

Abstract
The therapeutic potential of neurotrophic factors has been hampered by their inability to achieve adequate tissue penetration. Brain blood vessels, however, could be an alternative target for neuro-salvage therapies by virtue of their close proximity to neurons. Here we show that hemizygous deletion of Rac1 in mouse endothelial cells (ECs) attenuates brain injury and edema after focal cerebral ischemia. Microarray analysis of Rac1+/− ECs revealed enrichment of stress response genes, basement membrane components, and neurotrophic factors that could affect neuronal survival. Consistent with these expression profiles, endothelial Rac1 hemizygosity enhanced antioxidative and endothelial barrier capacities and potentiated paracrine neuroprotective activities through the up-regulation of the neurotrophic factor, artemin. Endothelial Rac1, therefore, could be an important therapeutic target for promoting endothelial barrier integrity and neurotrophic activity.
INTRODUCTION
Vascular endothelial cells (ECs) are emerging targets for neuroprotection (1). For the maintenance of vascular integrity, ECs, together with the underlying basal lamina and astrocyte endfeet constitute the blood-brain barrier (BBB) (2). In the early phases of many neurological diseases and ischemic stroke, increased production of reactive oxygen species (ROS) and activation of proteases cause disruption of the BBB and transudation of plasma components. This leads to edema, which exacerbates neuronal damage (3). Therefore, protection of BBB integrity is an important component of neuroprotective therapies. The small guanosine triphosphatase (GTPase) Rac1 is a critical mediator of various aspects of EC function. In addition to its effects on gene expression, Rac1 regulates ROS production, endothelial permeability, and cell adhesion (4–6), all of which have been implicated in the deterioration of neurovascular integrity in neurological diseases.
In animal studies, neurotrophic factors prevent the neuronal death associated with various neurological disorders. However, the efficacy of these agents in humans has been limited by their inability to penetrate target neural tissues, necessitating their invasive delivery (7–9). Emerging evidence suggests that neurons, glial cells, pericytes, and ECs, by virtue of their close proximity, form a topographical compartment termed the neurovascular unit, in which they are functionally coupled to maintain brain homeostasis (10, 11). This functional coupling involves reciprocal trophic signaling. Indeed, recent evidence indicates that ECs can support neuronal survival through paracrine signals (12, 13). Given the pharmacological accessibility of vascular endothelium in vivo, the exploitation of EC-derived neurotrophic activity could provide a feasible strategy for safe and efficacious neuroprotection. However, this approach has been hampered by the lack of information on the regulatory mechanisms that direct EC paracrine signaling and coordinate them with EC-mediated vascular functions. In this study, we tested the hypothesis that endothelial Rac1 provides a therapeutic target for promoting vascular integrity and neuronal survival.
RESULTS
Neuroprotection in endothelial Rac1 haploinsufficient mice
To determine the endothelium-specific role of Rac1, we generated mice with conditional deletion of the Rac1 gene in ECs (14). Rac1 null mutation in ECs was achieved by crossing Tie2 promoter and enhancer-driven Cre Tg with Rac1 floxed allele knock-in mice (15, 16). Because EC-Rac1 null homozygosity results in embryonic lethality, we used EC-Rac1 haploinsufficient (EC-Rac1+/−) and control (Rac1flox/+) mice. The EC-Rac1+/− mice showed a 50% decrease in endothelial Rac1 protein abundance and activity; however, body and organ development was similar to that of control mice.
To investigate the effect of reducing endothelial Rac1 on neuroprotection, mice were subjected to 2-hour middle cerebral artery occlusion (MCAo) followed by 22-hour reperfusion, a model of acute nonthrombotic focal ischemia (Fig. 1A) (17). Infarct size, which reflects the extent of ischemic cell death, was 37% smaller in EC-Rac1+/− mice than in controls (Fig. 1B). Edema volume, which depends on BBB disruption, was 63% smaller in EC-Rac1+/− mice than in controls (Fig. 1C). Consistent with these anatomical findings, neurological deficits were less severe in EC-Rac1+/− mice than in control mice (Fig. 1D). Neuroprotection in the transient MCAo model is thought to depend on two major factors: residual cerebral blood flow (CBF) in the ischemic region and CBF-independent, direct neuroprotective components, such as BBB functionality and growth factor–mediated neurosurvival. The decrease in CBF in the ischemic hemisphere during MCAo was comparable in EC-Rac1+/− and control mice (Fig. 1E). Furthermore, the mean arterial blood pressure and blood gas profiles in the two groups were similar (Table 1).
Fig. 1.
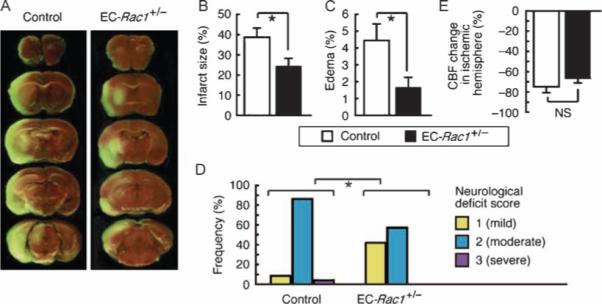
Haploinsufficiency of endothelial Rac1 in mice leads to neuroprotection. EC-Rac1+/− and control mice underwent 2-hour MCAo followed by 22-hour reperfusion. (A) Representative brain sections stained with 2,3,5-triphenyltetrazolium chloride visualizing the infarct area. (B and C) Infarct (B) and edema (C) volumes of the ischemic hemisphere. (D) Neurological deficit score (n = 19 to 23 mice; *P < 0.05, Mann-Whitney test). (E) Change in CBF in the ischemic hemisphere at the end of 2-hour MCAo. (B, C, and E) Values are percent of the nonischemic hemisphere and shown as means ± SEM of 7 to 10 mice (*P < 0.05, ANOVA). NS, not significant.
Table 1.
Hemodynamic profile and arterial blood gas analysis. MABP, mean arterial blood pressure; rCBF (regional cerebral blood flow) and blood gas were measured under inhalation anesthesia in the MCAo perioperative period. Pre, before MCAo; Post, after MCAo; Paco2, partial pressure of arterial CO2; Pao2, partial pressure of arterial O2. Control, n = 7 mice; EC-Rac1+/−, n = 6 mice.
| Hemodynamic parameter | Control | EC-Rac1+/− |
|---|---|---|
| MABP (mmHg) | ||
| Pre | 91.1 ± 4.3 | 99.8 ± 3.2 |
| Post | 95.8 ± 6.0 | 102.1 ± 5.0 |
| rCBF (%) | ||
| Pre | 102.6 ± 2.5 | 102.6 ± 1.5 |
| MCAo | 8.3 ± 1.1 | 6.0 ± 1.0 |
| pH (arterial) | ||
| Pre | 7.40 ± 0.01 | 7.37 ± 0.04 |
| MCAo | 7.29 ± 0.02 | 7.25 ± 0.04 |
| Post | 7.28 ± 0.03 | 7.23 ± 0.03 |
| Paco2 (mmHg) | ||
| Pre | 29.3 ± 1.1 | 30.4 ± 3.5 |
| MCAo | 32.9 ± 3.2 | 35.5 ± 3.5 |
| Post | 48.6 ± 4.4 | 51.9 ± 4.2 |
| Pao2 (mmHg) | ||
| Pre | 120.6 ± 6.9 | 120.3 ± 16.9 |
| MCAo | 124.9 ± 9.2 | 127.5 ± 12.7 |
| Post | 127.1 ± 13.1 | 119.6 ± 7.1 |
Expression profiling of Rac1 haploinsufficient ECs
The independence from differences in blood flow of the reduction of infarct size in EC-Rac1+/− mice compared to controls suggested that decreasing EC Rac1 had a direct neuroprotective effect. To explore this possibility, we performed a genome-wide microarray analysis in which we compared Rac1+/− and Rac1+/+ mouseprimary ECs. The integrity of the EC preparation was verified by >98% PECAM-1 (platelet endothelial cell adhesion molecule 1)–positive labeling and the 50% reduction of total and active Rac1 abundance in Rac1+/− versus Rac1+/+ ECs (Fig. 2A and fig. S1). Tetraplicate hybridizations on the Massachusetts General Hospital 19,549 oligo chips revealed differential expression of 299 gene features (∼1.5%) in Rac1+/− versus Rac1+/+ ECs (P < 0.05, fold increase greater than 1.5 or less than −1.5) (Fig. 2, B and C, and table S4). To characterize the global features of these genes, we assessed functional annotation clustering of genes whose expression was down-regulated or up-regulated in Rac1+/− ECs (Fig. 2, D and E) (18). Additionally, the small but cumulatively significant difference in gene expression between Rac1+/− and Rac1+/+ ECs across the whole array data was determined by enrichment of a priori–defined gene sets [gene set enrichment analysis (GSEA); false discovery rate <25%; see Supplementary Materials and Methods] (Fig. 2, F and G, and tables S1 and S2) (19). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of selected genes showed a good correlation (R = 0.859) between the fold increase measured by microarray and that measured by qRT-PCR (Fig. 2, H and I). In addition, crystallin αB immunolabeling was more intense with Rac1+/− than with Rac1+/+ ECs, consistent with the 4.24-fold up-regulation of the crystallin αB (Cryab) messenger RNA (mRNA) seen in the microarray data (Fig. 2J).
Fig. 2.
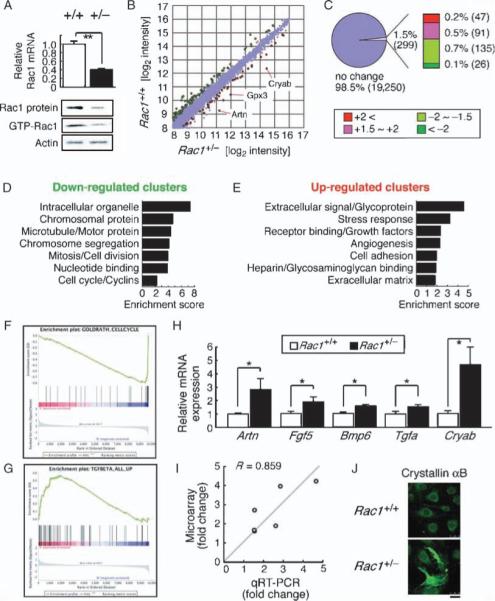
Expression profiling of Rac1+/− ECs. Differential mRNA expression between Rac1+/− and Rac1+/+ mouse ECs was explored by microarray analysis. (A) qRT-PCR, immunoblotting, and GST-PAK (glutathione S-transferase–p21-activated protein kinase) pull-down assays showing Rac1 expression and activity in Rac1+/− and Rac1+/+ ECs. (B and C) The up-regulated (red) and down-regulated (green) genes in Rac1+/− ECs. The scatter plot (B) shows 12,089 gene features with the signal intensity >200. (D and E) Functional annotation clustering of the down-regulated (D) and up-regulated (E) genes. (F and G) GSEA showing enrichment of cell cycle–related genes in Rac1+/+ (F) and TGF-β–inducible genes in Rac1+/− ECs (G). Each plot displays (top) progression of the running enrichment score; (middle) “hits” in the gene set against the ranked list of all genes in the data; (bottom) histogram for the ranked list. (H and I) qRT-PCR (H) and correlation analysis (I) to validate the microarray data. (J) Crystallin αB immunostaining of Rac1+/− versus Rac1+/+ ECs. Scale bar, 25 μm. Values are means ± SEM of three to four replicates (*P < 0.05, **P < 0.01; ANOVA).
Functional clustering and GSEA revealed that many genes whose expression was decreased in Rac1+/− ECs are associated with mitotic cell division (Fig. 2, D and F, Supplementary text, Table 2, and tables S1 and S3). The expression of many of these genes is tightly controlled in a cell cycle–dependent fashion, suggesting that their down-regulation represents an overall growth-inhibitory state of Rac1+/− ECs. Indeed, Rac1+/− ECs exhibited decreased proliferation and capillary formation compared to Rac1+/+ ECs (fig. S2A). These findings are consistent with the recognized roles of Rac1 in mediating cell cycle progression, further validating our microarray data (20).
Table 2.
Selected genes down-regulated in Rac1+/− mouse ECs. Genes are categorized by the GO annotation.
| Symbol | Gene name | Fold |
|---|---|---|
| Cell cycle | ||
| Fos | FBJ osteosarcoma oncogene | −3.02 |
| Myb | Myeloblastosis oncogene | −1.57 |
| Cdc2a | Cell division cycle 2 homolog A (Schizosaccharomyces pombe) | −2.11 |
| Ccnb2 | Cyclin B2 | −1.88 |
| Ccna2 | Cyclin A2 | −1.59 |
| Nek2 | NIMA (never in mitosis gene a)–related expressed kinase 2 | −1.85 |
| Mad2l1 | MAD2 (mitotic arrest deficient, homolog)–like 1 (yeast) | −1.60 |
| Cdca5 | Cell division cycle associated 5 | −1.54 |
| Stk6 | Aurora kinase A | −1.64 |
| Tacc3 | Transforming, acidic coiled-coil containing protein 3 | −1.93 |
| Bard1 | BRCA1 associated RING domain 1 | −1.57 |
| Gadd45g | Growth arrest and DNA-damage–inducible 45 gamma | −1.70 |
| Ect2 | ect2 oncogene | −1.65 |
| Mki67 | Ki-67 | −2.23 |
| Cytoskeleton organization and biogenesis | ||
| Kif2c | Kinesin family member 2C | −1.65 |
| Kif11 | Kinesin-related mitotic motor protein | −2.05 |
| Kif18a | Kinesin family member 18A | −1.74 |
| Kif20a | Kinesin family member 20A | −1.76 |
| Kif20b | Kinesin family member 20B | −1.73 |
| Kif22 | Kinesin family member 22 | −1.58 |
| Kif23 | Kinesin family member 23 | −1.59 |
| Tpx2 | TPX2, microtubule-associated protein homolog (Xenopus laevis) | −1.75 |
| Birc5 | Baculoviral IAP repeat–containing 5, survivin | −1.59 |
| Mapt | Microtubule-associated protein tau | −2.06 |
| Tubb6 | Tubulin, beta6 | −1.57 |
| Pfn3 | Profilin3 | −1.58 |
| Diap3 | Diaphanous homolog 3 | −1.97 |
| Chromosome organization and biogenesis | ||
| Hist1h1a | Histone 1, H1A | −1.77 |
| Hist1h1b | Histone 1, H1B | −1.99 |
| Hist1h2ab | Histone 1, H2AB | −1.57 |
| Hist2h2aa1 | Histone 2, H2AA1 | −1.52 |
| Hist1h2bb | Histone 1, H2BB | −1.74 |
| Hist2h2bb | Histone 2, H2BB | −1.73 |
| Hist1h3f | Histone 1, H3F | −1.83 |
| Hist1h3g | Histone 1, H3G | −1.94 |
| Hmgb2 | High-mobility group box 2 | −1.52 |
| Top2a | Topoisomerase (DNA) II alpha | −1.87 |
Rac1+/− ECs show increased expression of gene clusters relevant to neurovascular protection
Functional clustering revealed enrichment of Rac1+/− ECs in three gene categories that were potentially relevant to their neurotrophic properties. There was increased expression of genes encoding proteins implicated in the stress response (“stress response genes”), genes encoding proteins associated with cell adhesion and the extracellular matrix (ECM) (“cell adhesion genes” and “ECM genes”), and genes encoding growth factors (Fig. 2E and Table 3). The functional relevance of these expression profiles was examined by testing appropriate cellular phenotypes (Figs. 3 to 5). The stress response genes included Gpx3, which encodes glutathione peroxidase 3 (Gpx3), an extracellular scavenger of ROS that catalyzes the reduction of hydrogen peroxide and lipid peroxides (21). Decreased Gpx3 activity is associated with familial child stroke (22). Up-regulated stress response genes also included those encoding three heat shock proteins (HSPs), crystallin αB, hsp25, and hsp70, which protect cells from apoptosis under various stress conditions (23–25). Various neurological diseases are associated with increased generation of ROS in the vascular wall, a process that both leads to the proteolytic disruption of the BBB and decreases neurovascular cell survival. EC death, in turn, causes further deterioration of BBB function. We therefore examined how Rac1 haploinsufficiency affected the ROS-generating capacity of ECs in hypoxia-reoxygenation (H/R), a condition that mimics ischemia-reperfusion injury in MCAo. In Rac1+/+ ECs, H/R elicited a 1.9-fold increase in the activity of NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) oxidase, the major EC source of ROS, over its activity under baseline (normoxic) conditions (Fig. 3A). The NADPH oxidase activity of Rac1+/− ECs under H/R was comparable to that of Rac1+/+ ECs under normoxic conditions, consistent with the known role of Rac1 as an activator of the NADPH oxidase complex (4). H/R caused a 1.6-fold increase in Rac1+/+ ECs of the overall oxidative stress (OS), including hydrogen peroxide. OS of Rac1+/− ECs under H/R was substantially decreased compared to that of Rac1+/+ ECs; indeed, OS of Rac1+/− ECs subjected to H/R was ∼30% lower than that of Rac1+/+ ECs at baseline. The decrease in OS in Rac1+/− ECs was more pronounced than the decrease in NADPH oxidase activity, suggesting the involvement of additional oxidoreductases, including Gpx3. In line with the decrease in ROS and the increase in HSP abundance, apoptotic death of Rac1+/− ECs, at both baseline and H/R, was ∼50% less than that of Rac1+/+ ECs (Fig. 3B).
