

Abstract
To spatially control the delivery of multiple viral vectors from biomaterial scaffolds, digoxigenin (DIG) was conjugated to adenoviral capsid proteins as an antigenic determinant for antibody immobilization. The infectivity, toxicity, specificity and immobilization stability of DIG-modified adenovirus were examined to investigate the feasibility and effectiveness of this viral surface modification. Anti-DIG antibody conjugated on chitosan surfaces was able to immobilize DIG-modified adenovirus and could be stably bound on the material for at least two weeks, yet the modification was mild enough that viral infectivity was maintained. To immobilize two different adenoviruses, wax masking was applied to conjugate anti-DIG and anti-adenovirus antibodies in two discrete regions of a chitosan film, respectively. The distribution of these two viral vectors expressing different reporter genes was examined after cell culture. Fluorescent protein expression from transduced cells illustrated that the infection distribution could be controlled: one gene was delivered to the entire region of the biomaterial, and another was only delivered to defined regions. Compared to three other cardiac glycosides, ATPase inhibition was undetectable when DIG was conjugated on the adenovirus, suggesting that the method may be safe for in vivo application. This dual viral vector delivery system should be capable of generating distinct interfaces between cell signaling viruses to control tissue regeneration from a range of different biomaterials.
Keywords: Antibody, Tissue engineering, Adenovirus, Digoxigenin, Gene delivery
Introduction
To facilitate tissue regeneration in wound sites, appropriate biological signals are required to recruit and induce the desired cells to proliferate and differentiate within scaffolds [1]. Regenerative gene therapy is an alternative to protein-based therapy and may offer improvements by transducing cells that are capable of expressing bioactive factors in vivo [2]. In order to fully achieve complex organ or tissue regeneration via a tissue engineering approach, more than one bioactive factor may be required to regulate new tissue growth in vivo [3-5]. In the gene therapy paradigm, the delivery of multiple viral vectors could transduce host cells in defect sites to express defined bioactive factors. While multiple viral vectors are capable of transducing host cells in tissue defects, how to precisely deliver these transgenes at the target sites remains a significant challenge.
Bolus and substrate-mediated gene delivery methods are two major strategies for in vivo gene therapy [6, 7]. With bolus virus administration, direct injection into target sites or indirect delivery via polymer carriers have been used to transfer genes to induce new tissue growth [8-13]. However, this delivery may lead to virus diffusion from target sites. Therefore, a higher viral titer becomes necessary to achieve therapeutic levels, which may be cytotoxic or elicit serious immune responses [14]. Virus that diffuses from the target site may also induce systemic infection [15]. Furthermore, it is difficult to restrict gene transfer to only the target sites due to virus dispersion. Consequently, a substrate-mediated strategy has become a compelling alternative strategy for controlling virus delivery. In this method virus can be complexed within, or on, a biomaterial that also serves as a substrate for cell adhesion [7, 16, 17]. Antibody immobilization is a frequently used substrate-mediated method, by which anti-virus antibodies tether viral particles to a scaffold, yet the viruses remain capable of being internalized by adherent cells [18]. This approach has been shown to successfully deliver adenovirus to cells without diffusing from scaffolds [19-22].
Although anti-virus antibodies can effectively immobilize virus, they are incapable of spatially controlling multiple viral vector delivery to specific sites within a scaffold because anti-virus antibodies cannot distinguish between viral vectors with different transgenes. The application of different viral vector strains with their antibodies may circumvent this difficulty. However, the administration of different vectors may lead to inconsistencies in the length of time in which transgenes are expressed. For example, the use of retrovirus would likely provide continuous expression during the lifetime of a cell, whereas adenovirus would only offer transient gene expression. In addition, different viral vectors may have interactions with each other, such as adeno-associated viral vectors being rescued to proliferate in host cells if they are co-infected with adenovirus. These risks make the co-administration of different types of viral vectors impractical. Therefore, we sought to tag the capsid proteins of adenovirus with different antigenic determinants that are capable of being distinguished by different antibodies.
Digoxigenin (DIG) is a steroid extracted from the plants Digitalis purpurea and D. lanata. It is commonly used for labeling DNA probes for in situ hybridization. N-hydroxysuccinimido-DIG (DIG-NHS) is a commerically available chemical disigned for conjugation to amine groups. This DIG modification has been applied to label red blood cells for in vivo aging studies [23]. Because DIG is a small chemical, we hypothesized that it should be able to tag the surface of a adenovirus without affecting viral infectivity. Furthermore, adenovirus is a broadly used viral vector that does not integrate into the host genome. Therefore, its use is appropriate for short-term expression during the therapeutic period [24]. For these reasons, we labeled the viral capsids of adenovirus with DIG. Chitosan was used as our biomaterial scaffold because it has intrinsic amines that can be used for bioconjugation. Additionally, chitosan has excellent biocompatiblity properties and its hydrophilic surface may promote cell adhesion, proliferation, and differentiation[11, 25]. Anti-DIG and anti-adenovirus antibodies were conjugated on chitosan surfaces and a wax masking technique was applied to control the antibody conjugation area. Finally, DIG-modified and non-modified adenoviruses were immobilized on two different antibody conjugated areas in one scaffold. We hypothesized that cells could be transduced in situ on specific sites of the biomaterial and thus develop a defined interface between the two cell signaling factors.
Materials and Methods
Virus modification by digoxigenin
Adenovirus encoding the bacterial β-galactosidase gene and nuclear localization sequence (AdLacZ) was prepared by the University of Michigan Vector Core. Digoxigenin-3-O-methylcarbonyl-ε-aminocaproic acid-N-hydroxysuccinimide ester (DIG-NHS, Roche, Indianapolis, IN, USA) was dissolved in phosphate buffered saline (PBS, Pierce, Rockford, IL, USA) prior to incubation with adenovirus. The conjugation reaction was performed at 4°C for 2 hours, and non-reacting, excess DIG-NHS was removed with a desalt spin column (Pierce, Rockford, IL, USA). The modified virus was sterilized by being passed through a 0.2 μm syringe filter (Nalgene, Rochester, NY).
To determine the appropriate concentration of DIG-NHS for adenovirus modification, AdLacZ was reacted with different concentrations of DIG-NHS. The level of DIG modification was then analyzed by an anti-DIG Fab fragment-conjugated alkaline phosphatase (anti-DIG Ab-AP, Roche, Indianapolis, IN, USA) sandwich enzyme-linked immunosorbent assay (ELISA). Goat anti-adenovirus antibody (1 μg/well) (Abcam, Cambridge, MA, USA) was coated on 96-well plates (Coring, Lowell, MA, USA) and the modified AdLacZ was added to the wells and incubated for 1 hr. After three washes with 0.05% Tween-20 in PBS (PBST, Teknova, Hollister, CA, USA), anti-DIG Ab-AP was added to label the DIG on the plate for 1 hour. Subsequently, the substrate p-nitrophenyl phosphate (PNPP, Pierce, Rockford, IL, USA) was added for 20 min, and enzyme activity was detected by optical density at 405nm (OD405nm).
