

Abstract
The perturbation of the racemic equilibrium of luminescent D3 terbium(III) complexes with chelidamic acid (CDA), a hydroxylated derivative of 2,6-pyridine-dicarboxylic acid (DPA), by added chiral biomolecules such as l-amino acids has been studied using circularly polarized luminescence and 13C NMR spectroscopy. It is shown in this work that the chiral-induced equilibrium shift of [Tb(CDA)3]6− by l-amino acids (i.e. l-proline or l-arginine) was largely influenced by the hydrogen-bonding networks formed between the ligand interface of racemic [Tb(CDA)3]6− and these added chiral agents. The capping of potential hydrogen-bonding sites by acetylation in l-proline led to a ∼100-fold drop in the induced optical activity of the [Tb(CDA)3]6−:N-acetyl-l-proline system. This result suggested that the hydrogen-bonding networks serve as the basis for further noncovalent discriminatory interactions between racemic [Tb(CDA)3]6− and added l-amino acids.
Keywords: circularly polarized luminescence, lanthanide, chirality, amino acid
INTRODUCTION
Crucial to modern drug discovery is the recognition of chiral molecules. To fully understand the structure–activity relationships (SAR) and structure–property relationships (SPR)1 that are often based on “pharmaceutical profiling”,2 one needs to develop a reliable method to measure the absolute stereochemistry of each analog prepared or separated. In the year 2009, it is estimated that ∼$15 billion in revenue will come from enantiopure drugs.3 Thus, the emergence of new techniques in chiral separation and resolution must be found to meet this growing demand.4
Currently, the most common tools used in drug discovery and elucidation include HPLC,5-8 circular dichroism (CD),9 NMR,10 X-ray diffraction,11,12 and molecular imprinting methods.13,14 X-ray diffraction is the most commonly used technique in resolving absolute structures; however, the caveat for this approach is the difficulty of obtaining suitable crystals from small amounts of material. NMR methods for determining stereochemical activity based on modified Mosher's Methods15,16 or the Trost-Type approach17,18 are commonly used in the resolution of primary amines, secondary alcohols, and carboxylic acids. A study by Marathias et al.,19 where Ibuprofen's enantiomers were identified using NMR and residual dipolar coupling's (RDC's), serves as an example of the shift in modern time to ab initio methods. This is due to the complexity in resolving ambiguous proton environments through routine NMR methods. More recently, CD has been used extensively when the structure and specific optical rotation values are known.9 This technique serves as a staple in current DNA structure analysis, where the absorption bands and intensity can yield substantial information on the structural activity. The use of CD has given rise to newly modified techniques including the spectroscopic approach of comparing observed and calculated vibrational circular dichroism (VCD) spectra, electronic circular dichroism (ECD), or optical rotation values which have gained popularity because of the commercially manufactured software and supercomputers, powerful PC's, and Linux clusters.20 However, CD and molecular mechanics calculations rarely provide unambiguous data21 as corroborated by the revised assignment of the helical handedness initially proposed of peptide nucleic acid double helices.22-24
Circularly polarized luminescence (CPL), the emission analog of CD, has been used in recent times with an attempt to resolve the absolute configuration of 9-coordinate lanthanide(III) complexes with 2,6-pyridine-dicarboxylate (DPA). These complexes possess almost exact D3 symmetry and as such occur in solution as racemic mixtures of complexes with Δ and Λ helicity.25,26 Much of this work has focused on the perturbation of the equilibrium between Δ- and Λ-[Ln(DPA)3]3− by an added chiral bio-molecule (e.g. tartrate substrates, amino acids, or sugar derivatives).26-28 The added chiral species may perturb the racemic equilibrium resulting in a nonracemic ground state (this is often referred to as the “Pfeiffer effect”,29-32 a term originated from a work on the enhancement of optical rotation of optically active alkaloids in the presence of labile racemic transition-metal complexes).33,34 These differences can be detected by measurement of the circular polarization in the luminescence. The sign of the CPL should correlate with a unique stereochemical environment associated with the chiral organic molecule.