Table 3.
Selected genes up-regulated in Rac1+/− mouse ECs. Genes are categorized by the GO annotation.
| Symbol | Gene name | Fold |
|---|---|---|
| Receptor binding | ||
| Artn | Artemin | 3.97 |
| Fgf5 | Fibroblast growth factor 5 | 2.73 |
| Cxcl2 | Chemokine (C-X-C motif) ligand 2 | 1.93 |
| Fgf14 | Fibroblast growth factor 14 | 1.91 |
| Bmp6 | Bone morphogenetic protein 6 | 1.82 |
| Npy | Neuropeptide Y | 1.69 |
| Tgfa | Transforming growth factor alpha | 1.68 |
| Frs3 | Fibroblast growth factor receptor substrate 3 | 1.55 |
| Edn1 | Endothelin 1 | 1.54 |
| Nts | Neurotensin | 1.53 |
| Cell surface receptor–linked signal transduction | ||
| Crhr2 | Corticotropin releasing hormone receptor 2 | 2.00 |
| Adcy7 | Adenylate cyclase 7 | 1.63 |
| Robo4 | Roundabout homolog 4 (Drosophila) | 1.58 |
| Cxcr4 | Chemokine (C-X-C MOTIF) receptor 4 | 1.56 |
| Ltbp1 | Latent transforming growth factor beta binding protein 1 | 1.55 |
| Adamts1 | A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 1 | 1.52 |
| Response to stimulus | ||
| Cryab | Crystallin, alpha B | 4.24 |
| Gpx3 | Glutathione peroxidase 3 | 2.86 |
| Hspb1 | Heat shock protein 1 (hsp25) | 1.68 |
| Hspa1a | Heat shock protein 1B (hsp70) | 1.62 |
| Cell adhesion | ||
| Dsc3 | Desmocollin 3 | 2.85 |
| Pcdh10 | Protocadherin 10 | 2.18 |
| Col2a1 | Procollagen, type II, alpha 1 | 1.74 |
| Col8a1 | Procollagen, type VIII, alpha 1 | 1.53 |
| Fn1 | Fibronectin 1 | 1.64 |
| Thbs2 | Thrombospondin 2 | 1.68 |
| Spp1 | Osteopontin | 1.52 |
Fig. 3.
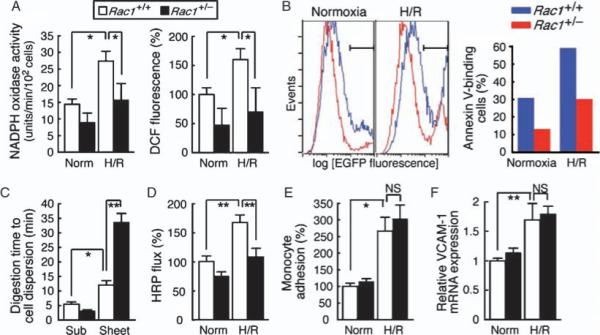
Phenotypic profiling of Rac1+/− ECs. (A, B, D, E, and F) Rac1+/+ and Rac1+/− mouse ECs were sequentially exposed to 4-hour hypoxia and 2-hour (A and D) or 20-hour (B, E, and F) reoxygenation (H/R) or treated with normoxia (Norm). (A) NADPH oxidase activity (left) and ROS production (right) as determined by DPI-inhibitable lucigenin chemiluminescence and DCF fluorescence, respectively. (B) Apoptotic death was assessed by flow cytometry (left) as the Annexin V–EGFP–labeled EC fraction (right). (C)ECs at subconfluence (Sub) or prolonged confluence (>4 weeks) (Sheet) were subjected to standard trypsinization until single-cell suspensions were obtained. (D) The permeability of confluent EC monolayers grown on 0.4-μm Transwell inserts was assessed as HRP leak across the membrane during normoxia and H/R. (E) After exposure to normoxia or H/R, ECs were co-cultured with [3H]thymidine-labeled monocytes (THP-1) for 3 hours. After washing, monocyte-EC binding was assessed as the cell lysate radioactivity. (F) VCAM-1 mRNA abundance was determined by qRT-PCR. (A, C to F) Values are means ± SEM of triplicates (*P < 0.05, **P < 0.01; ANOVA).
Fig. 5.
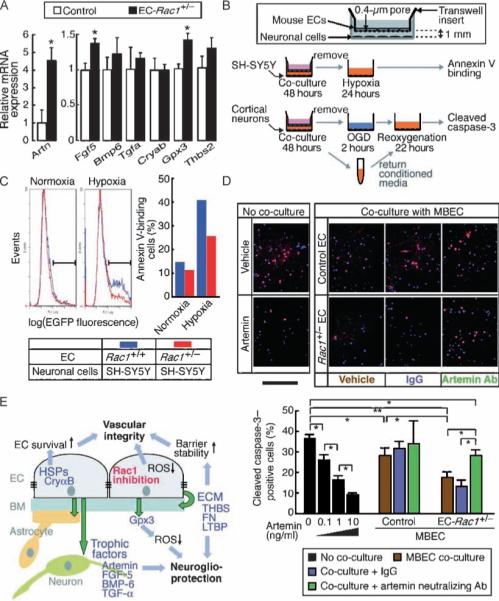
Rac1+/− ECs show enhanced neurotrophic activity through paracrine mechanisms. (A) mRNA expression of the potential neurotrophic factors was assessed in the EC-Rac1+/− and control mouse whole brains by qRT-PCR (n = 7 mouse brains). (B) Schematic of EC-neuron co-culture. (C) Co-culture with Rac1+/− versus Rac1+/+ ECs mitigates hypoxia-induced apoptotic death of SH-SY5Y cells, as assessed by Annexin V-EGFP labeling and flow cytometry. (D) Cortical neurons isolated from fetal mouse brains (∼E16) were co-cultured with EC-Rac1+/− or control MBECs in the presence of artemin-neutralizing antibody (Ab), control IgG, or vehicle in the lower chamber. Control neurons were treated with blank inserts and recombinant artemin proteins. Upper panel, after OGD and reoxygenation, the neurons were double-labeled with DAPI (blue) and cleaved caspase 3 (red). Scale bar, 1 mm. Lower panel, quantification of the cleaved caspase 3–positive fractions. (E) A proposed model of neuroprotection through targeting endothelial Rac1. Values are means ± SEM (*P < 0.05, **P < 0.01; ANOVA).
In the neurovascular unit, the basement membrane (BM) surrounds blood vessels and astrocyte endfeet and supports BBB integrity (2). In the early phases of stroke, the BM is attacked by proteases, such as matrix metalloproteinase 2 (MMP-2) and MMP-9 (3). Thus, enhancement of BM structure and function may help preserve BBB function. The ECM genes showing increased expression in Rac1+/− ECs included a subset that encodes BM constituents (Table 3 and table S4). These BM constituents are composed of structural [collagens (Col2a1, Col8a1), fibronectin (Fn1)] and regulatory proteins [thrombospondin 2 (Thbs2), osteopontin (Spp1)]. Additionally, a weak but coordinate increase in expression was noted for genes encoding perlecan [heparan sulfate proteoglycan 2 (Hspg2), 1.29-fold], collagens (Col1a1, 1.48-fold; Col4a1, 1.19-fold; Col4a2, 1.25-fold), thrombospondin 1 (Thbs1, 1.32-fold), and SPARC (secreted acidic cysteine-rich glycoprotein) (Sparc, 1.40-fold). Many of these proteins enhance collagen fibrillogenesis and matrix-cell adhesion, processes that stabilize the BM (26). Specifically, thrombospondin 1 and 2 bind to and inhibit MMP-2, a constitutively expressed protease that initiates the degradation of BM early in MCAo (3, 26, 27).
GSEA showed broad enrichment of Rac1+/− ECs in transforming growth factor–β (TGF-β)–responsive genes, including those encoding many ECM proteins (Fig. 2G and table S2). In the array data, the genes encoding TGF-β1−3 and TGF-β receptors 1 to 3 were not differentially expressed in Rac1+/− and Rac1+/+ ECs; rather, genes encoding proteins that coordinately enhance activation of latent TGF-β complex, such as LTBP1 (latent TGF-β binding protein 1), LTBP3 (1.46-fold), thrombospondin 1, SPARC, and fibronectin, were enriched in Rac1+/− ECs (Table 3 and table S4) (28, 29). Because TGF-β reportedly enhances neuronal survival and BBB function (2, 30), the possible increase in the TGF-β signal may play a role in neuroprotection in EC-Rac1+/− mice.
Resistance of Rac1+/− ECs to hypoxia-induced endothelial permeability
The cell adhesion gene cluster also included genes encoding components of adherence junctions (protocadherin 10), desmosomes (desmocollin 3), and tight junctions (claudin 3, 1.35-fold) (Table 3 and table S4) (2). The increased abundance of BM and intercellular adhesion proteins in Rac1+/− ECs suggested that Rac1+/− EC monolayers would show a less permeable and dissociation-resistant phenotype. Indeed, maintenance of confluent Rac1+/− EC monolayers for more than 4 weeks reproducibly yielded firm cell sheets hardly dissociable with trypsin digestion (Fig. 3C). In contrast, Rac1+/+ EC monolayers maintained under comparable conditions readily underwent dispersion by trypsin treatment. H/R increased the paracellular permeability of Rac1+/+ ECs to horseradish peroxidase (HRP) 1.7-fold, but only affected that of Rac1+/− ECs minimally (Fig. 3D). Rac1+/− ECs showed additional features consistent with enhanced ability to act as a barrier: up-regulation of the gene encoding anti-edemagenic protein thrombospondin 2 (31) and the circumferential redistribution of F-actin together with a decrease in its abundance [a cytoskeletal change typically associated with reduced endothelial permeability (32)] (Fig. 4). The decrease in F-actin abundance in Rac1+/− ECs is consistent with the decrease in Rac1 and mDia2 (Diap3, Table 2), which cooperatively promote actin microfilament formation (6, 33, 34).
Fig. 4.
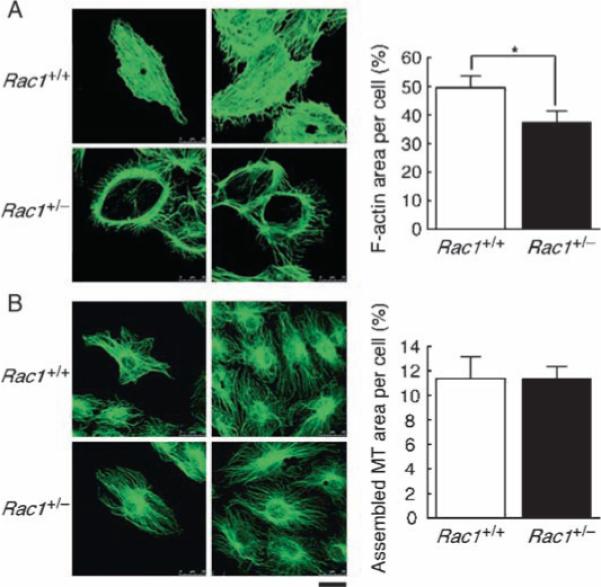
Decreased actin polymerization and circumferential redistribution of F-actin to the cortical areas in Rac1+/− ECs. Mouse ECs cultured in the presence of 20% serum were fixed and stained with (A) phalloidin to visualize F-actin and (B) antibody against β-tubulin for microtubules (MT). The F-actin and MT-labeled areas were quantified and represented as percent of total cell areas. Scale bar, 25 μm. Values are means ± SEM of five to seven replicates (*P < 0.05; ANOVA).
Leucocyte-EC adhesion is an important component of vascular injury in MCAo. However, the similar increase in monocyte adhesion (Fig. 3E) and vascular cell adhesion molecule–1 (VCAM-1) (Fig. 3F) mRNA abundance produced by H/R in Rac1+/− and Rac1+/+ ECs argues against the involvement of this mechanism for the neuroprotection in EC-Rac1+/− mice. Overall, decreased Rac1 promotes the antioxidative, survival, and barrier properties of ECs, thereby imparting resistance to ischemic insult.
Rac1+/− ECs exert neuroprotection through paracrine mechanisms
The expression of genes encoding 13 growth factors was up-regulated in Rac1+/− ECs. Four of these growth factors [artemin, fibroblast growth factor 5 (FGF-5), bone morphogenetic protein 6 (BMP-6), and TGF-α] are endowed with potential neurotrophic capacity (Table 3) (35–39). The genes encoding these factors are expressed basally or inducibly in vascular endothelium and in the central nervous system (CNS). We first determined the mRNA abundance of these neurotrophic factors, along with that of crystallin αB, Gpx3, and thrombospondin 2, in EC-Rac1+/− and control whole mouse brains (Fig. 5A). Of these, artemin (4.5-fold), FGF-5 (1.4-fold), and Gpx3 (1.4-fold) were significantly up-regulated in the EC-Rac1+/− brain compared to control. All of the genes examined are reportedly expressed in nonvascular cells such as neurons and glia; thus, background expression would likely mask their possible local enrichment in the vascular endothelium of EC-Rac1+/− brain.
Given the enrichment of neurotrophic factors, we hypothesized that Rac1+/− ECs support neuronal survival through paracrine mechanisms. To address this question, we performed noncontiguous co-culture experiments in which ECs were grown on a porous insert over a compartment in which neuronal cells were cultured (Fig. 5B). Rac1+/− and Rac1+/+ mouse ECs were grown to confluence on Transwell insert membranes with 0.4-μm pores, which allow passage of humoral factors to the lower compartment. After reaching confluence, ECs on inserts were co-cultivated with SH-SY5Y cells, a neuroblastoma cell line that expresses functional receptors to glial cell line–derived neurotrophic factor (GDNF) ligands, including artemin (35). After 2-day co-culture, SH-SY5Y cells left in conditioned media were exposed to hypoxic conditions for 24 hours. Hypoxia-induced apoptosis, as assessed by Annexin V labeling, was reduced in cells co-cultured with Rac1+/− ECs (14%) compared to those co-cultured with Rac1+/+ ECs (26%), suggesting increased production of neurotrophic factors by Rac1+/− versus Rac1+/+ ECs (Fig. 5C). To extend this finding, we focused on artemin, the most highly up-regulated neurotrophic factor in Rac1+/− ECs. Cortical neurons isolated from fetal mouse brain at around embryonic day 16 (E16) were co-cultured with mouse brain ECs (MBECs) derived from EC-Rac1+/− and control mice (Fig. 5B and fig S3). After 2-day co-culture, the neurons were challenged with 2-hour oxygen and glucose deprivation (OGD) followed by 22-hour reoxygenation (Fig. 5D). Administration of recombinant artemin (with blank inserts) elicited a dose-dependent inhibition of neuronal apoptosis, determined by the fraction of cells containing cleaved caspase 3. Endothelial co-culture attenuated neuronal apoptosis by 23% with control MBECs and by 52% with EC-Rac1+/− MBECs, indicating that EC secretion of neurotrophic factors is increased by Rac1 haploinsufficiency. Next, we tested the effect of artemin-neutralizing antibody or control immunoglobulin G (IgG) in neuron-EC co-cultures. Artemin-neutralizing antibodies had little effect on neuroprotection by control MBECs, but reduced neuroprotection by EC-Rac1+/− MBECs by half. These results suggest that the potentiation of EC-derived neurotrophic activity by Rac1 haploinsufficiency depends in part on increased production of artemin. The residual antiapoptotic activity that persists in the presence of artemin-neutralizing antibody implicates additional neurotrophic factors in the neurotrophic effects of EC-Rac1+/− MBECs.
DISCUSSION
We have shown that Rac1 haploinsufficiency in ECs is neuroprotective through the release of neurotrophic factors. The neuroprotective mechanism is due, in part, to increased release of artemin from haploinsufficient Rac1 ECs. Artemin is a member of the GDNF family of ligands (GFLs) that is expressed in various neural and nonneural tissues, including vascular walls (smooth muscle cells and ECs) (35, 40, 41). GFLs exert antiapoptotic signals on neurons by binding to a receptor complex composed of GDNF receptor α (GFRα) and RET (rearranged during transfection) proto-oncogene (8). The principal receptor for artemin is GFRα3, which is expressed mainly in nonneural tissues and in the peripheral nervous system (42). Studies with mice lacking artemin uncovered its role as a vascular-derived guidance factor for sympathetic neuron axonal projections (40). In the CNS, artemin is expressed in low abundance during development and adulthood in distinct regions, such as the basal ganglia and thalamus, whereas GFRα3 expression is almost nonexistent (35, 42). Despite the low abundance and restricted distribution of endogenous artemin in the CNS, administration of exogenous artemin potently promotes the survival of neurons from various CNS regions including midbrain dopaminergic neurons, cortical GABAergic neurons, and hippocampal neurons (43–45). Reportedly, artemin can also signal through GFRα1, which is the principal receptor for GDNF and shows widespread, ischemia-inducible CNS expression and neuroprotective capacity in MCAo (35, 37, 39). Therefore, we consider that the reduction in infarct size in EC-Rac1+/− mice was likely exerted through EC-derived artemin acting through the GFRα1 system.
The mechanism whereby inhibition of Rac1 leads to differential expression of the genes encoding a subset of neurotrophic factors is unclear. A recent study showed that integrin-linked kinase up-regulates brain-derived neurotrophic factor in ECs (13). Given that Rac1 is critical for outside-in signaling in response to cell adhesion, our observation that Rac1 insufficiency promotes the expression of ECM components and cell adhesion molecules may be pertinent to ECM regulation of neurotrophic activity in ECs.The TGF-β pathway may also play a role in up-regulating neurotrophic factors in Rac1+/− ECs, because the expression of artemin and TGF-α is reportedly inducible by TGF-β in pancreatic stellate cells and ECs (46, 47).