Virus infectivity before and after DIG modification was determined by in vitro cell culture infection. A human gingival fibroblast (HGF) cell line was cultured in alpha-minimal essential medium (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco) and penicillin (100 unit/ml)-streptomycin (100 mg/ml) (Gibco) at a density of 5×104 cells/well in 24-well culture plates (Coring, Lowell, MA, USA) for 24 hr. Subsequently, AdLacZ before and after DIG-NHS treatment was added to the culture wells for a 48 hr infection. The transduction efficiency of each group was determined by the expression of β-galactosidase, which was detected using a sandwich ELISA kit (Roach, Indianapolis, IN, USA).
The influence of the DIG on ATPase
As an initial test to investigate if digoxigenin conjugated on a virus surface would be safe for in vivo application, an adenosine 5′-triphosphatase (ATPase) activity assay was performed to evaluate the ATPase inhibition of the modified virus. ATPase activity was determined using a Quamtichrom ATPase/GTPase assay kit (Bioassay, Hayward, CA, USA). DIG modified AdLacZ was diluted to different concentrations and 5 μl/well was placed in 96-well microplates with equal volumes of ATPase (Sigma-Aldrich, St Louis, MO) for 15 min at room temperature. Subsequently, 10 μl 4mM adenosine 5′-triphosphate (ATP, Sigma-Aldrich, St Louis, MO) was added and the plate was incubated at room temperature for 30 min. Finally, 200 μl kit reagent was added per well and incubated for 30 min before reading the OD620nm. The same process was also performed for three different ATPase inhibitors ouabain, digoxigenin, and digoxin (Sigma-Aldrich). These inhibitors were used as positive control groups to compare the inhibitory effects to DIG-modified adenovirus.
Antibody conjugation on chitosan surfaces
Chitosan was coated on 24-well culture plates [17]. To conjugate antibody on the surface, chitosan was modified using N-(γ-maleimidobutyryloxy) succinimide ester (Sulfo-GMBS, Pierce) to functionalize a layer of maldimide, that could react with sulfhydryl groups. After dissolving in PBS, Sulfo-GMBS (0.5 mg/well) was added at room temperature for 2 hours and then removed with several PBS washes. Simultaneously, 12.5 nmole Tris (2-carboxyethyl) phosphine hydrochloride (TCEP-HCl, Pierce) was dissolved in PBS with 10 mM ethylenediaminetetraacetic acid (EDTA, Sigma-Aldrich) and reacted with 1 mg goat anti-adenovirus or sheep anti-DIG IgG (AbD Serotec, Kidlington, Oxford, UK). With the TCEP treatment, the labile disulfides between heavy chains in the hinge region of IgG molecules were selectively reduced to get two half-IgG fragments with sulfhydryls. Such partial reduction of IgG disulfides usually results in sulfhydryl group attachment points that will not sterically hinder antigen binding. After one hour incubation at room temperature, the TCEP was removed using desalt spin columns. The antibody (20μg/well) was then added at room temperature for overnight incubation. Finally, the unbound antibody was washed out and the wells were sterilized with 70% ethanol.
DIG-modified virus immobilization on material surfaces
DIG-modified AdLacZ (DIG-AdLacZ) was diluted in 0.5% gelatin (w/v in PBS) at different virus concentrations and was then placed on anti-DIG IgG conjugated chitosan surfaces at 4°C for 2 hours. To determine the virus immobilization efficiency on chitosan, an indirect sandwich ELISA assay was performed: 100 μl supernatant with unbound DIG-AdLacZ was applied to goat anti-adenovirus antibody coated 96-microwell plates for 1 hour. The bound virus was labeled with rabbit anti-adenovirus IgG (Abcam, Cambridge, MA, USA), followed by anti-rabbit IgG antibody conjugated alkaline phosphatase (Abcam), for one hour each. Finally, PNPP substrate was added to quantify the virus in the supernatant. The extent of immobilized virus was determined by subtracting the amount of supernatant virus from the total amount of virus present before the reaction. Additionally, the immobilized DIG-AdLacZ on chitosan surfaces was visualized using scanning electron microscopy (SEM) to demonstrate the virus distribution on the material surface [17].
To investigate the stability of immobilized adenovirus on biomaterial surfaces, a time course experiment was performed to determine adenovirus release. After immobilizing 1 × 109 viral particles on chitosan surfaces, 1 ml PBS was added on each surface at 37 °C. These samples were collected at different time points and quantified by sandwich ELISA.
Antibody conjugation with spatial control using wax masking
Because conjugated anti-DIG IgG was capable of immobilizing viral particles on material surfaces, the tethered virus should be able to transduce cells that attach and proliferate on chitosan surfaces. To investigate the spatial control of the antibody conjugation, low melting point wax was applied to mask defined regions of the material surface so that the antibody could only be conjugated on the non-masked area. Polyester wax (EMS, Hatfield, PA, USA) was melted at 40 °C and then added (200 μl/well) to cover the right side of the well. After the wax solidified, anti-DIG IgG was conjugated on exposed chitosan surfaces. Finally, the wax was removed by incubation in absolute ethanol at 37 °C for 1 hr.
To illustrate the region of antibody conjugation, a secondary antibody (Rabbit anti-sheep IgG conjugated FITC, Millipore, Billerica, MA, USA) was used to label the sheep IgG (anti-DIG antibody) conjugated on the chitosan surfaces. The FITC labeled region was observed under a fluorescent microscope (Eclipse TE300, Nikon, Melville, NY, USA).
The feasibility of in situ cell transduction by immobilized AdLacZ was examined. DIG-AdLacZ was diluted in 0.5% gelatin (w/v in PBS), and was added to culture wells with spatially conjugated anti-DIG IgG. The virus was incubated at 4 °C for 2 hours and the wells were then washed 3 times with PBS. Fibroblasts were seeded at 1 × 105 cells/well for 2 days and stained with 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside (X-gal) to demonstrate the infection cells [16]. Counter staining was performed by incubating the cells in crystal violet. Samples were observed under an SMZ-U stereoscopic zoom microscope (Nikon).
The specificity of antibody to DIG modified virus
To determine the extent to which DIG modification would ensure virus immobilization to only anti-DIG antibodies, antibody conjugated wells were examined to determine the specificity between the viruses and antibodies. Anti-DIG and anti-adenovirus antibodies were conjugated on chitosan surfaces individually. These surfaces were used to bind DIG-AdLacZ and AdLacZ in different concentrations, separately. After two hours incubation at 4 °C, unbound virus was washed out with PBS and the surface virus was examined by an indirect ELISA [17].
