Gaudio E, Carpino G, Benedetti A, Alpini G. Prolactin stimulates the proliferation of normal female cholangiocytes by differential regulation of Ca2+-dependent PKC isoforms. BMC Physiol. 2007;7:6. doi: 10.1186/1472-6793-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.LeSage G, Alvaro D, Benedetti A, Glaser S, Marucci L, Baiocchi L, Eisel W, Caligiuri A, Phinizy JL, Rodgers R, Francis H, Alpini G. Cholinergic system modulates growth, apoptosis, and secretion of cholangiocytes from bile duct-ligated rats. Gastroenterology. 1999;117:191–199. doi: 10.1016/s0016-5085(99)70567-6. [DOI] [PubMed] [Google Scholar]
- 34.Sheng G, Guo J, Warner BW. Epidermal growth factor receptor signaling modulates apoptosis via p38alpha MAPK-dependent activation of Bax in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2007;293:G599–G606. doi: 10.1152/ajpgi.00182.2007. [DOI] [PubMed] [Google Scholar]
- 35.Glaser S, Rodgers RE, Phinizy JL, Robertson WE, Lasater J, Caligiuri A, Tretjak Z, LeSage G, Alpini G. Gastrin inhibits secretin-induced ductal secretion by interaction with specific receptors on rat cholangiocytes. Am J Physiol Gastrointest Liver Physiol. 1997;273:G1061–G1070. doi: 10.1152/ajpgi.1997.273.5.G1061. [DOI] [PubMed] [Google Scholar]
- 36.Rutenburg AM, Kim H, Fischbein JW, Hanker JS, Wasserkrug HL, Seligman AM. Histochemical and ultrastructural demonstration of g-glutamyl transpeptidase activity. J Histochem Cytochem. 1969;17:517–526. doi: 10.1177/17.8.517. [DOI] [PubMed] [Google Scholar]
- 37.Ishii M, Vroman B, LaRusso NF. Isolation and morphologic characterization of bile duct epithelial cells from normal rat liver. Gastroenterology. 1989;97:1236–1247. doi: 10.1016/0016-5085(89)91695-8. [DOI] [PubMed] [Google Scholar]
- 38.Chen MF, Keng PC, Lin PY, Yang CT, Liao SK, Chen WC. Caffeic acid phenethyl ester decreases acute pneumonitis after irradiation in vitro and in vivo. BMC Cancer. 2005;5:158. doi: 10.1186/1471-2407-5-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tille JC, Wang X, Lipson KE, McMahon G, Ferrara N, Zhu Z, Hicklin DJ, Sleeman JP, Eriksson U, Alitalo K, Pepper MS. Vascular endothelial growth factor (VEGF) receptor-2 signaling mediates VEGF-C(deltaNdeltaC)- and VEGF-A-induced angiogenesis in vitro. Exp Cell Res. 2003;285:286–298. doi: 10.1016/s0014-4827(03)00053-3. [DOI] [PubMed] [Google Scholar]
- 40.Kirkin V, Mazitschek R, Krishnan J, Steffen A, Waltenberger J, Pepper MS, Giannis A, Sleeman JP. Characterization of indolinones which preferentially inhibit VEGF-C- and VEGF-D-induced activation of VEGFR-3 rather than VEGFR-2. Eur J Biochem. 2001;268:5530–5540. doi: 10.1046/j.1432-1033.2001.02476.x. [DOI] [PubMed] [Google Scholar]
- 41.Lazaridis KN, Strazzabosco M, LaRusso NF. The cholangiopathies: disorders of biliary epithelia. Gastroenterology. 2004;127:1565–1577. doi: 10.1053/j.gastro.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 42.Alpini G, Lenzi R, Zhai WR, Slott PA, Liu MH, Sarkozi L, Tavoloni N. Bile secretory function of intrahepatic biliary epithelium in the rat. Am J Physiol Gastrointest Liver Physiol. 1989;257:G124–G133. doi: 10.1152/ajpgi.1989.257.1.G124. [DOI] [PubMed] [Google Scholar]
- 43.Kus I, Colakoglu N, Pekmez H, Seckin D, Ogeturk M, Sarsilmaz M. Protective effects of caffeic acid phenethyl ester (CAPE) on carbon tetrachloride-induced hepatotoxicity in rats. Acta Histochem. 2004;106:289–297. doi: 10.1016/j.acthis.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 44.Alvaro D, Onori P, Alpini G, Franchitto A, Jefferson DM, Torrice A, Cardinale V, Stefanelli F, Mancino MG, Strazzabosco M, Angelico M, Attili A, Gaudio E. Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. Am J Pathol. 2008;172:321–332. doi: 10.2353/ajpath.2008.070293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alvaro D, Mancino MG, Glaser S, Gaudio E, Marzioni M, Francis H, Alpini G. Proliferating cholangiocytes: a neuroendocrine compartment in the diseased liver. Gastroenterology. 2007;132:415–431. doi: 10.1053/j.gastro.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 46.Fabris L, Cadamuro M, Libbrecht L, Raynaud P, Spirli C, Fiorotto R, Okolicsanyi L, Lemaigre F, Strazzabosco M, Roskams T. Epithelial expression of angiogenic growth factors modulate arterial vasculo-genesis in human liver development. Hepatology. 2008;47:719–728. doi: 10.1002/hep.22015. [DOI] [PubMed] [Google Scholar]
- 47.Kim HS, Lee YK, Kim JW, Baik SK, Kwon SO, Jang HI. [Modulation of colon cancer cell invasiveness induced by deoxycholic acid]. Korean J Gastroenterol. 2006;48:9–18. [PubMed] [Google Scholar]
- 48.Soma T, Kaganoi J, Kawabe A, Kondo K, Tsunoda S, Imamura M, Shimada Y. Chenodeoxycholic acid stimulates the progression of human esophageal cancer cells: a possible mechanism of angiogenesis in patients with esophageal cancer. Int J Cancer. 2006;119:771–782. doi: 10.1002/ijc.21917. [DOI] [PubMed] [Google Scholar]