In animal models of neurological diseases, neurotrophic factors show therapeutic potential when delivered locally by surgical procedures. In clinical application, less invasive delivery through blood flow and cerebrospinal fluid was ineffective because of the poor bioavailability at target tissues due, in part, to poor BBB penetration, low tissue diffusion, and short half-life (7–9). Moreover, administration of high-dose neurotrophic factors frequently led to adverse effects, because of their systemic, pleiotropic actions, and interference with normal brain functions. Therefore, augmentation of endogenous neurotrophic activity within the confines of the neurovascular unit could represent an alternative therapeutic strategy that circumvents many, if not all, of these hurdles. We showed that inhibition of EC Rac1 potentiated endothelial expression of neurotrophic factors severalfold. Small increases in neurotrophic factor production may be therapeutically efficacious, as suggested by a recent study showing that a threefold increase in the abundance of GDNF protein was effective in a primate model of Parkinson's disease (48).
Our data suggest that EC Rac1 haploinsufficiency is neuroprotective through two mechanisms: It enhances vascular barrier integrity and independently promotes neuronal survival (Fig. 5E). Accumulating evidence indicates that targeting a single pathological process does not suffice for neuroprotection and points to the importance of combination therapies (49). In this regard, the multiple neuroprotective mechanisms potentially elaborated by Rac1+/− ECs may be beneficial. Considering the well-established roles of matrix-cell interaction in cell survival, increased expression of BM components may not only enhance BBB integrity, but also could directly contribute to preventing neuronal cell death. Indeed, fibronectin provides neuroprotection in mice MCAo (50). Conversely, the neurotrophic factors up-regulated in Rac1+/− ECs also have gliotrophic functions that could enhance the vascular barrier (2). For example, FGF-5 regulates the astrocyte endfeet coverage of blood vessels and enhances BBB function (51). Furthermore, it is becoming increasingly clear that cytokines that are ineffective in isolation can become neurotrophically active when acting together (30). The small but coordinate up-regulation of various growth factors in Rac1+/− ECs conforms to this notion and may underlie the neuroprotection seen in MCAo.
Because ECs represent a highly drug-accessible brain compartment, targeting endothelial Rac1 may provide a therapeutically feasible approach for intervention of the cross talk between ECM and cellular components within the neurovascular unit. The therapeutic time window of neurotrophic factor therapy for acute ischemic stroke is typically within 1 hour of reperfusion (37). Furthermore, BM disruption commences within hours of the stroke onset (3). Therefore, prophylactic inhibition of EC Rac1 may be beneficial for effective neuroprotection. However, Rac1 deletion in ECs could also lead to decreased angiogenic response (Supplementary text and fig. S2). It remains to be determined, therefore, what the effect of sustained inhibition of endothelial Rac1 is on neurogenesis, which is coupled to angiogenesis within the neurovascular niche (52, 53).
In conclusion, our study provides a previously unexplored therapeutic strategy for neuroprotection by targeting EC signaling. Our study is somewhat limited by the examination of transcriptional profile of Rac1 deletion in heart rather than brain ECs. However, our finding that EC-Rac1+/− mice showed a reduction in brain edema volume after ischemic stroke agrees with the notion that inhibition of Rac1 in brain ECs is protective of BBB function. Moreover, increased expression of the neurotrophic factor artemin was validated with Rac1 deletion in brain ECs. In addition, expression of the genes encoding artemin and FGF-5, as well as that of the stress-related gene Gpx3, was up-regulated in whole brains of EC-Rac1+/− mice. Thus, the emerging possibility that paracrine signals from endothelium could have a global role in tissue differentiation (and organ formation and function) indicates the broad applicability of exploiting EC signals for therapies targeting adjacent parenchymal tissues (54).
MATERIALS AND METHODS
Expanded materials and methods are available in Supplementary Materials.
Generation of EC-Rac1+/− mice and the transient MCAo model
All animal protocols were approved by the Harvard Medical School's Standing Committee on Animal Welfare and Protection and are in accordance with the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health. Male littermates (12 to 20 weeks) were used for experiments. Rac1 conditional allele knock-in mice (Rac1flox/flox) were developed as described (16) and crossed with Tie2-Cre Tg mice (Jackson Laboratory) (15) to generate EC-Rac1+/− (Tie2-Cre Rac1flox/+) and control (Rac1flox/+) mice. Transient MCAo and assessment of cerebral infarct volume, neurological deficit, and absolute CBF was conducted as described (17).
Isolation and culture of mouse primary ECs
Mouse primary ECs were isolated from heart as described (55), using an affinity selection method with Dynabeads conjugated with sheep antibody against rat IgG (Invitrogen), and rat antibodies against PECAM-1, and intercellular adhesion molecule–2 (ICAM2) (BD Biosciences). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 20% fetal calf serum (FCS), heparin, penicillin, streptomycin, and EC growth factor (ECGF). The preparation of Rac1+/− and Rac1+/+ ECs was verified by guanosine triphosphate–Rac1 pull-down assay and immunoblotting with Rac1 (BD Biosciences) and actin (Sigma) antibodies.
Microarray and gene cluster analysis
Total RNA was prepared from subconfluent Rac1+/− and Rac1+/+ mouse ECs at passage 4 through the sequential use of Trizol reagent (Invitrogen) and RNeasy columns (Qiagen). Complementary DNA (cDNA) labeling, hybridization, and image acquisition was performed at the Massachusetts General Hospital Microarray Core. The cDNA was reverse transcribed from 8 μg of RNA, coupled to Cy3 or Cy5 dye, and hybridized in four replicates to a microarray containing 19,549 70-mer oligonucleotides. The fold ratio of Rac1+/− versus Rac1+/+ for each reporter was statistically analyzed with one-sample Student's t test and Wilcoxon signed rank test and considered significant at P < 0.05. Reporters with the fold ratio of greater than 1.5 (up-regulated) or less than −1.5 (down-regulated) were considered differentially expressed and were uploaded onto DAVID 2007 (http://david.abcc.ncifcrf.gov/home.jsp) for gene annotation analysis (18). Enrichment of a given gene annotation was determined by EASE score, a modified Fisher's exact P value. Functional annotation clusters were determined by the degrees of gene co-association between annotation terms and were ranked by the Enrichment Score, the geometric mean (in –log scale) of member's EASE scores. To test for related genes systematically altered in Rac1+/− ECs, we used GSEA, a method that combines information from previously defined gene sets (www.broad.mit.edu/gsea/index.jsp) (19). Genes from the microarray were first ranked according to the expression difference (signal-to-noise ratio) between Rac1+/− and Rac1+/+ ECs. The extent of association was then measured by a nonparametric running sum statistic. The complete set of microarray data is available at Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/) with an accession number GSE15003.
Quantitative RT-PCR
Total RNA (50 to 100 ng) was used for real-time RT-PCR with a QuantiTect SYBR Green RT-PCR kit (Qiagen). The mRNA abundance was normalized with GAPDH mRNA or 18S ribosomal RNA.
Immunofluorescence
Mouse ECs were fixed with 3.7% paraformaldehyde, permeabilized with 0.1% Triton X-100, and probed with crystallin αB antibody (Stressgen) and Alexa488-conjugated secondary antibody (Molecular Probe). Images were acquired by a confocal microscope (Leica).
Assessment of oxidative stress
Hypoxic condition was introduced by modular chambers (Billups-Rothenberg) filled with 95% N2 and 5% CO2 at 37°C. After hypoxia, cells were returned to a regular incubator for reoxygenation (H/R). ECs were exposed to H/R (4 hours/2 hours) and treated with 3.3 μM lucigenin and 67 μM NADPH. The NADPH oxidase activity was determined as diphenylene iodonium (DPI)–inhibitable lucigenin chemiluminescence by a luminometer (PerkinElmer). Cells preloaded with 100 μM 2′,7′-dichlorofluorescein diacetate (DCFH-DA) were exposed to H/R and the OS was determined as DCF fluorescence by 485-nm excitation and 530-nm emission filters.
Annexin V binding assay
Cells were trypsinized, labeled with Annexin V-EGFP (10 μg/ml) in binding buffer containing 2.5 mM CaCl2 for 15 min and analyzed by flow cytometry.
Assessment of cell adhesion and EC barrier function
Cell adhesion was assessed by the time required for obtaining single-cell suspensions in 0.25% trypsin/1 mM EDTA solution (Invitrogen) at 37°C. Confluent ECs on Transwell inserts (0.4-μm pore size) were treated with HRP (20 μg/ml) in the upper chamber, and the medium in the lower chamber was incubated in buffer (0.5 mM guaiacol, 50 mM Na2HPO4, and 0.6 mM H2O2) to assess the HRP activity that passed across the EC layer as absorbance at 470 nm.
Assessment of EC-leucocyte attachment
ECs were co-cultured with [3H]thymidine-labeled THP-1 cells (106 cells/ml) for 3 hours. The adherent cells were lysed with 0.5 M NaOH and analyzed in a scintillation counter.
Isolation and culture of neuronal cells and MBECs
SH-SY5Y cells (American Type Culture Collection) were cultured in Eagle's minimum essential medium/F12 medium with 10% FCS. Cortical neurons were isolated from fetal mouse brains (∼E16) with the use of trypsin and deoxyribonuclease I (DNase I) and plated at 105 cells/cm2 on poly-d-lysine–coated coverslips in Neurobasal medium (Invitrogen) with 1 mM glutamine, 2% B27 supplement, and antibiotics. The medium was half changed every 3 days. For MBEC isolation, cortices from 6 to 10 adult mice were homogenized, deprived of lipid debris by centrifugation in 15% dextran (10,000g), and digested with Collagenase/Dispase (Roche) and DNase I. After centrifugation in 45% Percoll (20,000g), the top layer was plated in DMEM/F12 with 10% horse serum, 10% FCS, heparin, antibiotics, and ECGF.
EC-neuron co-culture and assessment of neuronal cell death
For co-culture, ECs grown on Transwell inserts (0.4-μm pore) were applied to neuronal cell cultures for 48 hours and thereafter removed. SH-SY5Y cells were exposed to 24-hour hypoxia, followed by Annexin V-EGFP labeling. The cortical neurons (day 14) were co-cultured with MBECs (passage 3) in the presence of vehicle, goat anti-mouse artemin-neutralizing antibody or goat IgG (0.5 μg/ml, R&D) in the lower compartment. Some neurons were treated with blank inserts and recombinant mouse artemin (0 to 10 ng/ml). After co-culture, the neurons were deprived of the conditioned media (CM), treated with glucose-free DMEM under hypoxia for 2 hours (OGD), and resupplied with the CM and reoxygenated for 22 hours. The neurons were immunostained with cleaved caspase-3 (Asp175) (5A1) rabbit monoclonal antibody (Cell Signaling) overnight, and Alexa568-conjugated antibody against rabbit IgG and 4′,6-diamino-2-phenylindole (DAPI) for 1 hour.
Statistical analysis
Unless otherwise noted, all statistical analysis was carried out by analysis of variance (ANOVA) followed by Fisher's test. P < 0.05 was considered to be significant.
Supplementary Material
Supplementary Materials for
Rac1 Is a Critical Mediator of Endothelium-Derived Neurotrophic Activity
Naoki Sawada, Hyung-Hwan Kim, Michael A. Moskowitz, James K. Liao*
*To whom correspondence should be addressed. E-mail: jliao@rics.bwh.harvard.edu
Published 10 March 2009, Sci. Signal. 2, ra10 (2009)
DOI: 10.1126/scisignal.2000162
This PDF file includes:
Materials and Methods
Supplementary Text
Fig. S1. CD31 labeling of Rac1+/− and Rac1+/+ ECs.
Fig. S2. Rac1+/− ECs show retarded proliferation, migration, and capillary formation.
Fig. S3. CD31 labeling of MBECs.
Table S1. Representative gene sets enriched in Rac1+/+ mouse ECs.
Table S2. Representative gene sets enriched in Rac1+/− mouse ECs.
Table S3. Transcription factors differentially expressed between Rac1+/− and Rac1+/+ mouse ECs.
Table S4. List of differentially expressed genes between Rac1+/− and Rac1+/+ mouse ECs.
Supplementary material
Supplementary Materials and Methods
Generation of EC-Rac1+/− mice.
All experimental procedures on animals were performed by using protocols approved by the Harvard Medical School's Standing Committee on Animal Welfare and Protection and are in accordance with the Guide for the Care and Use of Laboratory Animals, published by NIH. Mice were bred and maintained with a 12/12-hour light/dark cycle and fed standard chow with tap water available ad libitum. Age-matched male littermates (12−20 wk) were used for test and control groups in all experiments. Rac1 conditional allele knock-in mice were developed as described (1), and backcrossed at least eight times onto the C57BL/6 strain. Tie2-Cre transgenic mice were obtained from Jackson Laboratory and maintained on C57BL/6 background (2) . Rac1flox/flox mice were crossed with Tie2-Cre Tg mice to generate EC-Rac1+/− (Tie2-Cre Rac1flox/+) and control (Rac1flox/+) mice. For some experiments, global Rac1 haploinsufficient (Rac1+/−) and control (Rac1+/+) mice were also used (1). The genotyping PCR primers for Rac1 conditional and null alleles are described previously (1). The genotyping primers used for Tie2-Cre transgenic mice are: 5’-TGA TGA GGT TCG CAA GAA CC; 5’-ACC AGT TTA GTT ACC CCC AG.
Transient middle cerebral artery occlusion model.
Mice were anesthetized with 2% isoflurane mixed in 70% N2O and 30% O2, and then maintained on 1.5% to 2% isoflurane in a similar gaseous mixture. Transient focal cerebral ischemia was performed using an 8−0 nylon monofilament coated with silicone, which was introduced into the internal carotid artery via the external carotid artery, and then advanced 10 mm distal to the carotid bifurcation to occlude the MCA as described (3). Laser Doppler flowmetry of relative CBF was used to verify successful occlusion (<20% baseline value). The MCA was occluded 2 hr, followed by withdrawal of filament and reperfusion for 22 hr. Relative CBF returned to >95% of baseline values, indicating almost complete reperfusion without residual occlusion.
Cerebral infarct volume was measured 22 hr after reperfusion. Infarct area was measured in 2-mm-thick coronal brain sections stained with 2,3,5-triphenyltetrazolium chloride and quantitated with MCID Elite M6 image-analysis software (Interfocus Imaging). Cerebral infarct volume was determined by summing up the infarcted areas. The infarct and edema volumes relative to the non-ischemic hemisphere were calculated as follows: Infarct volume (%) = (non-ischemic hemisphere vol. (mm3) – ischemic hemisphere non-infarcted region vol. (mm3))/(non-ischemic hemisphere vol. (mm3)) × 100. Edema volume (%) = (ischemic hemisphere vol. (mm3) – non-ischemic hemisphere vol. (mm3))/(non-ischemic hemisphere vol. (mm3)) × 100.
The neurological deficit score (NDS) was determined at 20 hr post-reperfusion by two observers, who were blinded to the identity of the mice or treatment protocol. The following scoring system was used: 0, no motor deficits (normal); 1, flexion of the contralateral torso and forelimb on lifting the animal by the tail (mild); 2, circling to the contralateral side but normal posture at rest (moderate); 3, leaning to the contralateral side at rest (severe); and 4, no spontaneous movement (critical).
Absolute CBF in ischemic and non-ischemic hemispheres was measured at the end of 2 hr MCAo using an indicator fractionation technique as described, with slight modifications (3). Briefly, the right jugular vein and left femoral artery of anesthetized mice were cannulated. The mice received 1 μCi of N-isopropyl-[methyl 1,3-14C] p-iodoamphetamine from the right jugular vein as a bolus. A volume of 100 μL of arterial blood samples from the left femoral artery was collected for 20 sec (0.3 ml/min), and then the animal was decapitated and the whole brain was removed. The brain was immediately cut into the 2 hemispheres and frozen in chilled isopentane solution chilled with dry ice immediately. The frozen hemispheres were weighed and digested with Scintigest (Fischer Scientific) at 50°C for 6 h. Scintillation fluid and H2O2 were added to the sample and they were shaken together for 12 h. The radioactivity in the blood and brain samples was measured by liquid scintillation spectrometry (RackBeta 1209). Cerebral blood flow in each hemisphere was calculated as follows: CBF (ml/100 g per min) = (hemisphere count (c.p.m.) × 0.3 (ml/min))/(blood count (c.p.m.) × hemisphere weight (g)) × 100. The relative CBF change in ischemic hemisphere was determined as: CBF change (%) = (ischemic hemisphere CBF (ml/100 g per min) – non-ischemic hemisphere CBF (ml/100 g per min))/(non-ischemic hemisphere CBF (ml/100 g per min)) × 100.
Isolation and culture of mouse primary ECs.
Mouse primary ECs were isolated from the heart by an affinity selection method using Dynabeads sheep anti-rat IgG (Invitrogen) and anti-PECAM1 antibody (BD Biosciences) as described (4). The cells were cultivated in DMEM containing 20% FCS, heparin (100 μg/ml), penicillin (100 U/ml), streptomycin (100 μg/ml) and endothelial cell growth factor (ECGF, 50 μg/ml), and grown on dishes coated with 0.1% gelatin. After 8 days, the cells were trypsinized and underwent the second selection using Dynabeads plus anti-ICAM2 antibody (BD Biosciences). Mouse ECs were used for experiments at passage 3−5.