Dual adenoviral vector immobilization for in situ transduction on chitosan surfaces
To tether two different adenoviruses on a material surface, and thus create an interface between cell signaling viruses, anti-DIG and anti-adenovirus antibodies were conjugated on surfaces to demonstrate the spatial control of virus immobilization. Low melting point wax was added to the right region of chitosan coated wells, and anti-adenovirus IgG was then conjugated on the exposed material surfaces. After a 2 hour incubation period, the wax was dissolved with ethanol washes and the anti-DIG IgG was conjugated on the entire chitosan surface. The second conjugation of anti-DIG IgG may not be completely restricted to the right side because there was no physical masking. However, because most of the reactive maldimide groups on chitosan were already saturated by the first anti-adenovirus IgG, most of anti-DIG IgG conjugation should be distributed on the right side.
Adenovirus conjugated blue fluorescent protein (AdBFP) and green fluorescent protein (AdGFP) were prepared by the Vector Core at the University of Michigan. The AdBFP was modified by DIG (DIG-AdBFP) and AdGFP was not modified. Both viruses were diluted in 0.5% gelatin (w/v in PBS) to a final concentration of 1×108 pfu and were incubated together at 4 °C for immobilization. The unbound virus was removed by PBS washes and fibroblasts were cultured on the modified surface for 2 days. A red fluorescent dye that stains for nucleic acid (SYTO 62, Invitrogen, Carlsbad, CA, USA) was used to illustrate cell distribution.
Results
Adenoviral infectivity is preserved after DIG-NHS modification
The level of DIG modification was determined by a sandwich ELISA (Fig 1a). The amount of digoxigenin on viral surfaces increased with increasing concentrations of DIG-NHS. The level of DIG modification was saturated when the number of DIG-NHS molecules exceeded 0.075 nmole per 109 viral particles. We therefore used this concentration for the remainder of our experiments.
Figure 1.

Adenovirus modified by DIG-NHS maintains its infectivity. (a) DIG-NHS treated AdLacZ was captured on ELISA plates to detect and quantify DIG molecules on viral surfaces with anti-DIG Ab-AP. The data suggest that the DIG modification was dose dependent and saturated when there were more than 0.075 nmole DIG-NHS per 109 viral particles. (b) The β-galactosidase expression from cells infected with AdLacZ with (solid line) and without (dashed line) DIG modification were compared by sandwich ELISA. The results demonstrate that there were no significant differences between these two groups with different virus concentrations, suggesting that this modification was mild, and that viral infectivity could be maintained.
Fibroblasts were infected with AdLacZ before and after DIG modification to determine if DIG modification affected virus infectivity. After two days infection, the transduced cells were examined by a β-galactosidase ELISA assay (Fig 1b). This experiment demonstrated that the β-galactosidase expression of these two groups was similar and without significant differences in the range of virus concentrations studied. These findings indicated that the DIG modification was a mild chemical reaction that did not adversely affect adenovirus infectivity.
Digoxigenin conjugated on viral surface reduces its inhibition of ATPase
Digoxigenin is derived from digitalis which is also well-known as a cardiac glycoside. Cardiac glycosides have inotropic effects on heart muscles, that can be cardiotoxic following in vivo administration [26]. To investigate the potential risk raised by DIG modification, the toxicity of the modified virus was evaluated. The inotropic mechanism of cardiac glycosides is mediated through the blocking of Na+/K+ transporting ATPase transmembrane pump activity in the sarcolemma [27]. Therefore, an ATPase inhibition assay was performed to assess the extent of blockage caused by cardiac glycosides and the DIG-modified virus [28]. Three different cardiac glycosides: ouabain, digoxigenin, and digoxin, served as positive control groups in this inhibition assay. The levels of phosphate ion released significantly decreased with increasing cardiac glycoside concentration, suggesting these inhibitors blocked ATPase activity (Fig 2a).
Figure 2.

An ATPase inhibition assay was performed to investigate the potential toxicity caused by DIG modification. ATPase was reacted with different concentrations of DIG-AdLacZ (cross), and the enzymatic activities of ATPase were determined by the freeing of phosphate ions released from ATP. Three different cardiac glycosides, ouabain (triangle), digoxigenin (square), and digoxin (diamond), were compared as positive controls. (a) The phosphate ion concentrations decreased with increasing inhibitors because the enzymatic activity of ATPase was blocked. DIG-AdLacZ only slightly reduced phosphate release. (b) Three cardiac glycoside molecules demonstrated a dose dependent inhibition of ATPase activity, while an equivalent dose of DIG-AdLacZ exhibited nearly undetectable levels.
Concentrations of DIG-AdLacZ, ranging from 0 to 2 × 1010 viral particles, were also examined in this assay. The equivalent numbers of DIG molecules conjugated to the viral surfaces were converted by the assumption that the conjugation rate was perfect. For example, because the modification of DIG-NHS resulted in a concentration of 0.075 nmole per 109 viral particles, the equivalent concentration of DIG conjugated to 2 × 1010 viral particles in a 5 μL volume would be 300 μM. The levels of phosphate ion released in the DIG-AdLacZ group was significantly higher than the other three cardiac glycoside groups, suggesting that the ATPase supported ATP hydrolysis without interference (Fig 2a).
The inhibition results were based on the ratio of reducing phosphate ion release to the ideal amount which should be released without inhibitors (Fig 2b). Ouabain, digoxigenin, and digoxin significantly inhibited the ATPase activity. Even at the lowest concentration, 0.2 μM, they had inhibition effects of 18 %, 8 %, and 11 %, respectively. In contrast, DIG-modified adenovirus displayed essentially no inhibition except at the highest concentration of DIG-AdLacZ (300 μM, i.e. 2 × 1010 viral particles), where inhibition was only about 10% of maximum. These data suggest that DIG molecules conjugated to viral surfaces significantly reduced their reactivity to ATPase by about 1,500-fold (Fig 2b).
Digoxigenin-modified viruses are effectively and stably immobilized by anti-DIG IgG conjugation to biomaterial surfaces
The efficiency of DIG-AdLacZ immobilized by anti-DIG IgG conjugated to chitosan surfaces was determined by indirectly detecting unbound virus in supernatants after conjugation. The immobilized DIG-AdLacZ on chitosan surfaces increased with increasing DIG-AdLacZ concentrations (Fig 3a). This amount of immobilized DIG-AdLacZ was compared to the amount of DIG-AdLacZ unbound in solution to determine the conjugation rates (Fig 3b). The conjugation rates were perfect in the low concentrations and began to decline when the DIG-AdLacZ concentration was 109 viral particles per well. These data suggest that adenovirus with digoxigenin modification can be effectively bound to chitosan surfaces.
Figure 3.
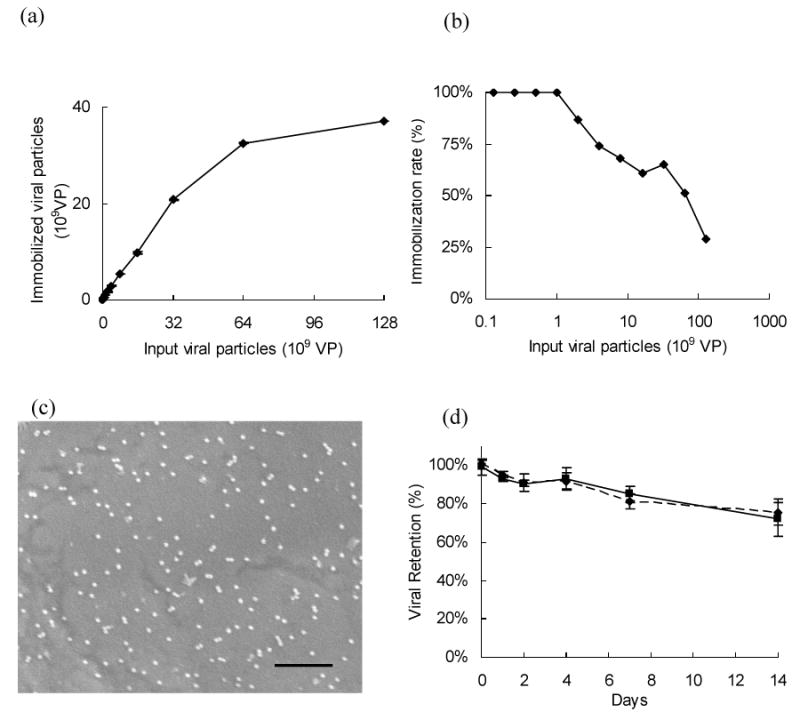
The binding capacity of conjugated anti-DIG IgG on chitosan and the virus immobilization stability were determined by indirect sandwich ELISA assay. (a) The DIG-AdLacZ immobilized on chitosan was proportional to the concentration of incubated DIG-AdLacZ. (b) The immobilization rate of DIG-modified adenovirus was 100% when the concentration was equal to or less than 109 viral particles per well. Higher virus concentrations led to lower immobilization rates due to a limited substrate area. (c) The distribution of immobilized DIG-AdLacZ on anti-DIG conjugated chitosan surfaces was illustrated by SEM examination. The scale bar in the picture is 1 μm. (d) Adenovirus with and without DIG modification were placed on anti-DIG IgG (solid line) and anti-adenovirus IgG (dashed line) conjugated surfaces, respectively. The released viral particles were detected at different time points to determine retention rates. There were no differences between these two groups and more than 75% of the virus could be stably maintained for two weeks.
The immobilized DIG-AdLacZ viral particles were examined by SEM to determine the distribution patterns on the material surface. The adenovirus was found to be evenly bound to conjugated antibodies on chitosan surfaces (Fig 3c). The stability of adenovirus with and without DIG modification was compared by incubation in PBS at 37°C. Sandwich ELISA results demonstrated that the viral retention rates on chitosan surfaces were similar in these two groups, suggesting the adenovirus immobilization mediated by DIG did not affect the stability (Fig 3d). In addition, more than 80% of the viral particles, both with and without DIG modification, bound to the surfaces for 1 week. Approximately 70% of viral particles were present for 2 weeks. These results indicated that virus with and without DIG modification, could be stably tethered by antibodies conjugated on biomaterial surfaces.
Wax masking spatially controls antibody conjugation for adenovirus immobilization
Low melting point polyester wax (37° C) is an inert polymer that dissolves in ethanol and can thus serve as an excellent material for physical masking. After antibody was conjugated to the non-masked region of chitosan, the surface wax was removed with ethanol washes at 40° C. Secondary antibody conjugated FITC was used to label the area of antibody immobilization (Fig 4a). Only exposed chitosan surfaces were identified by green fluorescence, indicating that antibody conjugation was spatially controlled.
Figure 4.

Anti-DIG IgG immobilization was spatially controlled by a low melting point wax masking technique. (a) Anti-sheep IgG antibody conjugated FITC was used to label the surface antibody. Only the exposed area without wax protection illustrated green fluorescent expression. (b) HGF cells were cultured on chitosan surfaces for 2 days. The transduced cells turned blue after X-gal staining. Cells grew to confluence on the material surfaces; however, cell transduction was restricted to the non-masked area. These findings are consistent with the results of the fluorescent labeling.
To investigate the extent to which this antibody immobilization strategy could be applied to spatially control cell transduction directly from the surface of biomaterials, anti-DIG IgG was conjugated to chitosan surfaces in which defined areas were controlled by wax masking. After removing the wax, DIG-AdLacZ was immobilized on chitosan surfaces and fibroblasts were cultured on the material for 2 days. The transduced cells were identified as blue after X-gal staining. Non-transduced cells were only identified after counter staining with crystal violet (Fig 4b). These data demonstrated that HGF cells proliferated to confluence on chitosan surfaces, and the infected cells were restricted to the non-masked. This suggested that adenovirus modified by DIG could be immobilized in discrete sites on biomaterials and could still be released for in situ cell transduction.
Anti-DIG IgG specifically immobilizes DIG-modified adenovirus on material surfaces
In this study, adenovirus was modified by DIG to distinguish it from non-modified adenovirus for antibody binding. To investigate the extent to which this modification could control virus immobilization at specific sites, two different antibodies, anti-adenovirus IgG and anti-DIG IgG, were separately conjugated on chitosan surfaces to demonstrate the specificity of antibodies to adenovirus with or without DIG modification.
On chitosan surfaces conjugated with anti-DIG IgG, DIG-modified adenovirus was dose-dependently immobilized on the biomaterial. The bound DIG-modified adenovirus increased with increasing virus concentration and was saturated when the virus concentration exceeded 3.2×1010 viral particles/well. This was likely due to the limited area of the biomaterial surface. In contrast, for non-modified adenovirus, there was only minor physical adsorption at the same concentrations, suggesting that only adenovirus modified by DIG could be recognized and tethered on conjugated anti-DIG IgG (Fig 5a).
Figure 5.