GTP-Rac1 pull down assay and immunoblotting analysis.
pGEX-2T-PAK-CRIB domain construct is a gift from Dr. John G. Collard. Preparation of GST-PAK-CD and GTP-Rac1 pull down assay was performed as described (5). The GTP-Rac1, total Rac1 and actin protein levels in mouse EC lysate were determined by immunoblotting analysis with Rac1 (BD Biosciences) and actin (Sigma) antibodies. The blots were subsequently probed with HRP-conjugated secondary antibody and visualized by Enhanced Chemiluminescence (GE Health Care).
Microarray expression profiling.
Rac1+/− and Rac1+/+ mouse primary ECs were isolated from 6 mice/genotype derived from 2 age-matched litters (12 wk). Following the first and the second sorting, subconfluent ECs at passage 4 were used for the isolation of RNA. To validate the purity of the EC population, a portion of the cells was immunolabeled with FITC-conjugated anti-mouse CD31 antibody (eBioscience). Flow cytometric analysis revealed that > 98% of the Rac1+/− and Rac1+/+ ECs were positive for CD31 (fig. S1). High quality total RNA was prepared by 2-step purification through the sequential use of Trizol reagent (Invitrogen) and RNeasy columns (Qiagen) according to the manufacturer's instructions. The quality control of RNA was conducted by measuring the 28S/18S rRNA ratio using Agilent 2100 BioAnalyzer. cDNA labeling, hybridization and image acquisition was performed at Massachusetts General Hospital Microarray Core. Briefly, fluorescent dye labeling was performed using the Atlas Powerscript Fluorescent Labeling Kit (Clontech). Isolated total RNA (8 μg) was reverse transcribed into amine modified cDNA using random hexamers and oligo (dT) primers in 4 replicates per each genotype. The cDNA was labeled via a coupling reaction to the Cy3 (2 replicates) or Cy5 (2 replicates) dye, then purified and concentrated. Equimolar mixtures of Cy3-labeled Rac1+/+ and Cy5-labeled Rac1+/− probes (or dye-swapped combinations) were independently hybridized to 4 microarray slides. The microarray was a glass-based pre-printed array containing PGA Mouse v1.1 probe set (19,549 70-mer oligonucleotides), which provides complete coverage of the 2002 Mouse genome, built off the GenPept database. Images were acquired using Axon (Molecular Devices) 4000B 2-color single-slide scanner. Filtering, normalization of the raw data and basic statistical tests were conducted using BASE software. All spots were sequentially subjected to background subtraction, Lowess normalization, and brightness filtering that removed spots with intensities less than 200. This processing resulted in 12,089 reporters (gene features) comprising 45,311 spots from the 4 slides. The averaged fold ratio of Rac1+/− versus Rac1+/+ was calculated for each reporter and statistically analyzed using 1 sample Student t-test and Wilcoxon signed rank test, yielding 1,431 reporters considered significant at a confidence level of 95% (P < 0.05). The averaged fold ratios distributed with 2 SD of 1.56. Gene reporters with the fold ratio of > 1.5 or < −1.5 were considered differentially expressed between Rac1+/+ and Rac1+/− ECs.
Gene cluster and pathway analysis.
To characterize the enrichment profiles of gene clusters and pathways in Rac1+/− and Rac1+/+ ECs, we conducted 2 different analyses.
DAVID Functional Annotation Clustering (6).
The 138 upregulated (fold ratio > 1.5) and 161 downregulated (fold ratio < −1.5) reporters were imported into DAVID 2007 software (http://david.abcc.ncifcrf.gov/home.jsp) and collapsed to 130 and 138 unique genes, respectively, using the DAVID accession conversion tool. The upregulated and downregulated gene lists were separately uploaded for gene annotation analysis. Enrichment of a given gene annotation in each gene list against the default Mus Musculus background was determined by EASE score, a modified Fisher Exact P-Value. The DAVID default setting was used for the source databases of gene annotation: Gene Ontology (GOTERM_BP_ALL, GOTERM_CC_ALL, GOTERM_MF_ALL), Protein Domains (INTERPRO, PIR_SUPERFAMILY, SMART), Pathways (BIOCARTA, KEGG_PATHWAY), and Functional Categories (COG_ONTOLOGY, SP_PIR_KEYWORDS, UP_SEQ_FEATURE). Subsequently, Functional Annotation Clustering was performed to measure relationships among the annotation terms based on the degrees of their co-association genes to group the similar, redundant, and heterogenous contents from the same or different resources into annotation groups. The Functional Annotation Clustering integrates Kappa statistics to measure the common genes between two annotations, and fuzzy heuristic clustering to classify the groups of similar annotations according to kappa values. The biological significance of functional annotation clusters was ranked by the Enrichment Score, which is the geometric mean (in –log scale) of member's EASE scores within each cluster.
Gene Set Enrichment Analysis (GSEA) (7).
To test for sets of related genes that might be systematically altered in Rac1+/− ECs, we used Gene Set Enrichment Analysis (GSEA), a method that combines information from members of previously defined sets of genes to increase signal relative to noise and improve statistical power to detect subtle changes. Complete details on the method for this analysis, the software and information on the biological data sets are available at http://www.broad.mit.edu/gsea/index.jsp. Briefly, genes from the microarray were first ranked according to the expression difference (signal to noise ratio) between Rac1+/− and Rac1+/+ ECs. The standard GSEA null hypothesis is that the rank ordering of the genes in a given comparison is random and not associated with the order of the genes and/or treatment. The extent of association was then measured by a non-parametric running sum statistic termed the enrichment score (ES), and the maximum ES (MES) was recorded over each gene set. Phenotype permutation testing was used to assess the statistical significance of the MES, which was calculated as the fraction of 1,000 random permutations of the gene list. The unadjusted nominal P value estimated the statistical significance of a gene set without adjusting for gene set size or multiple hypothesis testing, whereas the false discovery rate (FDR) statistic adjusts for both. In the present study, we used curated gene sets (C2), archived at Molecular Signature Database, which were collected from various sources such as online pathway databases, publications in PubMed, and knowledge of domain experts. Among these gene sets, 1782 sets with the gene set size > 15 were used for analysis, and 161 gene sets were significantly upregulated in Rac1+/−, whereas 145 sets were enriched in Rac1+/+ ECs at FDR < 25%.
Quantitative RT-PCR.
Total RNA was extracted from the mice brains and ECs using RNeasy mini kit (Qiagen) with DNase I treatment, and 50−100 ng of total RNA was used for one-step real-time RT-PCR using QuantiTect SYBR Green RT-PCR kit (Qiagen) according to the manufacturer's instruction. The reaction was performed with 7900HT Fast Real-Time PCR System (Applied Biosystems). The mRNA level was normalized with that of either GAPDH mRNA or 18S rRNA. The PCR primers used are: Rac1, 5’-GAG ACG GAG CTG TTG GTA AAA, 5’- ATA GGC CCA GAT TCA CTG GTT; Artemin, 5’-CCC TAG CTG TTC TAG CCC TG, 5’-AGG GTT CTT TCG CTG CAC AA; FGF5, 5’-AAG TAG CGC GAC GTT TTC TTC, 5’- CTG GAA ACT GCT ATG TTC CGA G; BMP6, 5’-AGA AGC GGG AGA TGC AAA AGG, 5’-GAC AGG GCG TTG TAG AGA TCC; TGFα, 5’-CAC TCT GGG TAC GTG GGT G, 5’-CAC AGG TGA TAA TGA GGA CAG C; Crystallin αB, 5’-GTT CTT CGG AGA GCA CCT GTT, 5’-GAG AGT CCG GTG TCA ATC CAG; VCAM-1, 5’-AGT TGG GGA TTC GGT TGT TCT, 5’-CCC CTC ATT CCT TAC CAC CC; Gpx3, 5’-CCT TTT AAG CAG TAT GCA GGC A, 5’-CAA GCC AAA TGG CCC AAG TT; Thrombospondin 2, 5’-CTG GGC ATA GGG CCA AGA G, 5’-GCT TGA CAA TCC TGT TGA GAT CA; GAPDH, 5’-AGG TCG GTG TGA ACG GAT TTG, 5’-TGT AGA CCA TGT AGT TGA GGT CA; 18S, 5’-AAA TCA GTT ATG GTT CCT TTG GTC, 5’-GCT CTA GAA TTA CCA CAG TTA TCC AA.
Immunofluorescence staining.
Mouse ECs were cultured to subconfluence on Labtek II chamber slides (Nunc) under standard culture conditions. Cells were fixed with 3.7% paraformaldehyde in PBS for 10 min on ice, and then free aldehyde was reduced with 50 mM NH4Cl. The cells were subsequently permeabilized with 0.1% Triton X-100 in PBS for 5 min, and blocked with 1% BSA in PBS for 30 min at room temperature. For F-actin staining, the cells were incubated with 1U/ml Alexa488-conjugated phalloidin (Molecular Probe) for 20 min. For immunostaining, the cells were incubated with rabbit anti-ß tubulin (Lab Vision) or mouse anti-crystallin αB antibody (Stressgen) at 4°C overnight. After wash, the cells were labeled with the corresponding secondary antibody conjugated with Alexa488 (Molecular Probe) for 1 hr at room temperature. The fluorescent images were acquired by TCS SP5 confocal microscope (Leica). F-actin and ßtubulin fluorescence was quantified with Photoshop and Image J softwares.
Hypoxia treatment.
Cells were placed without culture dish covers in humidified airtight chambers (Billups-Rothenberg) perfused with 95% N2 and 5% CO2 at 2 psi for 10 min. The chambers were sealed and maintained at 37°C for indicated time periods. At the end of hypoxia exposure, cells were removed from the chamber and returned to a regular incubator for reoxygenation. Control cultures were incubated in a regular incubator under normoxic condition for the corresponding hypoxic duration.
Assessment of NADPH oxidase activity.
Mouse ECs were grown to confluence on 96-well plates. The cells were treated without or with 10 μM Diphenyleneiodonium chloride (DPI), a NADPH oxidase inhibitor, and then exposed to 4-hr hypoxia and 2-hr reoxygenation. The cells were sequentially pre-equilibrated with Hanks’ Balanced Salt Solution (HBSS) containing 10 mM Sodium diethyldithiocarbamate trihydrate (DETC) and 1 mM DTT for 30 min, and then with 3.3 μM lucigenin for 5 min. The cells were treated with 67 μM NADPH, and the chemiluminescence was captured for 1 min with a luminometer (Victor3, PerkinElmer). The NADPH oxidase activity was determined as DPI-inhibitable lucigenin chemiluminescence.
Assessment of oxidative stress.
Mouse ECs were grown to confluence on 96-well plates. The cells were loaded with 100 μM 2’,7’-dichlorofluorescein diacetate (DCFH-DA) in Krebs-Ringer-HEPES (KRH) buffer, pH 7.4, containing 4% bovine serum albumin and 0.55 mM glucose for 30 min. After the loading buffer was removed, the cells in KRH buffer were exposed to 4-hr hypoxia and 2-hr reoxygenation. The fluorescence caused by oxidation of DCFH-DA to DCF was measured in a plate reader by 485-nm excitation and 530-nm emission filters.
Annexin V binding assay.
Following various apoptosis-inducing conditions, 1−5 × 105 cells were detached with trypsin/EDTA and washed with Annexin V binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). The cells were labeled in 0.2 ml Annexin V binding buffer containing 10 μg/ml Annexin V-EGFP in the dark for 15 min. The cells were pelleted by centrifugation, resuspended in 1 ml binding buffer, and analyzed by flow cytometry with the excitation beam of 488 nm through FL1 detector (Cytomics FC 500, Beckman Coulter). Unstained cells were used as a negative control for background subtraction.
Cell adhesion assay.
Mouse ECs were grown to subconfluence, or kept at confluence for > 4 weeks. Then, the cells were washed 2 times with PBS (Ca++-free), and subjected to standard trypsinization using 0.25% trypsin/1 mM EDTA solution (Invitrogen) at 37°C. The cells were pipetted and observed under light microscope at the intervals of 3−5 min. The strength of cell adhesion was assessed by the time required for obtaining single cell suspensions (8).
Assessment of EC barrier function.
Mouse ECs grown to confluence on Transwell inserts (0.4-μm pore size) were maintained thereafter for ∼10 days. The medium in the upper compartment was replaced with fresh medium containing HRP (20 μg/ml), and the cells were exposed to 4-hr hypoxia and 2-hr reoxygenation. Medium from the bottom well was incubated with assay buffer containing 0.5 mM guaiacol, 50 mM Na2HPO4 and 0.6 mM H2O2. HRP activity was assessed as the formation of O-phenylenediamine determined by absorbance at 470 nm (9).
Assessment of EC-leucocyte attachment.
Monocyte cell line THP-1 was cultured in RPMI 1640 supplemented with 10% FCS. The THP-1 cells were labeled with 1 μCi/ml [3H]-thymidine for 48 hr and washed. Confluent mouse ECs in 6-well plates were subjected to 4-hr hypoxia and 20-hr reoxygenation. The ECs were co-cultured with the radiolabelled THP-1 cells (1 million cells per ml) at a static condition for 3 hr. At the end of incubation, non-adherent THP-1 cells were removed and the ECs were washed 3 times with PBS. The remaining adherent cells were lysed with 1mL of 0.5 M NaOH, and the radioactivity was measured in a scintillation counter (10).
Isolation and culture of neuronal cells and mouse brain ECs.
The human neuroblastoma cell line SH-SY5Y was purchased from ATCC and maintained in a 1:1 mixture of Eagle's Minimum Essential Medium (EMEM) and F-12 medium, supplemented with 10% FCS and penicillin/streptomycin. Primary cortical neurons were isolated from C57/BL6 fetal mouse brains (∼E16). Briefly, dissected cortices were digested with 0.25% trypsin at 37°C for 10min, and DNase I (25 U/ml) for 5 min. The digested cortices were triturated and passed through a 40-μm strainer. The neurons were plated on cover glasses pre-coated with poly-D-lysine (0.1 mg/ml) at a density of 1−1.5 × 106 cells/well in 6-well plates. The cells were maintained in serum-free Neurobasal medium (Invitrogen) supplemented with 1 mM glutamine, 2% B27 supplement and penicillin/streptomycin, and treated with 25 μg/ml glutamate (day 0−3) and 10 μM Ara-C (day 3−4). Thereafter, half of the medium was replaced with fresh medium every 3 days. The neurons were used for experiments at days 14−21. Mouse brain microvascular endothelial cells (MBEC) were isolated as described with modifications (11–13). Age-matched adult EC-Rac1+/− and control mice (n = 6−10 per genotype) from 2−3 litters were used for cell isolation. Dissected cortices were homogenized in ice-cold DMEM/F12 (1:1) using a Dounce tissue grinder (loose pestle) with 5 strokes and further minced by scissors. The homogenate was centrifuged (200 g, 5min, 4°C), and the pellets were resuspended with 15% dextran (MW ∼100,000) in PBS (−) and centrifuged at 10,000 g, 15 min, 4°C. The pellets were suspended and digested in DMEM/F12 containing Collagenase/Dispase (1mg/ml) (Roche) and DNase I (25 μg/ml) at 37°C for 90 min, and centrifuged at 400g, 10 min, room temperature. The pellets were suspended with 45% Percoll in PBS, centrifuged at 20,000 g, 15 min, 4°C. The top layer containing microvessels and ECs were transferred to a new tube, centrifuged at 400 g, and washed once with complete media (DMEM/F12 with 10% horse serum, 10% FCS, heparin, penicillin/streptomycin and ECGF). Cells were plated and grown on gelatin-coated T25 plates, and split at 1:2. More than 95% of MBECs at passage 1 were positive for CD31 staining (fig. S3). MBECs were used for experiments at passage 3−5.
EC-neuron co-culture and assessment of neuronal cell death.
ECs were plated on Transwell culture inserts (0.4-μm pore size) and grown to confluence. ECs and neuronal cells (SH-SY5Y or cortical neurons) were maintained in respective complete media (ECs, upper compartment, 1.5 ml; neuronal cells, lower compartment, 2.6 ml). Following the change of media, co-culture was started by adding inserts with ECs to neuronal cell cultures in 6-well plates. Co-culture proceeded for 48 hr and then the inserts with ECs were removed. SH-SY5Y cells left in the conditioned media were exposed to hypoxic conditions for 24 hr, and the apoptotic cell fraction was assessed through Annexin V-EGFP labeling. Cortical neurons were co-cultured with MBEC in the presence of vehicle, goat anti-mouse artemin neutralizing antibody (0.5 μg/ml), or goat IgG (0.5 μg/ml, R&D) in the lower compartment. Some neurons were treated with blank inserts and recombinant mouse artemin (0−10 ng/ml). The cortical neurons, after co-culture with MBEC, were subjected to oxygen/glucose deprivation (OGD). Briefly, the conditioned media (CM) was removed from the neurons and stored separately. The cells were simultaneously treated with glucose-free DMEM and hypoxia for 2 hr, re-supplied with CM, and returned to normoxic conditions (reoxygenation) for 22 hr. Neurons on cover slips were fixed in 3.7% paraformaldehyde in PBS on ice for 15 min, washed, and permeabilized with methanol at −20°C for 10 min. The cells were washed, blocked with 1% BSA for 30 min, and probed with cleaved caspase-3 (Asp175) (5A1) rabbit mAb (Cell Signaling) at 4°C overnight. Following wash, the cells were stained with Alexa568-conjugated anti-rabbit Ab and 4’ 6’-diamino-2-phenylindole (DAPI) for 1 hr at room temperature. Images were captured using an epifluorescence microscope. The cleaved caspase 3 positive fraction was analyzed by the Photoshop software.