Adenovirus, with (solid line) or without (dashed line), DIG modification were placed on antibody conjugated chitosan surfaces to investigate the specificity of the conjugated antibody to adenovirus. (a) On anti-DIG IgG conjugated chitosan surfaces, DIG-modified virus was immobilized by surface antibodies in a dose dependent manner, whereas there was only a slight adsorption of non-modified adenovirus due to nonspecific binding. (b) On anti-adenovirus IgG conjugated chitosan, both adenovirus with and without DIG modification were bound to the surfaces with a similar affinity.
When these two adenoviruses were individually placed on anti-adenovirus IgG conjugated surfaces, the ELISA results demonstrated that the binding affinities of these two groups were almost identical. They all increased with virus concentration and were saturated when the virus concentration was greater than 3.2×1010 viral particles per well (Fig 5b). This indicated that virus modified by DIG preserved the epitopes that are recognized by anti-adenovirus IgG for immobilization.
Dual viral vector delivery spatially controls cell transduction with different genes
Because the anti-DIG IgG conjugated surfaces only binds DIG-modified adenovirus, while anti-adenovirus IgG can immobilize adenovirus with or without modification, we further investigated the possibility of utilizing these properties to spatially control different bioactive factor expression on material surfaces (Fig 6a). In this experiment, wax masking was applied to restrict the conjugation of anti-adenovirus IgG to only the non-masked area, and anti-DIG IgG was conjugated after removing the wax mask (Fig 6b). The virus-immobilized surfaces were seeded with fibroblasts to demonstrate the distribution of cell transduction. After 2 days in culture, the cells were stained with SYTO 62 and observed under a fluorescent microscope with a TRITC filter. The red fluorescence observed throughout the entire material surface demonstrated that the cells grew to confluence (Fig 6c). Using a FTIR and DAPI filter, green and blue fluorescence were identified as expressed from cells transduced by AdGFP and DIG-AdBFP, respectively (Fig 6c). The BFP expression was distributed throughout the material surface because DIG-AdBFP could be bound to both anti-DIG and anti-adenovirus IgG. In contrast, the GFP expression was restricted to the left side of the surface because anti-adenovirus IgG was only conjugated to the non-masked area. There were no cells that expressed both blue and green fluorescence, suggesting that co-infection did not occur on the modified material surface. These results demonstrated that two different transgenes could be spatially controlled by the dual viral vector immobilization method to generate a defined interface between the cell signaling viruses.
Figure 6.
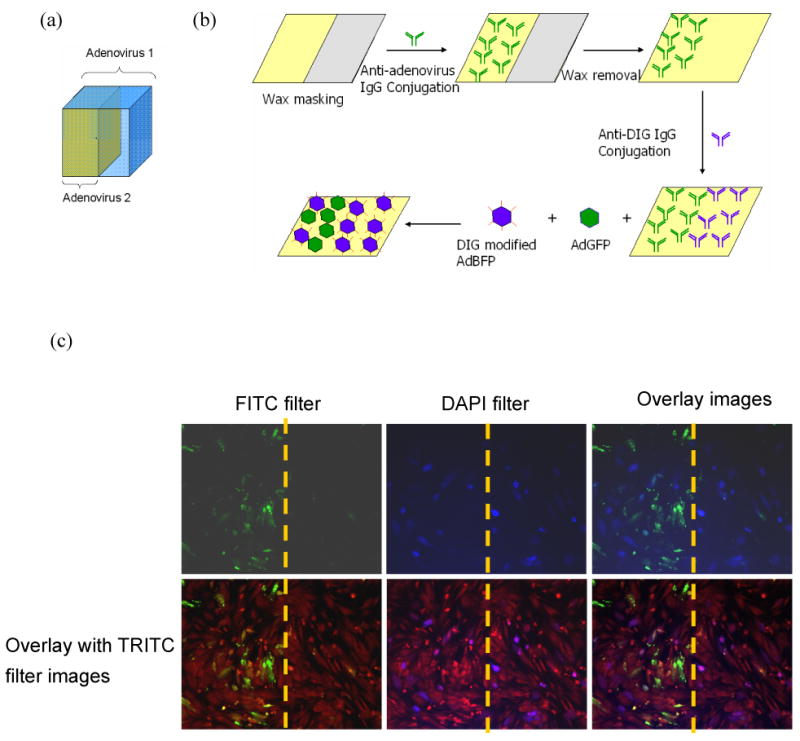
Dual adenoviral vector immobilization was performed to demonstrate the spatial control of in situ transduction. (a) Schematic of the viral delivery model. By conjugating two different antibodies against DIG and adenovirus using the wax masking technique, one viral vector is tethered on the entire scaffold, whereas the other virus is restricted to specific regions. (b) The scheme of the dual viral vector immobilization method to control two antibody conjugations on biomaterial surfaces. (c) The transduced cell distribution visualized under fluorescent microscopy. Red fluorescent staining demonstrated that cells grew to confluence on material surfaces (TRITC filter). Cells expressing BFP were distributed over the entire surface, whereas GFP expression was restricted to the left side, where AdGFP was bound by anti-adenovirus IgG
Discussion
To effectively regenerate tissues using tissue engineering strategies, several different bioactive factors may be required to regulate tissue growth and differentiation at target sites. To address this complex issue, several methods have been developed to deliver multiple bioactive factors from biomaterial scaffolds [3, 4, 29-31]. Most of these approaches use protein-based delivery methods to induce new tissue formation in scaffolds, but their short half-lives may limit their therapeutic effectiveness. In addition, the high price of producing large quantities of growth factors makes clinical administration problematic. Through regenerative gene therapy approaches, sustainable and stable bioactive factor delivery from transduced cells may be possible. However, this strategy may result in the rapid diffusion of vectors from the intended site and elicit systemic infection or immune responses. In contrast, substrate-mediated delivery is an improved method in which vectors are immobilized on biomaterial surfaces [7, 17, 32]. Virus bound to material surfaces can transduce cells in situ within deigned biomaterial scaffolds. Because the virus infection is limited to cells directly in contact with the scaffold, bioactive factor delivery is restricted to the scaffolds without dispersion. Furthermore, the viral titer can be reduced due to concentrated transgene localization [16, 17].
Virus immobilization by antibodies conjugated to biomaterial surfaces represents an effective strategy for binding viral vectors to scaffold surfaces. By covalently linking anti-virus IgG on biomaterials, viral vectors can be stably tethered for site-specific delivery. This strategy has been applied for the immobilization of viruses on biomaterials to deliver genes in micro-coils, stents, and intra-aortic implants [19-22]. While this method was applied to effectively control single gene delivery from scaffolds, this strategy would be incapable of transferring multiple genes in defined regions of scaffolds because anti-virus antibodies cannot distinguish viral vectors with different transgenes. Therefore, we sought to tag an antigenic determinant on viral surfaces so that the modified virus could be bound by antibodies against this antigen. Small chemicals were used for tagging because they proved effective in modifying viral surfaces, and because they could easily be conjugated without inhibiting virus infectivity [17]. For example, biotinylation has been applied to modify adenoviral surfaces that tether viral vectors to avidin immobilized surfaces [17, 33]. Using a similar strategy, we developed a new method for modifying viral surfaces by DIG conjugation.
In our study, DIG-NHS effectively modified adenovirus surfaces and we were able to maintain the virus titer after this reaction (Fig 1b). These findings demonstrated that the grafting modification was a mild chemical reaction that preserved viral integrity. In addition, DIG should not inhibit the recognition of coxsackie-adenovirus receptors on host cells for internalization due to its small size. The conjugation of DIG-NHS to amine groups on viral proteins forms amide bonds, which is a covalent bonding and thus DIG can be stably maintained on viral surfaces. The MW of DIG-NHS is only 659 Da, which is extremely small compared to adenovirus (MW=180 × 106 Da). Also, the entry of adenovirus into the host cell is initiated by the knob domain of the fiber protein binding to the cell receptor. The molecular weight of fiber protein is 62 kDa, which is approximately 100 times larger than DIG-NHS. Due to the small size of the conjugated DIG, the infectivity of modified virus can be preserved after reaction.
Patient safety is an important concern in regenerative gene therapy. Although digoxigenin is broadly applied as a tag for labeling in different in vitro analyses, it is rarely used for in vivo studies because of its potential for cardiotoxicity. This cardiotoxicity is caused by ATPase inhibition [26, 27]. Therefore, as an initial measure of safety, an ATPase inhibition assay was performed in which DIG-modified adenovirus was compared with digoxigenin and two other common cardiac glycosides, digoxin and ouabain. Assuming the DIG-NHS molecules perfectly grafted on viral surfaces, the ATPase inhibition assay demonstrated that the DIG-modified virus was 1500 times less active than the three positive control groups (Fig 2b). This may be caused, in part, by the overestimation of DIG molecules on the viral surfaces, resulting from an imperfect conjugation rate of DIG-NHS. In addition, the interactions between grafted DIG and ATPase may be inactivated because of an increase in steric hindrance. Because adenovirus is a very large molecule, DIG covalently linked on adenoviral surface may be too large to engage its ATPase binding sites. ATPase is located on the transmembrane Na+/K+ pump and steric hindrance would likely be even more significant in vivo, suggesting that inhibition should be even less than that observed in our experimental tests. Furthermore, it would be difficult for the bound DIG-modified adenovirus to affect cardiac muscle function via bloodstream transportation. Although extensive in vivo analyses would need to be performed prior to use in humans, the experiments described here suggest that this method may be safe and without serious cardiac implications.
The functionalization of chitosan surfaces was achieved by treating the material with Sulfo-GMBS. These thiol-reactive surfaces conjugated with the sulfhydryls of half-IgG derived from reducing disulfide bonds in the antibody hinge region. By this method, IgG was conjugated to the material surface by the specific sulfhydryl groups of Fc. Therefore, this treatment forced Fab to face out from the biomaterial surface to reduce steric hindrance. The DIG-modified adenovirus immobilizations were dependent on the titer of viral vectors, suggesting that the disulfide reduction of IgG was mild, and that IgG maintained its ability to bind adenovirus (Fig 3a). In addition, the conjugated anti-DIG half-IgG perfectly immobilized DIG-modified adenovirus at concentrations as high as 1×109 viral particles per well (Fig 3b). Higher virus concentrations caused the immobilization rate to decline due to the limited substrate area, but over 50% of the virus was still captured on the surface when virus concentration was less than 1011 viral particles per well. This suggests that DIG-modified adenovirus can be effectively tethered by surface anti-DIG IgG. The DIG-modified adenovirus maintained a similar stability pattern as non-modified virus (Fig 3d). At last 75% of both of these two adenoviral vectors were bound on the biomaterial for 2 weeks; evidence that virus remains stable on material surfaces without spreading from target sites and eliciting unwanted systemic infection.
To further investigate the potential of this model for in situ cell transduction, a low melting point wax technique was applied to spatially control anti-DIG IgG conjugation on chitosan surfaces. Secondary antibody and X-gal staining results illustrated that viruses immobilized by conjugated antibodies specifically transduced cells on the target sites (Fig 4a & b). These results demonstrated that DIG modification is feasible for mediating adenoviral vector immobilization and for in situ cell transduction on biomaterial surfaces. Although this study was performed on 2-D surfaces, wax or other masking techniques can easily be used to infiltrate 3-D structures to spatially control antibody conjugation. Therefore, this method should be able to successfully deliver virus to specific sites on biomaterial scaffolds with the goal of generating specific interfaces between cell signaling vectors and eventually used to engineer multi-tissue interfaces.
Several different bioactive factors have been applied to scaffolds to induce new tissue growth. For example, vascular endothelial growth factor (VEGF) can induce angiogenesis to facilitate new tissue growth [34, 35]. On the other hand, bone morphogenetic proteins (BMPs) are osteogenic growth factors that have a robust ability to induce bone regeneration in skeletal defects [36]. The combined release of both VEGF and BMP signaling proteins in scaffolds can greatly improve bone regeneration over the delivery of the individual growth factor [4]. For some complex clinical situations, such as orthopedic interfaces, the functional integration of subchondral bone with cartilage still poses a significant regenerative challenge because uncontrolled BMPs delivery may lead to excessive bone formation outside of the defect margins [37]. Composite scaffolds loaded with different cell types have been applied to control the regeneration at tissue interfaces [38, 39]. The loaded cells formed different tissues conducted by biomaterials, but this method requires the harvest of different cells from patients for in vitro cell culture followed by in vivo transplantation, which is very laborious and may not be cost effective.
In vivo regenerative gene therapy is a method to induce new tissue growth without the need for cell transplantation and repeated surgery [16]. Immobilizing viral vectors on biomaterials has been shown to be capable of delivering specific growth factors in scaffolds [16, 22]. This method spatially controls the region of transgene expression, something that is currently not possible with conventional in vivo gene transfer. Due to the difficulty in distinguishing viral vectors with different transgenes, we developed a viral surface modification method to differentiate multiple viral vectors. DIG grafts on viral surfaces were utilized as antigen determinants for antibody immobilization. Both adenovirus with and without DIG modification were immobilized on anti-adenovirus IgG conjugated surfaces, suggesting that DIG grafts were small enough that the epitopes maintained their antigenic properties after modification (Fig 5b). Likewise, anti-DIG IgG specifically captured DIG-modified adenovirus on the surface of the biomaterial (Fig 5a).