EC proliferation, migration and capillary formation assays.
For proliferation assay, mouse ECs were plated at ∼30% confluency on 24-well plates, and allowed to grow in complete medium (20% FCS). On day 1 and 3, cells were fixed in neutral buffered 10% formalin for 15 min, washed with water, and stained with 0.1% crystal violet in 200 mM MES (pH6.0) for 1 hr. Following extensive washing with water, plates were dried and cell-associated dye was extracted with 10% acetic acid, and quantitated by absorbance at OD590 (14). Cell migration was assessed by scratch assay of the monolayer. Briefly, ECs were grown to confluence, and a linear acellular wound 1.5 mm wide was made with a scraper. The wound edge was marked by a cover slip attached to the bottom of the cultureware. The migrating cells were observed under light microscope and photographed at day 2. The capillary forming capacity of mouse ECs was evaluated in aortic explants ex vivo. Aortic segments from mice were embedded in 0.4 ml per well of Matrigel (BD Biosciences) in 24 well plates. After the gel solidified, 0.5 ml of culture media was added and replaced every other day. At day 8, photographs were taken to evaluate capillary outgrowth.
Statistical analysis.
Unless otherwise noted, all statistical analysis was carried out by ANOVA followed by Fisher's test. P < 0.05 was considered to be significant.
Supplementary text
Global downregulation of cell cycle-related genes in Rac1+/− ECs.
Functional annotation clustering and GSEA revealed that the genes downregulated in Rac1+/− are globally associated with mitotic cell division (Fig. 2D, 2F and Tables 2, S1, S3). These included transcription factors (c-fos (15), c-myb (16), Egr1 (17), Runx1 (18)), cyclins (cyclin A2, cyclin B2), mitotic kinases (Cdc2, Aurora A (19)), regulators of mitotic spindle assembly (Tacc3 (20), Tpx2 (21), BARD1 (22)), mitotic checkpoint (Mad2l1 (23), survivin (24)), centrosome cycle (Nek2 (25)), cytokinesis (Ect2 (26)), sister chromatid cohesion (sororin (27)), G2/M checkpoint (GADD45γ (28)) and cell proliferation index (Ki-67). The downregulated genes further comprised mitotic kinesin-like motor proteins (29) associated with spindle assembly (Kif11 (30), Kif22 (31), Kif23 (32)), chromosome alignment (Kif2c (33), Kif18a (34)) and cytokinesis (Kif20a (35)); chromosomal proteins such as linker histone H1, nucleosome histones (H2A, H2B, H3) along with topoisomerase IIα (Top2a). Importantly, many of these genes are induced in a tightly cell cycle-dependent manner (c-myb, Runx1 (36), cyclins, Nek2 (37), Ect2 (38), Aurora A (39), Ki-67, histones (40), Top2a (41)), suggesting that the downregulation of these genes represent overall growth inhibitory state of Rac1+/− ECs (fig. S2A). These observations conform well to the previously appreciated roles of Rac1 in critically mediating cell cycle progression (42–44), thus confirming the validity of our microarray data.
Anti-angiogenic phenotype of Rac1+/− ECs.
The anti-angiogenic phenotype of Rac1+/− ECs (fig. S2) is correlated with the growth inhibition and coordinate upregulation of angiostatic genes, Robo4 (45), ADAMTS1 (46) and thrombospondin 2 (47) (Table 3), and consistent with the retardation in ischemia-induced angiogenesis of EC-Rac1+/− mice (48).
Fig. S1. CD31 labeling of Rac1+/− and Rac1+/+ and Rac1+/− mouse ECs were immunolabeled with anti-CD31 (PECAM1) antibody or control IgG. The fraction of Cd31-stained Rac1+/+ (red) and Rac1+/− (yellow) ECs was determined by FACS analysis in comparison to the IgG-stained control ECs (green).
Fig. S2. Rac1+/− ECs show retarded proliferation, migration and capillary formation. (A) Time-course assessment by crystal violet staining of the cell number of Rac1+/+ and Rac1+/− mouse endothelial cells cultured in the presence of 20% serum. Values are means ± SEM triplicates (**, P < 0.01; ANOVA). (B) EC confluent monolayer was scratched at the width of 1.5 mm. Thereafter, the cells were allowed to migrate in the presence of 20% serum. Photographs were taken at 48 hr. Scale bar 1 mm. (C) Rac1+/+ and Rac1+/− mouse aortic segments were embedded in Matrigel, and cultured under the presence of 20% serum to allow for capillary sprouting. Photographs were taken at day 8. Scale bar, 1 mm.
Fig. S3. CD31 labeling of mouse brain ECs (MBECs). MBECs were double stained with Hoechst 33342 (nulcear stain) and anti-CD31 (PECAM1) antibody. CD31 was visualized with Alexa 488-conjugated anti-rat IgG antibody. The fluorescence images were captured with confocal microscope. Scale bar = 50 μm.
Table S1. Representative gene sets enriched in Rac1+/+ mouse endothelial cells
Table S2. Representative gene sets enriched in Rac1+/− mouse endothelial cells
Table S3. Transcription factors differentially expressed between Rac1+/− and Rac1+/+ mouse endothelial cells
Table S4. List of genes differentially expressed in Rac1+/− and Rac1+/+ mouse endothelial cells.
Supplementary References and Notes
1. M. Glogauer, C. C. Marchal, F. Zhu, A. Worku, B. E. Clausen, I. Foerster, P. Marks, G. P. Downey, M. Dinauer, D. J. Kwiatkowski, Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol 170, 5652−5657 (2003).
2. P. A. Koni, S. K. Joshi, U. A. Temann, D. Olson, L. Burkly, R. A. Flavell, Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med 193, 741−754 (2001).
3. H. H. Kim, N. Sawada, G. Soydan, H. S. Lee, Z. Zhou, S. K. Hwang, C. Waeber, M. A. Moskowitz, J. K. Liao, Additive effects of statin and dipyridamole on cerebral blood flow and stroke protection. J Cereb Blood Flow Metab, (2008).
4. Y. C. Lim, G. Garcia-Cardena, J. R. Allport, M. Zervoglos, A. J. Connolly, M. A. Gimbrone, Jr., F. W. Luscinskas, Heterogeneity of endothelial cells from different organ sites in T-cell subset recruitment. Am J Pathol 162, 1591−1601 (2003).
5. E. E. Sander, S. van Delft, J. P. ten Klooster, T. Reid, R. A. van der Kammen, F. Michiels, J. G. Collard, Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J Cell Biol 143, 1385−1398 (1998).
6. G. Dennis, Jr., B. T. Sherman, D. A. Hosack, J. Yang, W. Gao, H. C. Lane, R. A. Lempicki, DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4, P3 (2003).
7. A. Subramanian, P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, A. Paulovich, S. L. Pomeroy, T. R. Golub, E. S. Lander, J. P. Mesirov, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545−15550 (2005).
8. M. P. Quinlan, Rac regulates the stability of the adherens junction and its components, thus affecting epithelial cell differentiation and transformation. Oncogene 18, 6434−6442 (1999).
9. R. A. Stockton, E. Schaefer, M. A. Schwartz, p21-activated kinase regulates endothelial permeability through modulation of contractility. J Biol Chem 279, 46621−46630 (2004).
10. H. Gao, M. Liang, A. Bergdahl, A. Hamren, M. W. Lindholm, K. Dahlman-Wright, B. O. Nilsson, Estrogen attenuates vascular expression of inflammation associated genes and adhesion of monocytes to endothelial cells. Inflamm Res 55, 349−353 (2006).
11. Z. Wu, F. M. Hofman, B. V. Zlokovic, A simple method for isolation and characterization of mouse brain microvascular endothelial cells. J Neurosci Methods 130, 53−63 (2003).
12. L. Song, J. S. Pachter, Culture of murine brain microvascular endothelial cells that maintain expression and cytoskeletal association of tight junction-associated proteins. In Vitro Cell Dev Biol Anim 39, 313−320 (2003).
13. C. Weidenfeller, S. Schrot, A. Zozulya, H. J. Galla, Murine brain capillary endothelial cells exhibit improved barrier properties under the influence of hydrocortisone. Brain Res 1053, 162−174 (2005).
14. S. Orsulic, Y. Li, R. A. Soslow, L. A. Vitale-Cross, J. S. Gutkind, H. E. Varmus, Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell 1, 53−62 (2002).
15. J. R. Brown, E. Nigh, R. J. Lee, H. Ye, M. A. Thompson, F. Saudou, R. G. Pestell, M. E. Greenberg, Fos family members induce cell cycle entry by activating cyclin D1. Mol Cell Biol 18, 5609−5619 (1998).
16. Y. Nakata, S. Shetzline, C. Sakashita, A. Kalota, R. Rallapalli, S. I. Rudnick, Y. Zhang, S. G. Emerson, A. M. Gewirtz, c-Myb contributes to G2/M cell cycle transition in human hematopoietic cells by direct regulation of cyclin B1 expression. Mol Cell Biol 27, 2048−2058 (2007).
17. V. P. Sukhatme, Early transcriptional events in cell growth: the Egr family. J Am Soc Nephrol 1, 859−866 (1990).
18. F. Bernardin, A. D. Friedman, AML1 stimulates G1 to S progression via its transactivation domain. Oncogene 21, 3247−3252 (2002).
19. T. Hirota, N. Kunitoku, T. Sasayama, T. Marumoto, D. Zhang, M. Nitta, K. Hatakeyama, H. Saya, Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell 114, 585−598 (2003).
20. L. Schneider, F. Essmann, A. Kletke, P. Rio, H. Hanenberg, W. Wetzel, K. Schulze-Osthoff, B. Nurnberg, R. P. Piekorz, The transforming acidic coiled coil 3 protein is essential for spindle-dependent chromosome alignment and mitotic survival. J Biol Chem 282, 29273−29283 (2007).
21. R. Bayliss, T. Sardon, I. Vernos, E. Conti, Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell 12, 851−862 (2003).
22. V. Joukov, A. C. Groen, T. Prokhorova, R. Gerson, E. White, A. Rodriguez, J. C. Walter, D. M. Livingston, The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell 127, 539−552 (2006).
23. G. Fang, H. Yu, M. W. Kirschner, The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev 12, 1871−1883 (1998).
24. A. Giodini, M. J. Kallio, N. R. Wall, G. J. Gorbsky, S. Tognin, P. C. Marchisio, M. Symons, D. C. Altieri, Regulation of microtubule stability and mitotic progression by survivin. Cancer Res 62, 2462−2467 (2002).
25. A. M. Fry, P. Meraldi, E. A. Nigg, A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J 17, 470−481 (1998).
26. T. Tatsumoto, X. Xie, R. Blumenthal, I. Okamoto, T. Miki, Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol 147, 921−928 (1999).
27. J. Schmitz, E. Watrin, P. Lenart, K. Mechtler, J. M. Peters, Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Curr Biol 17, 630−636 (2007).
28. X. W. Wang, Q. Zhan, J. D. Coursen, M. A. Khan, H. U. Kontny, L. Yu, M. C. Hollander, P. M. O'Connor, A. J. Fornace, Jr., C. C. Harris, GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci U S A 96, 3706−3711 (1999).
29. H. Miki, Y. Okada, N. Hirokawa, Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol 15, 467−476 (2005).
30. A. Blangy, H. A. Lane, P. d'Herin, M. Harper, M. Kress, E. A. Nigg, Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 83, 1159−1169 (1995).
31. A. A. Levesque, D. A. Compton, The chromokinesin Kid is necessary for chromosome arm orientation and oscillation, but not congression, on mitotic spindles. J Cell Biol 154, 1135−1146 (2001).
32. R. Neef, U. R. Klein, R. Kopajtich, F. A. Barr, Cooperation between mitotic kinesins controls the late stages of cytokinesis. Curr Biol 16, 301−307 (2006).
33. P. D. Andrews, Y. Ovechkina, N. Morrice, M. Wagenbach, K. Duncan, L. Wordeman, J. R. Swedlow, Aurora B regulates MCAK at the mitotic centromere. Dev Cell 6, 253−268 (2004).
34. J. Stumpff, G. von Dassow, M. Wagenbach, C. Asbury, L. Wordeman, The kinesin-8 motor Kif18A suppresses kinetochore movements to control mitotic chromosome alignment. Dev Cell 14, 252−262 (2008).
35. E. Hill, M. Clarke, F. A. Barr, The Rab6-binding kinesin, Rab6-KIFL, is required for cytokinesis. EMBO J 19, 5711−5719 (2000).
36. F. Bernardin-Fried, T. Kummalue, S. Leijen, M. I. Collector, K. Ravid, A. D. Friedman, AML1/RUNX1 increases during G1 to S cell cycle progression independent of cytokine-dependent phosphorylation and induces cyclin D3 gene expression. J Biol Chem 279, 15678−15687 (2004).
37. S. J. Schultz, A. M. Fry, C. Sutterlin, T. Ried, E. A. Nigg, Cell cycle-dependent expression of Nek2, a novel human protein kinase related to the NIMA mitotic regulator of Aspergillus nidulans. Cell Growth Differ 5, 625−635 (1994).
38. H. Sakata, J. S. Rubin, W. G. Taylor, T. Miki, A Rho-specific exchange factor Ect2 is induced from S to M phases in regenerating mouse liver. Hepatology 32, 193−199 (2000).
39. M. Tanaka, A. Ueda, H. Kanamori, H. Ideguchi, J. Yang, S. Kitajima, Y. Ishigatsubo, Cell-cycle-dependent regulation of human aurora A transcription is mediated by periodic repression of E4TF1. J Biol Chem 277, 10719−10726 (2002).
40. W. F. Marzluff, R. J. Duronio, Histone mRNA expression: multiple levels of cell cycle regulation and important developmental consequences. Curr Opin Cell Biol 14, 692−699 (2002).
41. K. Kimura, M. Saijo, M. Ui, T. Enomoto, Growth state- and cell cycle-dependent fluctuation in the expression of two forms of DNA topoisomerase II and possible specific modification of the higher molecular weight form in the M phase. J Biol Chem 269, 1173−1176 (1994).
42. M. F. Olson, A. Ashworth, A. Hall, An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 269, 1270−1272 (1995).
43. D. Michaelson, W. Abidi, D. Guardavaccaro, M. Zhou, I. Ahearn, M. Pagano, M. R. Philips, Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J Cell Biol 181, 485−496 (2008).
44. C. S. Hill, J. Wynne, R. Treisman, The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81, 1159−1170 (1995).
45. C. A. Jones, N. R. London, H. Chen, K. W. Park, D. Sauvaget, R. A. Stockton, J. D. Wythe, W. Suh, F. Larrieu-Lahargue, Y. S. Mukouyama, P. Lindblom, P. Seth, A. Frias, N. Nishiya, M. H. Ginsberg, H. Gerhardt, K. Zhang, D. Y. Li, Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nat Med 14, 448−453 (2008).
46. F. Vazquez, G. Hastings, M. A. Ortega, T. F. Lane, S. Oikemus, M. Lombardo, M. L. Iruela-Arispe, METH-1, a human ortholog of ADAMTS-1, and METH-2 are members of a new family of proteins with angio-inhibitory activity. J Biol Chem 274, 23349−23357 (1999).
47. B. Lange-Asschenfeldt, W. Weninger, P. Velasco, T. R. Kyriakides, U. H. von Andrian, P. Bornstein, M. Detmar, Increased and prolonged inflammation and angiogenesis in delayed-type hypersensitivity reactions elicited in the skin of thrombospondin-2--deficient mice. Blood 99, 538−545 (2002).
48. N. Sawada, S. Salomone, H. H. Kim, D. J. Kwiatkowski, J. K. Liao, Regulation of endothelial nitric oxide synthase and postnatal angiogenesis by Rac1. Circ Res 103, 360−368 (2008).
Footnotes
www.sciencesignaling.org/cgi/content/full/2/61/ra10/DC1
Fig. S1. CD31 labeling of Rac1+/− and Rac1+/+ ECs.
Fig. S2. Rac1+/− ECs show retarded proliferation, migration, and capillary formation.
Fig. S3. CD31 labeling of MBECs.
Table S1. Representative gene sets enriched in Rac1+/+ mouse ECs.
Table S2. Representative gene sets enriched in Rac1+/− mouse ECs.
Table S3. Transcription factors differentially expressed between Rac1+/− and Rac1+/+ mouse ECs.
Table S4. List of differentially expressed genes between Rac1+/− and Rac1+/+ mouse ECs.
Citation: N. Sawada, H.-H Kim, M. A. Moskowitz, J. K. Liao, Rac1 is a critical mediator of endothelium-derived neurotrophic activity. Sci. Signal. 2, ra10 (2009).