To coordinately combine different bioactive factors for regenerative applications, a dual adenovirus immobilization model was developed in our study (Fig 6a). In this model, one adenovirus was immobilized on the entire scaffold, and another virus was restricted to specific regions controlled by wax masking. This dual viral vector immobilization model was performed on a 2-D surface (Fig 6b). Cells expressing green or blue fluorescent proteins were both expressed in the non-masked area because anti-adenovirus IgG binds to both AdGFP and DIG-AdBFP. In contrast, the masked area that was conjugated by anti-DIG IgG contained only cells expressing BFP, suggesting that DIG modification can be used for specific virus immobilization (Fig 6c). While DIG-modified adenovirus can be bound on both anti-DIG and anti-adenovirus IgG, the immobilization ratio of modified virus to two different antibody-conjugated surfaces was dependent on antibody affinity. These results demonstrate that this dual adenoviral vector immobilization method can spatially control the distribution of cell signaling viruses on biomaterials and may be further developed to engineer multi-tissue interfaces in vivo.
Conclusions
A novel virus modification method was developed to tag DIG grafts as different antigenic determinants for antibody immobilization. This DIG modification method preserved virus activity without eliciting ATPase inhibition. In addition, the DIG modified virus was effectively and stably immobilized on biomaterial surfaces, whereas these bound viral vectors could still be released to infect adherent cells. A wax masking technique facilitated the patterning of two different IgG conjugations on the biomaterial surfaces. By immobilizing adenovirus with and without DIG modification, cells were transduced in situ so that one transgene was expressed on the entire scaffold surface, while another existed only in defined regions. This dual adenoviral vector delivery system should prove beneficial for engineering tissue interfaces by regulating multiple bioactive factor expression with spatial control.
Acknowledgments
The research was supported by a grant from the National Institutes of Health grant RO1 DE018890 (PHK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sheridan MH, Shea LD, Peters MC, Mooney DJ. Bioabsorbable polymer scaffolds for tissue engineering capable of sustained growth factor delivery. J Control Release. 2000;64(13):91–102. doi: 10.1016/s0168-3659(99)00138-8. [DOI] [PubMed] [Google Scholar]
- 2.Yao F, Eriksson E. Gene therapy in wound repair and regeneration. Wound Repair Regen. 2000;8(6):443–451. doi: 10.1046/j.1524-475x.2000.00443.x. [DOI] [PubMed] [Google Scholar]
- 3.Holland TA, Tabata Y, Mikos AG. Dual growth factor delivery from degradable oligo(poly(ethylene glycol) fumarate) hydrogel scaffolds for cartilage tissue engineering. Journal of Controlled Release. 2005;101(13):111–125. doi: 10.1016/j.jconrel.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 4.Huang YC, Kaigler D, Rice KG, Krebsbach PH, Mooney DJ. Combined angiogenic and osteogenic factor delivery enhances bone marrow stromal cell-driven bone regeneration. Journal of Bone and Mineral Research. 2005;20(5):848–857. doi: 10.1359/JBMR.041226. [DOI] [PubMed] [Google Scholar]
- 5.Sohier J, Vlugt TJH, Cabrol N, Van Blitterswijk C, de Groot K, Bezemer JM. Dual release of proteins from porous polymeric scaffolds. Journal of Controlled Release. 2006;111(12):95–106. doi: 10.1016/j.jconrel.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 6.Jang JH, Houchin TL, Shea LD. Gene delivery from polymer scaffolds for tissue engineering. Expert Rev Med Devices. 2004;1(1):127–138. doi: 10.1586/17434440.1.1.127. [DOI] [PubMed] [Google Scholar]
- 7.Bengali Z, Shea LD. Gene delivery by immobilization to cell-adhesive substrates. Mrs Bulletin. 2005;30(9):659–662. doi: 10.1557/mrs2005.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alden TD, Beres EJ, Laurent JS, Engh JA, Das S, London SD, Jane JA, Jr, Hudson SB, Helm GA. The use of bone morphogenetic protein gene therapy in craniofacial bone repair. J Craniofac Surg. 2000;11(1):24–30. doi: 10.1097/00001665-200011010-00005. [DOI] [PubMed] [Google Scholar]
- 9.Ashinoff RL, Cetrulo CL, Galiano RD, Dobryansky M, Bhatt KA, Ceradini DJ, Michaels J, McCarthy JG, Gurtner GC. Bone morphogenic protein-2 gene therapy for mandibular distraction osteogenesis. Annals of Plastic Surgery. 2004;52(6):585–591. doi: 10.1097/01.sap.0000123023.28874.1e. [DOI] [PubMed] [Google Scholar]
- 10.Lindsey WH. Osseous tissue engineering with gene therapy for facial bone reconstruction. Laryngoscope. 2001;111(7):1128–1136. doi: 10.1097/00005537-200107000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Song J, Shi B, Wang Y, Chen X, Huang C, Yang X, Xu D, Cheng X, Chen X. Combination of scaffold and adenovirus vectors expressing bone morphogenetic protein-7 for alveolar bone regeneration at dental implant defects. Biomaterials. 2007;28(31):4635–4642. doi: 10.1016/j.biomaterials.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 12.Qiang B, Segev A, Beliard I, Nili N, Strauss BH, Sefton MV. Poly(methylidene malonate 2.1.2) nanoparticles: a biocompatible polymer that enhances peri-adventitial adenoviral gene delivery. J Control Release. 2004;98(3):447–455. doi: 10.1016/j.jconrel.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 13.Wieland JA, Houchin-Ray TL, Shea LD. Non-viral vector delivery from PEG-hyaluronic acid hydrogels. J Control Release. 2007;120(3):233–241. doi: 10.1016/j.jconrel.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marshall E. Gene therapy death prompts review of adenovirus vector. Science. 1999;286(5448):2244–2245. doi: 10.1126/science.286.5448.2244. [DOI] [PubMed] [Google Scholar]
- 15.Grossman PM, Han ZG, Palasis M, Barry JJ, Lederman RJ. Incomplete retention after direct myocardial injection. Catheterization and Cardiovascular Interventions. 2002;55(3):392–397. doi: 10.1002/ccd.10136. [DOI] [PubMed] [Google Scholar]