REFERENCES AND NOTES
- 1.Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat. Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- 2.Abbott NJ, Ronnback L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 3.Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50:329–339. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- 4.Gregg D, Rauscher FM, Goldschmidt-Clermont PJ. Rac regulates cardiovascular superoxide through diverse molecular interactions: More than a binary GTP switch. Am. J. Physiol. Cell Physiol. 2003;285:C723–C734. doi: 10.1152/ajpcell.00230.2003. [DOI] [PubMed] [Google Scholar]
- 5.Tzima E. Role of small GTPases in endothelial cytoskeletal dynamics and the shear stress response. Circ. Res. 2006;98:176–185. doi: 10.1161/01.RES.0000200162.94463.d7. [DOI] [PubMed] [Google Scholar]
- 6.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 7.Pardridge WM. Drug and gene delivery to the brain: The vascular route. Neuron. 2002;36:555–558. doi: 10.1016/s0896-6273(02)01054-1. [DOI] [PubMed] [Google Scholar]
- 8.Bespalov MM, Saarma M. GDNF family receptor complexes are emerging drug targets. Trends Pharmacol. Sci. 2007;28:68–74. doi: 10.1016/j.tips.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Wu D. Neuroprotection in experimental stroke with targeted neurotrophins. NeuroRx. 2005;2:120–128. doi: 10.1602/neurorx.2.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 12.Leventhal C, Rafii S, Rafii D, Shahar A, Goldman SA. Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol. Cell. Neurosci. 1999;13:450–464. doi: 10.1006/mcne.1999.0762. [DOI] [PubMed] [Google Scholar]
- 13.Guo S, Kim WJ, Lok J, Lee SR, Besancon E, Luo BH, Stins MF, Wang X, Dedhar S, Lo EH. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. U.S.A. 2008;105:7582–7587. doi: 10.1073/pnas.0801105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawada N, Salomone S, Kim HH, Kwiatkowski DJ, Liao JK. Regulation of endothelial nitric oxide synthase and postnatal angiogenesis by Rac1. Circ. Res. 2008;103:360–368. doi: 10.1161/CIRCRESAHA.108.178897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: Impaired lymphocyte migration to bone marrow. J. Exp. Med. 2001;193:741–754. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, Downey GP, Dinauer M, Kwiatkowski DJ. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J. Immunol. 2003;170:5652–5657. doi: 10.4049/jimmunol.170.11.5652. [DOI] [PubMed] [Google Scholar]
- 17.Kim HH, Sawada N, Soydan G, Lee HS, Zhou Z, Hwang SK, Waeber C, Moskowitz MA, Liao JK. Additive effects of statin and dipyridamole on cerebral blood flow and stroke protection. J. Cereb. Blood Flow Metab. 2008;28:1285–1293. doi: 10.1038/jcbfm.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dennis G, Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 19.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- 21.Bierl C, Voetsch B, Jin RC, Handy DE, Loscalzo J. Determinants of human plasma glutathione peroxidase (GPx-3) expression. J. Biol. Chem. 2004;279:26839–26845. doi: 10.1074/jbc.M401907200. [DOI] [PubMed] [Google Scholar]
- 22.Voetsch B, Jin RC, Bierl C, Benke KS, Kenet G, Simioni P, Ottaviano F, Damasceno BP, Annichino-Bizacchi JM, Handy DE, Loscalzo J. Promoter polymorphisms in the plasma glutathione peroxidase (GPx-3) gene: A novel risk factor for arterial ischemic stroke among young adults and children. Stroke. 2007;38:41–49. doi: 10.1161/01.STR.0000252027.53766.2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gabai VL, Sherman MY. Invited review: Interplay between molecular chaperones and signaling pathways in survival of heat shock. J. Appl. Physiol. 2002;92:1743–1748. doi: 10.1152/japplphysiol.01101.2001. [DOI] [PubMed] [Google Scholar]
- 24.Kamradt MC, Chen F, Sam S, Cryns VL. The small heat shock protein αB-crystallin negatively regulates apoptosis during myogenic differentiation by inhibiting caspase-3 activation. J. Biol. Chem. 2002;277:38731–38736. doi: 10.1074/jbc.M201770200. [DOI] [PubMed] [Google Scholar]
- 25.Beere HM. Stressed to death: Regulation of apoptotic signaling pathways by the heat shock proteins. Sci. STKE. 2001;2001:RE1. doi: 10.1126/stke.2001.93.re1. [DOI] [PubMed] [Google Scholar]
- 26.Bornstein P, Agah A, Kyriakides TR. The role of thrombospondins 1 and 2 in the regulation of cell-matrix interactions, collagen fibril formation, and the response to injury. Int. J. Biochem. Cell Biol. 2004;36:1115–1125. doi: 10.1016/j.biocel.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 27.Bornstein P. Thrombospondins as matricellular modulators of cell function. J. Clin. Invest. 2001;107:929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-beta by thrombospondin-1: Mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11:59–69. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]
- 29.Dallas SL, Sivakumar P, Jones CJ, Chen Q, Peters DM, Mosher DF, Humphries MJ, Kielty CM. Fibronectin regulates latent transforming growth factor-β (TGFβ) by controlling matrix assembly of latent TGFβ-binding protein-1. J. Biol. Chem. 2005;280:18871–18880. doi: 10.1074/jbc.M410762200. [DOI] [PubMed] [Google Scholar]
- 30.Unsicker K, Krieglstein K. Co-activation of TGF-ss and cytokine signaling pathways are required for neurotrophic functions. Cytokine Growth Factor Rev. 2000;11:97–102. doi: 10.1016/s1359-6101(99)00033-7. [DOI] [PubMed] [Google Scholar]
- 31.Lange-Asschenfeldt B, Weninger W, Velasco P, Kyriakides TR, von Andrian UH, Bornstein P, Detmar M. Increased and prolonged inflammation and angiogenesis in delayed-type hypersensitivity reactions elicited in the skin of thrombospondin-2–deficient mice. Blood. 2002;99:538–545. doi: 10.1182/blood.v99.2.538. [DOI] [PubMed] [Google Scholar]
- 32.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 33.Guo F, Debidda M, Yang L, Williams DA, Zheng Y. Genetic deletion of Rac1 GTPase reveals its critical role in actin stress fiber formation and focal adhesion complex assembly. J. Biol. Chem. 2006;281:18652–18659. doi: 10.1074/jbc.M603508200. [DOI] [PubMed] [Google Scholar]
- 34.Harris ES, Rouiller I, Hanein D, Higgs HN. Mechanistic differences in actin bundling activity of two mammalian formins, FRL1 and mDia2. J. Biol. Chem. 2006;281:14383–14392. doi: 10.1074/jbc.M510923200. [DOI] [PubMed] [Google Scholar]
- 35.Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H, Simburger KS, Leitner ML, Araki T, Johnson EM, Jr., Milbrandt J. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRα3-RET receptor complex. Neuron. 1998;21:1291–1302. doi: 10.1016/s0896-6273(00)80649-2. [DOI] [PubMed] [Google Scholar]
- 36.Reuss B, von Bohlen und Halbach O. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 2003;313:139–157. doi: 10.1007/s00441-003-0756-7. [DOI] [PubMed] [Google Scholar]
- 37.Harvey BK, Hoffer BJ, Wang Y. Stroke and TGF-β proteins: Glial cell line–derived neurotrophic factor and bone morphogenetic protein. Pharmacol. Ther. 2005;105:113–125. doi: 10.1016/j.pharmthera.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Junier MP. What role(s) for TGFα in the central nervous system? Prog. Neurobiol. 2000;62:443–473. doi: 10.1016/s0301-0082(00)00017-4. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Chang CF, Morales M, Chiang YH, Hoffer J. Protective effects of glial cell line–derived neurotrophic factor in ischemic brain injury. Ann. N. Y. Acad. Sci. 2002;962:423–437. doi: 10.1111/j.1749-6632.2002.tb04086.x. [DOI] [PubMed] [Google Scholar]
- 40.Honma Y, Araki T, Gianino S, Bruce A, Heuckeroth R, Johnson E, Milbrandt J. Artemin is a vascular-derived neurotropic factor for developing sympathetic neurons. Neuron. 2002;35:267–282. doi: 10.1016/s0896-6273(02)00774-2. [DOI] [PubMed] [Google Scholar]
- 41.Damon DH, Teriele JA, Marko SB. Vascular-derived artemin: A determinant of vascular sympathetic innervation? Am. J. Physiol. Heart Circ. Physiol. 2007;293:H266–H273. doi: 10.1152/ajpheart.00859.2006. [DOI] [PubMed] [Google Scholar]
- 42.Baloh RH, Gorodinsky A, Golden JP, Tansey MG, Keck CL, Popescu NC, Johnson EM, Jr., Milbrandt J. GFRα3 is an orphan member of the GDNF/neurturin/persephin receptor family. Proc. Natl. Acad. Sci. U.S.A. 1998;95:5801–5806. doi: 10.1073/pnas.95.10.5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenblad C, Grønborg M, Hansen C, Blom N, Meyer M, Johansen J, Dagø L, Kirik D, Patel UA, Lundberg C, Trono D, Björklund A, Johansen TE. In vivo protection of nigral dopamine neurons by lentiviral gene transfer of the novel GDNF-family member neublastin/artemin. Mol. Cell. Neurosci. 2000;15:199–214. doi: 10.1006/mcne.1999.0817. [DOI] [PubMed] [Google Scholar]
- 44.Ducray A, Krebs SH, Schaller B, Seiler RW, Meyer M, Widmer HR. GDNF family ligands display distinct action profiles on cultured GABAergic and serotonergic neurons of rat ventral mesencephalon. Brain Res. 2006;1069:104–112. doi: 10.1016/j.brainres.2005.11.056. [DOI] [PubMed] [Google Scholar]
- 45.Bonde C, Kristensen BW, Blaabjerg M, Johansen TE, Zimmer J, Meyer M. GDNF and neublastin protect against NMDA-induced excitotoxicity in hippocampal slice cultures. Neuroreport. 2000;11:4069–4073. doi: 10.1097/00001756-200012180-00032. [DOI] [PubMed] [Google Scholar]
- 46.Ceyhan GO, Bergmann F, Kadihasanoglu M, Erkan M, Park W, Hinz U, Giese T, Müller MW, Büchler MW, Giese NA, Friess H. The neurotrophic factor artemin influences the extent of neural damage and growth in chronic pancreatitis. Gut. 2007;56:534–544. doi: 10.1136/gut.2006.105528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Viñals F, Pouysségur J. Transforming growth factor β1 (TGF-β1) promotes endothelial cell survival during in vitro angiogenesis via an autocrine mechanism implicating TGF-α signaling. Mol. Cell. Biol. 2001;21:7218–7230. doi: 10.1128/MCB.21.21.7218-7230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eslamboli A, Georgievska B, Ridley RM, Baker HF, Muzyczka N, Burger C, Mandel RJ, Annett L, Kirik D. Continuous low-level glial cell line–derived neurotrophic factor delivery using recombinant adeno-associated viral vectors provides neuroprotection and induces behavioral recovery in a primate model of Parkinson's disease. J. Neurosci. 2005;25:769–777. doi: 10.1523/JNEUROSCI.4421-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White BC, Sullivan JM, DeGracia DJ, O'Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS. Brain ischemia and reperfusion: Molecular mechanisms of neuronal injury. J. Neurol. Sci. 2000;179:1–33. doi: 10.1016/s0022-510x(00)00386-5. [DOI] [PubMed] [Google Scholar]
- 50.Sakai T, Johnson KJ, Murozono M, Sakai K, Magnuson MA, Wieloch T, Cronberg T, Isshiki A, Erickson HP, Fässler R. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat. Med. 2001;7:324–330. doi: 10.1038/85471. [DOI] [PubMed] [Google Scholar]
- 51.Reuss B, Dono R, Unsicker K. Functions of fibroblast growth factor (FGF)–2 and FGF-5 in astroglial differentiation and blood-brain barrier permeability: Evidence from mouse mutants. J. Neurosci. 2003;23:6404–6412. doi: 10.1523/JNEUROSCI.23-16-06404.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J. Comp. Neurol. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 53.Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- 54.Cleaver O, Melton DA. Endothelial signaling during development. Nat. Med. 2003;9:661–668. doi: 10.1038/nm0603-661. [DOI] [PubMed] [Google Scholar]
- 55.Lim YC, Garcia-Cardena G, Allport JR, Zervoglos M, Connolly AJ, Gimbrone MA, Jr., Luscinskas FW. Heterogeneity of endothelial cells from different organ sites in T-cell subset recruitment. Am. J. Pathol. 2003;162:1591–1601. doi: 10.1016/S0002-9440(10)64293-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.We thank D. J. Kwiatkowski for providing us with conditional Rac1 mice. This work was supported by grants from the NIH (HL052233, HL080187, and NS101828). N.S. was supported by the fellowships from the Uehara Memorial Foundation, the Cell Science Research Foundation, and Mochida Memorial Foundation for Medical and Pharmaceutical Research. There are no disclosures or conflicts of interest for all of the authors.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials for
Rac1 Is a Critical Mediator of Endothelium-Derived Neurotrophic Activity
Naoki Sawada, Hyung-Hwan Kim, Michael A. Moskowitz, James K. Liao*
*To whom correspondence should be addressed. E-mail: jliao@rics.bwh.harvard.edu
Published 10 March 2009, Sci. Signal. 2, ra10 (2009)
DOI: 10.1126/scisignal.2000162
This PDF file includes:
Materials and Methods
Supplementary Text
Fig. S1. CD31 labeling of Rac1+/− and Rac1+/+ ECs.
Fig. S2. Rac1+/− ECs show retarded proliferation, migration, and capillary formation.
Fig. S3. CD31 labeling of MBECs.
Table S1. Representative gene sets enriched in Rac1+/+ mouse ECs.
Table S2. Representative gene sets enriched in Rac1+/− mouse ECs.
Table S3. Transcription factors differentially expressed between Rac1+/− and Rac1+/+ mouse ECs.
Table S4. List of differentially expressed genes between Rac1+/− and Rac1+/+ mouse ECs.
Supplementary material
Supplementary Materials and Methods
Generation of EC-Rac1+/− mice.
All experimental procedures on animals were performed by using protocols approved by the Harvard Medical School's Standing Committee on Animal Welfare and Protection and are in accordance with the Guide for the Care and Use of Laboratory Animals, published by NIH. Mice were bred and maintained with a 12/12-hour light/dark cycle and fed standard chow with tap water available ad libitum. Age-matched male littermates (12−20 wk) were used for test and control groups in all experiments. Rac1 conditional allele knock-in mice were developed as described (1), and backcrossed at least eight times onto the C57BL/6 strain. Tie2-Cre transgenic mice were obtained from Jackson Laboratory and maintained on C57BL/6 background (2) . Rac1flox/flox mice were crossed with Tie2-Cre Tg mice to generate EC-Rac1+/− (Tie2-Cre Rac1flox/+) and control (Rac1flox/+) mice. For some experiments, global Rac1 haploinsufficient (Rac1+/−) and control (Rac1+/+) mice were also used (1). The genotyping PCR primers for Rac1 conditional and null alleles are described previously (1). The genotyping primers used for Tie2-Cre transgenic mice are: 5’-TGA TGA GGT TCG CAA GAA CC; 5’-ACC AGT TTA GTT ACC CCC AG.
Transient middle cerebral artery occlusion model.
Mice were anesthetized with 2% isoflurane mixed in 70% N2O and 30% O2, and then maintained on 1.5% to 2% isoflurane in a similar gaseous mixture. Transient focal cerebral ischemia was performed using an 8−0 nylon monofilament coated with silicone, which was introduced into the internal carotid artery via the external carotid artery, and then advanced 10 mm distal to the carotid bifurcation to occlude the MCA as described (3). Laser Doppler flowmetry of relative CBF was used to verify successful occlusion (<20% baseline value). The MCA was occluded 2 hr, followed by withdrawal of filament and reperfusion for 22 hr. Relative CBF returned to >95% of baseline values, indicating almost complete reperfusion without residual occlusion.
Cerebral infarct volume was measured 22 hr after reperfusion. Infarct area was measured in 2-mm-thick coronal brain sections stained with 2,3,5-triphenyltetrazolium chloride and quantitated with MCID Elite M6 image-analysis software (Interfocus Imaging). Cerebral infarct volume was determined by summing up the infarcted areas. The infarct and edema volumes relative to the non-ischemic hemisphere were calculated as follows: Infarct volume (%) = (non-ischemic hemisphere vol. (mm3) – ischemic hemisphere non-infarcted region vol. (mm3))/(non-ischemic hemisphere vol. (mm3)) × 100. Edema volume (%) = (ischemic hemisphere vol. (mm3) – non-ischemic hemisphere vol. (mm3))/(non-ischemic hemisphere vol. (mm3)) × 100.
The neurological deficit score (NDS) was determined at 20 hr post-reperfusion by two observers, who were blinded to the identity of the mice or treatment protocol. The following scoring system was used: 0, no motor deficits (normal); 1, flexion of the contralateral torso and forelimb on lifting the animal by the tail (mild); 2, circling to the contralateral side but normal posture at rest (moderate); 3, leaning to the contralateral side at rest (severe); and 4, no spontaneous movement (critical).