- 16.Hu WW, Wang Z, Hollister SJ, Krebsbach PH. Localized viral vector delivery to enhance in situ regenerative gene therapy. Gene Ther. 2007;14(11):891–901. doi: 10.1038/sj.gt.3302940. [DOI] [PubMed] [Google Scholar]
- 17.Hu WW, Lang MW, Krebsbach PH. Development of adenovirus immobilization strategies for in situ gene therapy. The Journal of Gene Medicine. 2008;10(10):1102–1112. doi: 10.1002/jgm.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klugherz BD, Song C, DeFelice S, Cui X, Lu Z, Connolly J, Hinson JT, Wilensky RL, Levy RJ. Gene delivery to pig coronary arteries from stents carrying antibody-tethered adenovirus. Hum Gene Ther. 2002;13(3):443–454. doi: 10.1089/10430340252792576. [DOI] [PubMed] [Google Scholar]
- 19.Abrahams JM, Song C, DeFelice S, Grady MS, Diamond SL, Levy RJ. Endovascular microcoil gene delivery using immobilized anti-adenovirus antibody for vector tethering. Stroke. 2002;33(5):1376–1382. doi: 10.1161/01.str.0000014327.03964.c0. [DOI] [PubMed] [Google Scholar]
- 20.Fishbein I, Alferiev IS, Nyanguile O, Gaster R, Vohs JM, Wong GS, Felderman H, Chen IW, Choi H, Wilensky RL, Levy RJ. Bisphosphonate-mediated gene vector delivery from the metal surfaces of stents. Proc Natl Acad Sci U S A. 2006;103(1):159–164. doi: 10.1073/pnas.0502945102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fishbein I, Stachelek SJ, Connolly JM, Wilensky RL, Alferiev I, Levy RJ. Site specific gene delivery in the cardiovascular system. J Control Release. 2005;109(13):37–48. doi: 10.1016/j.jconrel.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 22.Stachelek SJ, Song C, Alferiev I, Defelice S, Cui X, Connolly JM, Bianco RW, Levy RJ. Localized gene delivery using antibody tethered adenovirus from polyurethane heart valve cusps and intra-aortic implants. Gene Ther. 2004;11(1):15–24. doi: 10.1038/sj.gt.3302129. [DOI] [PubMed] [Google Scholar]
- 23.Gifford SC, Yoshida T, Shevkoplyas SS, Bitensky MW. A high-resolution, double-labeling method for the study of in vivo red blood cell aging. Transfusion. 2006;46(4):578–588. doi: 10.1111/j.1537-2995.2006.00776.x. [DOI] [PubMed] [Google Scholar]
- 24.Tepper OM, Mehrara BJ. Gene therapy in plastic surgery. Plast Reconstr Surg. 2002;109(2):716–734. doi: 10.1097/00006534-200202000-00047. [DOI] [PubMed] [Google Scholar]
- 25.Suh JK, Matthew HW. Application of chitosan-based polysaccharide biomaterials in cartilage tissue engineering: a review. Biomaterials. 2000;21(24):2589–2598. doi: 10.1016/s0142-9612(00)00126-5. [DOI] [PubMed] [Google Scholar]
- 26.Soldin SJ. Digoxin--issues and controversies. Clin Chem. 1986;32(1 Pt 1):5–12. [PubMed] [Google Scholar]
- 27.Smith TW, Haber E. Digitalis. I. N Engl J Med. 1973;289(18):945–952. doi: 10.1056/NEJM197311012891805. [DOI] [PubMed] [Google Scholar]
- 28.Chan EL, Swaminathan R. A rapid assay for the measurement of Na+,K(+)-ATPase inhibitors. Clin Biochem. 1992;25(1):15–19. doi: 10.1016/0009-9120(92)80040-n. [DOI] [PubMed] [Google Scholar]
- 29.Chen RR, Silva EA, Yuen WW, Mooney DJ. Spatio-temporal VEGF and PDGF delivery patterns blood vessel formation and maturation. Pharmaceutical Research. 2007;24(2):258–264. doi: 10.1007/s11095-006-9173-4. [DOI] [PubMed] [Google Scholar]
- 30.Richardson TP, Peters MC, Ennett AB, Mooney DJ. Polymeric system for dual growth factor delivery. Nature Biotechnology. 2001;19(11):1029–1034. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- 31.Hao XJ, Silva EA, Mansson-Broberg A, Grinnemo KH, Siddiqui AJ, Dellgren G, Wardell E, Brodin LA, Mooney DJ, Sylven C. Angiogenic effects of sequential release of VEGF-A(165) and PDGF-BB with alginate hydrogels after myocardial infarction. Cardiovascular Research. 2007;75(1):178–185. doi: 10.1016/j.cardiores.2007.03.028. [DOI] [PubMed] [Google Scholar]
- 32.Segura T, Volk MJ, Shea LD. Substrate-mediated DNA delivery: role of the cationic polymer structure and extent of modification. J Control Release. 2003;93(1):69–84. doi: 10.1016/j.jconrel.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 33.Hobson DA, Pandori MW, Sano T. In situ transduction of target cells on solid surfaces by immobilized viral vectors. BMC Biotechnol. 2003;3:4. doi: 10.1186/1472-6750-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patel ZS, Mikos AG. Angiogenesis with biomaterial-based drug- and cell-delivery systems. Journal of Biomaterials Science-Polymer Edition. 2004;15(6):701–726. doi: 10.1163/156856204774196117. [DOI] [PubMed] [Google Scholar]
- 35.Kaigler D, Wang Z, Horger K, Mooney DJ, Krebsbach PH. VEGF scaffolds enhance angiogenesis and bone regeneration in irradiated osseous defects. J Bone Miner Res. 2006;21(5):735–744. doi: 10.1359/jbmr.060120. [DOI] [PubMed] [Google Scholar]
- 36.Bessa PC, Casal M, Reis RL. Bone morphogenetic proteins in tissue engineering: the road from laboratory to clinic, part II (BMP delivery) Journal of Tissue Engineering and Regenerative Medicine. 2008;2(23):81–96. doi: 10.1002/term.74. [DOI] [PubMed] [Google Scholar]
- 37.Peng H, Usas A, Hannallah D, Olshanski A, Cooper GM, Huard J. Noggin improves bone healing elicited by muscle stem cells expressing inducible BMP4. Mol Ther. 2005;12(2):239–246. doi: 10.1016/j.ymthe.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 38.Spalazzi JP, Dagher E, Doty SB, Guo XE, Rodeo SA, Lu HH. In vivo evaluation of a multiphased scaffold designed for orthopaedic interface tissue engineering and soft tissue-to-bone integration. Journal of Biomedical Materials Research Part A. 2008;86A(1):1–12. doi: 10.1002/jbm.a.32073. [DOI] [PubMed] [Google Scholar]
- 39.Schek RM, Taboas JM, Segvich SJ, Hollister SJ, Krebsbach PH. Engineered osteochondral grafts using biphasic composite solid free-form fabricated scaffolds. Tissue Engineering. 2004;10(910):1376–1385. doi: 10.1089/ten.2004.10.1376. [DOI] [PubMed] [Google Scholar]