Absolute CBF in ischemic and non-ischemic hemispheres was measured at the end of 2 hr MCAo using an indicator fractionation technique as described, with slight modifications (3). Briefly, the right jugular vein and left femoral artery of anesthetized mice were cannulated. The mice received 1 μCi of N-isopropyl-[methyl 1,3-14C] p-iodoamphetamine from the right jugular vein as a bolus. A volume of 100 μL of arterial blood samples from the left femoral artery was collected for 20 sec (0.3 ml/min), and then the animal was decapitated and the whole brain was removed. The brain was immediately cut into the 2 hemispheres and frozen in chilled isopentane solution chilled with dry ice immediately. The frozen hemispheres were weighed and digested with Scintigest (Fischer Scientific) at 50°C for 6 h. Scintillation fluid and H2O2 were added to the sample and they were shaken together for 12 h. The radioactivity in the blood and brain samples was measured by liquid scintillation spectrometry (RackBeta 1209). Cerebral blood flow in each hemisphere was calculated as follows: CBF (ml/100 g per min) = (hemisphere count (c.p.m.) × 0.3 (ml/min))/(blood count (c.p.m.) × hemisphere weight (g)) × 100. The relative CBF change in ischemic hemisphere was determined as: CBF change (%) = (ischemic hemisphere CBF (ml/100 g per min) – non-ischemic hemisphere CBF (ml/100 g per min))/(non-ischemic hemisphere CBF (ml/100 g per min)) × 100.
Isolation and culture of mouse primary ECs.
Mouse primary ECs were isolated from the heart by an affinity selection method using Dynabeads sheep anti-rat IgG (Invitrogen) and anti-PECAM1 antibody (BD Biosciences) as described (4). The cells were cultivated in DMEM containing 20% FCS, heparin (100 μg/ml), penicillin (100 U/ml), streptomycin (100 μg/ml) and endothelial cell growth factor (ECGF, 50 μg/ml), and grown on dishes coated with 0.1% gelatin. After 8 days, the cells were trypsinized and underwent the second selection using Dynabeads plus anti-ICAM2 antibody (BD Biosciences). Mouse ECs were used for experiments at passage 3−5.
GTP-Rac1 pull down assay and immunoblotting analysis.
pGEX-2T-PAK-CRIB domain construct is a gift from Dr. John G. Collard. Preparation of GST-PAK-CD and GTP-Rac1 pull down assay was performed as described (5). The GTP-Rac1, total Rac1 and actin protein levels in mouse EC lysate were determined by immunoblotting analysis with Rac1 (BD Biosciences) and actin (Sigma) antibodies. The blots were subsequently probed with HRP-conjugated secondary antibody and visualized by Enhanced Chemiluminescence (GE Health Care).
Microarray expression profiling.
Rac1+/− and Rac1+/+ mouse primary ECs were isolated from 6 mice/genotype derived from 2 age-matched litters (12 wk). Following the first and the second sorting, subconfluent ECs at passage 4 were used for the isolation of RNA. To validate the purity of the EC population, a portion of the cells was immunolabeled with FITC-conjugated anti-mouse CD31 antibody (eBioscience). Flow cytometric analysis revealed that > 98% of the Rac1+/− and Rac1+/+ ECs were positive for CD31 (fig. S1). High quality total RNA was prepared by 2-step purification through the sequential use of Trizol reagent (Invitrogen) and RNeasy columns (Qiagen) according to the manufacturer's instructions. The quality control of RNA was conducted by measuring the 28S/18S rRNA ratio using Agilent 2100 BioAnalyzer. cDNA labeling, hybridization and image acquisition was performed at Massachusetts General Hospital Microarray Core. Briefly, fluorescent dye labeling was performed using the Atlas Powerscript Fluorescent Labeling Kit (Clontech). Isolated total RNA (8 μg) was reverse transcribed into amine modified cDNA using random hexamers and oligo (dT) primers in 4 replicates per each genotype. The cDNA was labeled via a coupling reaction to the Cy3 (2 replicates) or Cy5 (2 replicates) dye, then purified and concentrated. Equimolar mixtures of Cy3-labeled Rac1+/+ and Cy5-labeled Rac1+/− probes (or dye-swapped combinations) were independently hybridized to 4 microarray slides. The microarray was a glass-based pre-printed array containing PGA Mouse v1.1 probe set (19,549 70-mer oligonucleotides), which provides complete coverage of the 2002 Mouse genome, built off the GenPept database. Images were acquired using Axon (Molecular Devices) 4000B 2-color single-slide scanner. Filtering, normalization of the raw data and basic statistical tests were conducted using BASE software. All spots were sequentially subjected to background subtraction, Lowess normalization, and brightness filtering that removed spots with intensities less than 200. This processing resulted in 12,089 reporters (gene features) comprising 45,311 spots from the 4 slides. The averaged fold ratio of Rac1+/− versus Rac1+/+ was calculated for each reporter and statistically analyzed using 1 sample Student t-test and Wilcoxon signed rank test, yielding 1,431 reporters considered significant at a confidence level of 95% (P < 0.05). The averaged fold ratios distributed with 2 SD of 1.56. Gene reporters with the fold ratio of > 1.5 or < −1.5 were considered differentially expressed between Rac1+/+ and Rac1+/− ECs.
Gene cluster and pathway analysis.
To characterize the enrichment profiles of gene clusters and pathways in Rac1+/− and Rac1+/+ ECs, we conducted 2 different analyses.
DAVID Functional Annotation Clustering (6).
The 138 upregulated (fold ratio > 1.5) and 161 downregulated (fold ratio < −1.5) reporters were imported into DAVID 2007 software (http://david.abcc.ncifcrf.gov/home.jsp) and collapsed to 130 and 138 unique genes, respectively, using the DAVID accession conversion tool. The upregulated and downregulated gene lists were separately uploaded for gene annotation analysis. Enrichment of a given gene annotation in each gene list against the default Mus Musculus background was determined by EASE score, a modified Fisher Exact P-Value. The DAVID default setting was used for the source databases of gene annotation: Gene Ontology (GOTERM_BP_ALL, GOTERM_CC_ALL, GOTERM_MF_ALL), Protein Domains (INTERPRO, PIR_SUPERFAMILY, SMART), Pathways (BIOCARTA, KEGG_PATHWAY), and Functional Categories (COG_ONTOLOGY, SP_PIR_KEYWORDS, UP_SEQ_FEATURE). Subsequently, Functional Annotation Clustering was performed to measure relationships among the annotation terms based on the degrees of their co-association genes to group the similar, redundant, and heterogenous contents from the same or different resources into annotation groups. The Functional Annotation Clustering integrates Kappa statistics to measure the common genes between two annotations, and fuzzy heuristic clustering to classify the groups of similar annotations according to kappa values. The biological significance of functional annotation clusters was ranked by the Enrichment Score, which is the geometric mean (in –log scale) of member's EASE scores within each cluster.
Gene Set Enrichment Analysis (GSEA) (7).
To test for sets of related genes that might be systematically altered in Rac1+/− ECs, we used Gene Set Enrichment Analysis (GSEA), a method that combines information from members of previously defined sets of genes to increase signal relative to noise and improve statistical power to detect subtle changes. Complete details on the method for this analysis, the software and information on the biological data sets are available at http://www.broad.mit.edu/gsea/index.jsp. Briefly, genes from the microarray were first ranked according to the expression difference (signal to noise ratio) between Rac1+/− and Rac1+/+ ECs. The standard GSEA null hypothesis is that the rank ordering of the genes in a given comparison is random and not associated with the order of the genes and/or treatment. The extent of association was then measured by a non-parametric running sum statistic termed the enrichment score (ES), and the maximum ES (MES) was recorded over each gene set. Phenotype permutation testing was used to assess the statistical significance of the MES, which was calculated as the fraction of 1,000 random permutations of the gene list. The unadjusted nominal P value estimated the statistical significance of a gene set without adjusting for gene set size or multiple hypothesis testing, whereas the false discovery rate (FDR) statistic adjusts for both. In the present study, we used curated gene sets (C2), archived at Molecular Signature Database, which were collected from various sources such as online pathway databases, publications in PubMed, and knowledge of domain experts. Among these gene sets, 1782 sets with the gene set size > 15 were used for analysis, and 161 gene sets were significantly upregulated in Rac1+/−, whereas 145 sets were enriched in Rac1+/+ ECs at FDR < 25%.
Quantitative RT-PCR.
Total RNA was extracted from the mice brains and ECs using RNeasy mini kit (Qiagen) with DNase I treatment, and 50−100 ng of total RNA was used for one-step real-time RT-PCR using QuantiTect SYBR Green RT-PCR kit (Qiagen) according to the manufacturer's instruction. The reaction was performed with 7900HT Fast Real-Time PCR System (Applied Biosystems). The mRNA level was normalized with that of either GAPDH mRNA or 18S rRNA. The PCR primers used are: Rac1, 5’-GAG ACG GAG CTG TTG GTA AAA, 5’- ATA GGC CCA GAT TCA CTG GTT; Artemin, 5’-CCC TAG CTG TTC TAG CCC TG, 5’-AGG GTT CTT TCG CTG CAC AA; FGF5, 5’-AAG TAG CGC GAC GTT TTC TTC, 5’- CTG GAA ACT GCT ATG TTC CGA G; BMP6, 5’-AGA AGC GGG AGA TGC AAA AGG, 5’-GAC AGG GCG TTG TAG AGA TCC; TGFα, 5’-CAC TCT GGG TAC GTG GGT G, 5’-CAC AGG TGA TAA TGA GGA CAG C; Crystallin αB, 5’-GTT CTT CGG AGA GCA CCT GTT, 5’-GAG AGT CCG GTG TCA ATC CAG; VCAM-1, 5’-AGT TGG GGA TTC GGT TGT TCT, 5’-CCC CTC ATT CCT TAC CAC CC; Gpx3, 5’-CCT TTT AAG CAG TAT GCA GGC A, 5’-CAA GCC AAA TGG CCC AAG TT; Thrombospondin 2, 5’-CTG GGC ATA GGG CCA AGA G, 5’-GCT TGA CAA TCC TGT TGA GAT CA; GAPDH, 5’-AGG TCG GTG TGA ACG GAT TTG, 5’-TGT AGA CCA TGT AGT TGA GGT CA; 18S, 5’-AAA TCA GTT ATG GTT CCT TTG GTC, 5’-GCT CTA GAA TTA CCA CAG TTA TCC AA.
Immunofluorescence staining.
Mouse ECs were cultured to subconfluence on Labtek II chamber slides (Nunc) under standard culture conditions. Cells were fixed with 3.7% paraformaldehyde in PBS for 10 min on ice, and then free aldehyde was reduced with 50 mM NH4Cl. The cells were subsequently permeabilized with 0.1% Triton X-100 in PBS for 5 min, and blocked with 1% BSA in PBS for 30 min at room temperature. For F-actin staining, the cells were incubated with 1U/ml Alexa488-conjugated phalloidin (Molecular Probe) for 20 min. For immunostaining, the cells were incubated with rabbit anti-ß tubulin (Lab Vision) or mouse anti-crystallin αB antibody (Stressgen) at 4°C overnight. After wash, the cells were labeled with the corresponding secondary antibody conjugated with Alexa488 (Molecular Probe) for 1 hr at room temperature. The fluorescent images were acquired by TCS SP5 confocal microscope (Leica). F-actin and ßtubulin fluorescence was quantified with Photoshop and Image J softwares.
Hypoxia treatment.
Cells were placed without culture dish covers in humidified airtight chambers (Billups-Rothenberg) perfused with 95% N2 and 5% CO2 at 2 psi for 10 min. The chambers were sealed and maintained at 37°C for indicated time periods. At the end of hypoxia exposure, cells were removed from the chamber and returned to a regular incubator for reoxygenation. Control cultures were incubated in a regular incubator under normoxic condition for the corresponding hypoxic duration.
Assessment of NADPH oxidase activity.
Mouse ECs were grown to confluence on 96-well plates. The cells were treated without or with 10 μM Diphenyleneiodonium chloride (DPI), a NADPH oxidase inhibitor, and then exposed to 4-hr hypoxia and 2-hr reoxygenation. The cells were sequentially pre-equilibrated with Hanks’ Balanced Salt Solution (HBSS) containing 10 mM Sodium diethyldithiocarbamate trihydrate (DETC) and 1 mM DTT for 30 min, and then with 3.3 μM lucigenin for 5 min. The cells were treated with 67 μM NADPH, and the chemiluminescence was captured for 1 min with a luminometer (Victor3, PerkinElmer). The NADPH oxidase activity was determined as DPI-inhibitable lucigenin chemiluminescence.
Assessment of oxidative stress.
Mouse ECs were grown to confluence on 96-well plates. The cells were loaded with 100 μM 2’,7’-dichlorofluorescein diacetate (DCFH-DA) in Krebs-Ringer-HEPES (KRH) buffer, pH 7.4, containing 4% bovine serum albumin and 0.55 mM glucose for 30 min. After the loading buffer was removed, the cells in KRH buffer were exposed to 4-hr hypoxia and 2-hr reoxygenation. The fluorescence caused by oxidation of DCFH-DA to DCF was measured in a plate reader by 485-nm excitation and 530-nm emission filters.
Annexin V binding assay.
Following various apoptosis-inducing conditions, 1−5 × 105 cells were detached with trypsin/EDTA and washed with Annexin V binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). The cells were labeled in 0.2 ml Annexin V binding buffer containing 10 μg/ml Annexin V-EGFP in the dark for 15 min. The cells were pelleted by centrifugation, resuspended in 1 ml binding buffer, and analyzed by flow cytometry with the excitation beam of 488 nm through FL1 detector (Cytomics FC 500, Beckman Coulter). Unstained cells were used as a negative control for background subtraction.
Cell adhesion assay.
Mouse ECs were grown to subconfluence, or kept at confluence for > 4 weeks. Then, the cells were washed 2 times with PBS (Ca++-free), and subjected to standard trypsinization using 0.25% trypsin/1 mM EDTA solution (Invitrogen) at 37°C. The cells were pipetted and observed under light microscope at the intervals of 3−5 min. The strength of cell adhesion was assessed by the time required for obtaining single cell suspensions (8).
Assessment of EC barrier function.
Mouse ECs grown to confluence on Transwell inserts (0.4-μm pore size) were maintained thereafter for ∼10 days. The medium in the upper compartment was replaced with fresh medium containing HRP (20 μg/ml), and the cells were exposed to 4-hr hypoxia and 2-hr reoxygenation. Medium from the bottom well was incubated with assay buffer containing 0.5 mM guaiacol, 50 mM Na2HPO4 and 0.6 mM H2O2. HRP activity was assessed as the formation of O-phenylenediamine determined by absorbance at 470 nm (9).
Assessment of EC-leucocyte attachment.
Monocyte cell line THP-1 was cultured in RPMI 1640 supplemented with 10% FCS. The THP-1 cells were labeled with 1 μCi/ml [3H]-thymidine for 48 hr and washed. Confluent mouse ECs in 6-well plates were subjected to 4-hr hypoxia and 20-hr reoxygenation. The ECs were co-cultured with the radiolabelled THP-1 cells (1 million cells per ml) at a static condition for 3 hr. At the end of incubation, non-adherent THP-1 cells were removed and the ECs were washed 3 times with PBS. The remaining adherent cells were lysed with 1mL of 0.5 M NaOH, and the radioactivity was measured in a scintillation counter (10).
Isolation and culture of neuronal cells and mouse brain ECs.
The human neuroblastoma cell line SH-SY5Y was purchased from ATCC and maintained in a 1:1 mixture of Eagle's Minimum Essential Medium (EMEM) and F-12 medium, supplemented with 10% FCS and penicillin/streptomycin. Primary cortical neurons were isolated from C57/BL6 fetal mouse brains (∼E16). Briefly, dissected cortices were digested with 0.25% trypsin at 37°C for 10min, and DNase I (25 U/ml) for 5 min. The digested cortices were triturated and passed through a 40-μm strainer. The neurons were plated on cover glasses pre-coated with poly-D-lysine (0.1 mg/ml) at a density of 1−1.5 × 106 cells/well in 6-well plates. The cells were maintained in serum-free Neurobasal medium (Invitrogen) supplemented with 1 mM glutamine, 2% B27 supplement and penicillin/streptomycin, and treated with 25 μg/ml glutamate (day 0−3) and 10 μM Ara-C (day 3−4). Thereafter, half of the medium was replaced with fresh medium every 3 days. The neurons were used for experiments at days 14−21. Mouse brain microvascular endothelial cells (MBEC) were isolated as described with modifications (11–13). Age-matched adult EC-Rac1+/− and control mice (n = 6−10 per genotype) from 2−3 litters were used for cell isolation. Dissected cortices were homogenized in ice-cold DMEM/F12 (1:1) using a Dounce tissue grinder (loose pestle) with 5 strokes and further minced by scissors. The homogenate was centrifuged (200 g, 5min, 4°C), and the pellets were resuspended with 15% dextran (MW ∼100,000) in PBS (−) and centrifuged at 10,000 g, 15 min, 4°C. The pellets were suspended and digested in DMEM/F12 containing Collagenase/Dispase (1mg/ml) (Roche) and DNase I (25 μg/ml) at 37°C for 90 min, and centrifuged at 400g, 10 min, room temperature. The pellets were suspended with 45% Percoll in PBS, centrifuged at 20,000 g, 15 min, 4°C. The top layer containing microvessels and ECs were transferred to a new tube, centrifuged at 400 g, and washed once with complete media (DMEM/F12 with 10% horse serum, 10% FCS, heparin, penicillin/streptomycin and ECGF). Cells were plated and grown on gelatin-coated T25 plates, and split at 1:2. More than 95% of MBECs at passage 1 were positive for CD31 staining (fig. S3). MBECs were used for experiments at passage 3−5.
EC-neuron co-culture and assessment of neuronal cell death.
ECs were plated on Transwell culture inserts (0.4-μm pore size) and grown to confluence. ECs and neuronal cells (SH-SY5Y or cortical neurons) were maintained in respective complete media (ECs, upper compartment, 1.5 ml; neuronal cells, lower compartment, 2.6 ml). Following the change of media, co-culture was started by adding inserts with ECs to neuronal cell cultures in 6-well plates. Co-culture proceeded for 48 hr and then the inserts with ECs were removed. SH-SY5Y cells left in the conditioned media were exposed to hypoxic conditions for 24 hr, and the apoptotic cell fraction was assessed through Annexin V-EGFP labeling. Cortical neurons were co-cultured with MBEC in the presence of vehicle, goat anti-mouse artemin neutralizing antibody (0.5 μg/ml), or goat IgG (0.5 μg/ml, R&D) in the lower compartment. Some neurons were treated with blank inserts and recombinant mouse artemin (0−10 ng/ml). The cortical neurons, after co-culture with MBEC, were subjected to oxygen/glucose deprivation (OGD). Briefly, the conditioned media (CM) was removed from the neurons and stored separately. The cells were simultaneously treated with glucose-free DMEM and hypoxia for 2 hr, re-supplied with CM, and returned to normoxic conditions (reoxygenation) for 22 hr. Neurons on cover slips were fixed in 3.7% paraformaldehyde in PBS on ice for 15 min, washed, and permeabilized with methanol at −20°C for 10 min. The cells were washed, blocked with 1% BSA for 30 min, and probed with cleaved caspase-3 (Asp175) (5A1) rabbit mAb (Cell Signaling) at 4°C overnight. Following wash, the cells were stained with Alexa568-conjugated anti-rabbit Ab and 4’ 6’-diamino-2-phenylindole (DAPI) for 1 hr at room temperature. Images were captured using an epifluorescence microscope. The cleaved caspase 3 positive fraction was analyzed by the Photoshop software.
EC proliferation, migration and capillary formation assays.
For proliferation assay, mouse ECs were plated at ∼30% confluency on 24-well plates, and allowed to grow in complete medium (20% FCS). On day 1 and 3, cells were fixed in neutral buffered 10% formalin for 15 min, washed with water, and stained with 0.1% crystal violet in 200 mM MES (pH6.0) for 1 hr. Following extensive washing with water, plates were dried and cell-associated dye was extracted with 10% acetic acid, and quantitated by absorbance at OD590 (14). Cell migration was assessed by scratch assay of the monolayer. Briefly, ECs were grown to confluence, and a linear acellular wound 1.5 mm wide was made with a scraper. The wound edge was marked by a cover slip attached to the bottom of the cultureware. The migrating cells were observed under light microscope and photographed at day 2. The capillary forming capacity of mouse ECs was evaluated in aortic explants ex vivo. Aortic segments from mice were embedded in 0.4 ml per well of Matrigel (BD Biosciences) in 24 well plates. After the gel solidified, 0.5 ml of culture media was added and replaced every other day. At day 8, photographs were taken to evaluate capillary outgrowth.
Statistical analysis.
Unless otherwise noted, all statistical analysis was carried out by ANOVA followed by Fisher's test. P < 0.05 was considered to be significant.
Supplementary text
Global downregulation of cell cycle-related genes in Rac1+/− ECs.
Functional annotation clustering and GSEA revealed that the genes downregulated in Rac1+/− are globally associated with mitotic cell division (Fig. 2D, 2F and Tables 2, S1, S3). These included transcription factors (c-fos (15), c-myb (16), Egr1 (17), Runx1 (18)), cyclins (cyclin A2, cyclin B2), mitotic kinases (Cdc2, Aurora A (19)), regulators of mitotic spindle assembly (Tacc3 (20), Tpx2 (21), BARD1 (22)), mitotic checkpoint (Mad2l1 (23), survivin (24)), centrosome cycle (Nek2 (25)), cytokinesis (Ect2 (26)), sister chromatid cohesion (sororin (27)), G2/M checkpoint (GADD45γ (28)) and cell proliferation index (Ki-67). The downregulated genes further comprised mitotic kinesin-like motor proteins (29) associated with spindle assembly (Kif11 (30), Kif22 (31), Kif23 (32)), chromosome alignment (Kif2c (33), Kif18a (34)) and cytokinesis (Kif20a (35)); chromosomal proteins such as linker histone H1, nucleosome histones (H2A, H2B, H3) along with topoisomerase IIα (Top2a). Importantly, many of these genes are induced in a tightly cell cycle-dependent manner (c-myb, Runx1 (36), cyclins, Nek2 (37), Ect2 (38), Aurora A (39), Ki-67, histones (40), Top2a (41)), suggesting that the downregulation of these genes represent overall growth inhibitory state of Rac1+/− ECs (fig. S2A). These observations conform well to the previously appreciated roles of Rac1 in critically mediating cell cycle progression (42–44), thus confirming the validity of our microarray data.
Anti-angiogenic phenotype of Rac1+/− ECs.
The anti-angiogenic phenotype of Rac1+/− ECs (fig. S2) is correlated with the growth inhibition and coordinate upregulation of angiostatic genes, Robo4 (45), ADAMTS1 (46) and thrombospondin 2 (47) (Table 3), and consistent with the retardation in ischemia-induced angiogenesis of EC-Rac1+/− mice (48).
Fig. S1. CD31 labeling of Rac1+/− and Rac1+/+ and Rac1+/− mouse ECs were immunolabeled with anti-CD31 (PECAM1) antibody or control IgG. The fraction of Cd31-stained Rac1+/+ (red) and Rac1+/− (yellow) ECs was determined by FACS analysis in comparison to the IgG-stained control ECs (green).
Fig. S2. Rac1+/− ECs show retarded proliferation, migration and capillary formation. (A) Time-course assessment by crystal violet staining of the cell number of Rac1+/+ and Rac1+/− mouse endothelial cells cultured in the presence of 20% serum. Values are means ± SEM triplicates (**, P < 0.01; ANOVA). (B) EC confluent monolayer was scratched at the width of 1.5 mm. Thereafter, the cells were allowed to migrate in the presence of 20% serum. Photographs were taken at 48 hr. Scale bar 1 mm. (C) Rac1+/+ and Rac1+/− mouse aortic segments were embedded in Matrigel, and cultured under the presence of 20% serum to allow for capillary sprouting. Photographs were taken at day 8. Scale bar, 1 mm.
Fig. S3. CD31 labeling of mouse brain ECs (MBECs). MBECs were double stained with Hoechst 33342 (nulcear stain) and anti-CD31 (PECAM1) antibody. CD31 was visualized with Alexa 488-conjugated anti-rat IgG antibody. The fluorescence images were captured with confocal microscope. Scale bar = 50 μm.
Table S1. Representative gene sets enriched in Rac1+/+ mouse endothelial cells
Table S2. Representative gene sets enriched in Rac1+/− mouse endothelial cells
Table S3. Transcription factors differentially expressed between Rac1+/− and Rac1+/+ mouse endothelial cells
Table S4. List of genes differentially expressed in Rac1+/− and Rac1+/+ mouse endothelial cells.
Supplementary References and Notes
1. M. Glogauer, C. C. Marchal, F. Zhu, A. Worku, B. E. Clausen, I. Foerster, P. Marks, G. P. Downey, M. Dinauer, D. J. Kwiatkowski, Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol 170, 5652−5657 (2003).
2. P. A. Koni, S. K. Joshi, U. A. Temann, D. Olson, L. Burkly, R. A. Flavell, Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med 193, 741−754 (2001).
3. H. H. Kim, N. Sawada, G. Soydan, H. S. Lee, Z. Zhou, S. K. Hwang, C. Waeber, M. A. Moskowitz, J. K. Liao, Additive effects of statin and dipyridamole on cerebral blood flow and stroke protection. J Cereb Blood Flow Metab, (2008).
4. Y. C. Lim, G. Garcia-Cardena, J. R. Allport, M. Zervoglos, A. J. Connolly, M. A. Gimbrone, Jr., F. W. Luscinskas, Heterogeneity of endothelial cells from different organ sites in T-cell subset recruitment. Am J Pathol 162, 1591−1601 (2003).
5. E. E. Sander, S. van Delft, J. P. ten Klooster, T. Reid, R. A. van der Kammen, F. Michiels, J. G. Collard, Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J Cell Biol 143, 1385−1398 (1998).
6. G. Dennis, Jr., B. T. Sherman, D. A. Hosack, J. Yang, W. Gao, H. C. Lane, R. A. Lempicki, DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4, P3 (2003).
7. A. Subramanian, P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, A. Paulovich, S. L. Pomeroy, T. R. Golub, E. S. Lander, J. P. Mesirov, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545−15550 (2005).
8. M. P. Quinlan, Rac regulates the stability of the adherens junction and its components, thus affecting epithelial cell differentiation and transformation. Oncogene 18, 6434−6442 (1999).
9. R. A. Stockton, E. Schaefer, M. A. Schwartz, p21-activated kinase regulates endothelial permeability through modulation of contractility. J Biol Chem 279, 46621−46630 (2004).
10. H. Gao, M. Liang, A. Bergdahl, A. Hamren, M. W. Lindholm, K. Dahlman-Wright, B. O. Nilsson, Estrogen attenuates vascular expression of inflammation associated genes and adhesion of monocytes to endothelial cells. Inflamm Res 55, 349−353 (2006).
11. Z. Wu, F. M. Hofman, B. V. Zlokovic, A simple method for isolation and characterization of mouse brain microvascular endothelial cells. J Neurosci Methods 130, 53−63 (2003).
12. L. Song, J. S. Pachter, Culture of murine brain microvascular endothelial cells that maintain expression and cytoskeletal association of tight junction-associated proteins. In Vitro Cell Dev Biol Anim 39, 313−320 (2003).
13. C. Weidenfeller, S. Schrot, A. Zozulya, H. J. Galla, Murine brain capillary endothelial cells exhibit improved barrier properties under the influence of hydrocortisone. Brain Res 1053, 162−174 (2005).
14. S. Orsulic, Y. Li, R. A. Soslow, L. A. Vitale-Cross, J. S. Gutkind, H. E. Varmus, Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell 1, 53−62 (2002).
15. J. R. Brown, E. Nigh, R. J. Lee, H. Ye, M. A. Thompson, F. Saudou, R. G. Pestell, M. E. Greenberg, Fos family members induce cell cycle entry by activating cyclin D1. Mol Cell Biol 18, 5609−5619 (1998).
16. Y. Nakata, S. Shetzline, C. Sakashita, A. Kalota, R. Rallapalli, S. I. Rudnick, Y. Zhang, S. G. Emerson, A. M. Gewirtz, c-Myb contributes to G2/M cell cycle transition in human hematopoietic cells by direct regulation of cyclin B1 expression. Mol Cell Biol 27, 2048−2058 (2007).
17. V. P. Sukhatme, Early transcriptional events in cell growth: the Egr family. J Am Soc Nephrol 1, 859−866 (1990).
18. F. Bernardin, A. D. Friedman, AML1 stimulates G1 to S progression via its transactivation domain. Oncogene 21, 3247−3252 (2002).
19. T. Hirota, N. Kunitoku, T. Sasayama, T. Marumoto, D. Zhang, M. Nitta, K. Hatakeyama, H. Saya, Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell 114, 585−598 (2003).
20. L. Schneider, F. Essmann, A. Kletke, P. Rio, H. Hanenberg, W. Wetzel, K. Schulze-Osthoff, B. Nurnberg, R. P. Piekorz, The transforming acidic coiled coil 3 protein is essential for spindle-dependent chromosome alignment and mitotic survival. J Biol Chem 282, 29273−29283 (2007).
21. R. Bayliss, T. Sardon, I. Vernos, E. Conti, Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell 12, 851−862 (2003).
22. V. Joukov, A. C. Groen, T. Prokhorova, R. Gerson, E. White, A. Rodriguez, J. C. Walter, D. M. Livingston, The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell 127, 539−552 (2006).
23. G. Fang, H. Yu, M. W. Kirschner, The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev 12, 1871−1883 (1998).
24. A. Giodini, M. J. Kallio, N. R. Wall, G. J. Gorbsky, S. Tognin, P. C. Marchisio, M. Symons, D. C. Altieri, Regulation of microtubule stability and mitotic progression by survivin. Cancer Res 62, 2462−2467 (2002).
25. A. M. Fry, P. Meraldi, E. A. Nigg, A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J 17, 470−481 (1998).
26. T. Tatsumoto, X. Xie, R. Blumenthal, I. Okamoto, T. Miki, Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol 147, 921−928 (1999).
27. J. Schmitz, E. Watrin, P. Lenart, K. Mechtler, J. M. Peters, Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Curr Biol 17, 630−636 (2007).
28. X. W. Wang, Q. Zhan, J. D. Coursen, M. A. Khan, H. U. Kontny, L. Yu, M. C. Hollander, P. M. O'Connor, A. J. Fornace, Jr., C. C. Harris, GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci U S A 96, 3706−3711 (1999).
29. H. Miki, Y. Okada, N. Hirokawa, Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol 15, 467−476 (2005).
30. A. Blangy, H. A. Lane, P. d'Herin, M. Harper, M. Kress, E. A. Nigg, Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 83, 1159−1169 (1995).
31. A. A. Levesque, D. A. Compton, The chromokinesin Kid is necessary for chromosome arm orientation and oscillation, but not congression, on mitotic spindles. J Cell Biol 154, 1135−1146 (2001).
32. R. Neef, U. R. Klein, R. Kopajtich, F. A. Barr, Cooperation between mitotic kinesins controls the late stages of cytokinesis. Curr Biol 16, 301−307 (2006).
33. P. D. Andrews, Y. Ovechkina, N. Morrice, M. Wagenbach, K. Duncan, L. Wordeman, J. R. Swedlow, Aurora B regulates MCAK at the mitotic centromere. Dev Cell 6, 253−268 (2004).
34. J. Stumpff, G. von Dassow, M. Wagenbach, C. Asbury, L. Wordeman, The kinesin-8 motor Kif18A suppresses kinetochore movements to control mitotic chromosome alignment. Dev Cell 14, 252−262 (2008).
35. E. Hill, M. Clarke, F. A. Barr, The Rab6-binding kinesin, Rab6-KIFL, is required for cytokinesis. EMBO J 19, 5711−5719 (2000).
36. F. Bernardin-Fried, T. Kummalue, S. Leijen, M. I. Collector, K. Ravid, A. D. Friedman, AML1/RUNX1 increases during G1 to S cell cycle progression independent of cytokine-dependent phosphorylation and induces cyclin D3 gene expression. J Biol Chem 279, 15678−15687 (2004).
37. S. J. Schultz, A. M. Fry, C. Sutterlin, T. Ried, E. A. Nigg, Cell cycle-dependent expression of Nek2, a novel human protein kinase related to the NIMA mitotic regulator of Aspergillus nidulans. Cell Growth Differ 5, 625−635 (1994).
38. H. Sakata, J. S. Rubin, W. G. Taylor, T. Miki, A Rho-specific exchange factor Ect2 is induced from S to M phases in regenerating mouse liver. Hepatology 32, 193−199 (2000).
39. M. Tanaka, A. Ueda, H. Kanamori, H. Ideguchi, J. Yang, S. Kitajima, Y. Ishigatsubo, Cell-cycle-dependent regulation of human aurora A transcription is mediated by periodic repression of E4TF1. J Biol Chem 277, 10719−10726 (2002).
40. W. F. Marzluff, R. J. Duronio, Histone mRNA expression: multiple levels of cell cycle regulation and important developmental consequences. Curr Opin Cell Biol 14, 692−699 (2002).
41. K. Kimura, M. Saijo, M. Ui, T. Enomoto, Growth state- and cell cycle-dependent fluctuation in the expression of two forms of DNA topoisomerase II and possible specific modification of the higher molecular weight form in the M phase. J Biol Chem 269, 1173−1176 (1994).
42. M. F. Olson, A. Ashworth, A. Hall, An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 269, 1270−1272 (1995).
43. D. Michaelson, W. Abidi, D. Guardavaccaro, M. Zhou, I. Ahearn, M. Pagano, M. R. Philips, Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J Cell Biol 181, 485−496 (2008).
44. C. S. Hill, J. Wynne, R. Treisman, The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell 81, 1159−1170 (1995).
45. C. A. Jones, N. R. London, H. Chen, K. W. Park, D. Sauvaget, R. A. Stockton, J. D. Wythe, W. Suh, F. Larrieu-Lahargue, Y. S. Mukouyama, P. Lindblom, P. Seth, A. Frias, N. Nishiya, M. H. Ginsberg, H. Gerhardt, K. Zhang, D. Y. Li, Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nat Med 14, 448−453 (2008).
46. F. Vazquez, G. Hastings, M. A. Ortega, T. F. Lane, S. Oikemus, M. Lombardo, M. L. Iruela-Arispe, METH-1, a human ortholog of ADAMTS-1, and METH-2 are members of a new family of proteins with angio-inhibitory activity. J Biol Chem 274, 23349−23357 (1999).
47. B. Lange-Asschenfeldt, W. Weninger, P. Velasco, T. R. Kyriakides, U. H. von Andrian, P. Bornstein, M. Detmar, Increased and prolonged inflammation and angiogenesis in delayed-type hypersensitivity reactions elicited in the skin of thrombospondin-2--deficient mice. Blood 99, 538−545 (2002).
48. N. Sawada, S. Salomone, H. H. Kim, D. J. Kwiatkowski, J. K. Liao, Regulation of endothelial nitric oxide synthase and postnatal angiogenesis by Rac1. Circ Res 103, 360−368 (2008).