Some early CPL studies25,26,28,35 have sometimes assumed that the “Pfeiffer effect” is a direct consequence of the absolute configuration of the added chiral molecule, i.e. all l-amino acids give the same sign for the CPL from selected transitions of the DPA complexes. It appears that, unfortunately, such a simple structural picture is not accurate. From the measurements performed in the last few years,33,34 one can conclude that simple substitutions of similar groups such as minor modifications of the alkyl group of l-histidine amino acid derivatives do not change the overall chirality of the perturbation, but no correlation has been observed with different underivatized amino acids. Further contradictions have been found in the literature regarding the source of this complex mechanism responsible for the equilibrium shift28,36 while attempting to correlate the sign of the CPL to some element of the chiral structure of simple or complex sugars,27,37 or in the use of mixed–ligand lanthanide complexes38,39 containing more than one type of ligand. For instance, opposite CPL signs have been found by distinct research groups concerning the addition of chiral sugars such as D-ribose or D-mannose to racemic solutions of [Eu(DPA)3]3− and [Tb(DPA)3]3−. One can conclude that the diverse outcomes reported could result from experimental conditions not precisely controlled,27,28,36,38 as pointed out by recent measurements.33,34,40,41 In particular, an instrument limitation has also been advanced as a possible reason for the observation of an opposite CPL sign which would have a consequent influence on the conclusions of the studies, especially for measurements of very small luminescence dissymmetry factors. Although most of the CPL studies published in the literature have been done with custom-made instruments,34 the detection of CPL can now be done with a high degree of sensitivity (∼1 part in 104–105) and reliability using modern instrumentation.42 It should also be mentioned that in the pioneering works, the main purpose was to determine whether or not the solution species formed exhibited circularly polarized luminescence.
Thus far, limited attention has been devoted to the understanding of the factors that govern the perturbation of the racemic lanthanide(III) complexes by addition of chiral molecules such as amino acid derivatives.25,26,40,41 From this standpoint, our preliminary results have shown that the CPL sign and its magnitude are dependent upon several factors and not only from the chirality of the enantiomerically pure amino acid.33,34 We have observed that (i) simple modifications in the chiral molecules added to the racemic system did not change the sign of the CPL signal (the same enantiomeric form was favored), and (ii) the magnitude of the CPL signal was influenced by the presence of additional aromatic groups in the chiral molecules. It is imperative to take into account the effect of the various noncovalent chiral discriminatory interactions such as hydrogen bonding, coulombic forces, π-stacking, hydrophobic effects, experimental conditions (i.e. pH, temperature, ratio of system of interest to amino acid), and steric effects on the CPL sign and magnitude. In this article, the goal is to explain the outer-sphere interactions between the racemic solutions of 9-coordinate terbium(III) complexes with chelidamic acid (CDA) and various amino acids (Fig. 1). We have chosen to use l-proline (l-Pro) and its derivatives N-acetyl-l-proline (N-Ac-l-Pro) and N-acetyl-l-prolinamide (N-Ac-l-Pro-NH2), l-arginine (l-Arg), l-asparagine (l-Asn), l-tyrosine (l-Tyr), and l-phenylalanine (l-Phe), as they have been found to be some of the most effective chiral agents in the perturbation of racemic solutions of [Ln(DPA)3]3− under physiological conditions (pH 6–7). The aim of this study is to discern the effects which dictate the perturbation of the racemic lanthanide(III) complexes according to the fundamental differences between the chiral agents previously mentioned, where electrostatics, ring stacking, hydrogen bonding, coulombic forces, pH, etc, all have a specific role in the interactions seen. This article is part of a series of ongoing efforts to provide a further understanding of the intermolecular forces which constitute the base of the “Pfeiffer effect” (see theory section later), so that better predictions in future CPL studies focused on the chiral recognition of amino acids may take place.
Fig. 1.

Structures of 2,6-pyridine-dicarboxylate (DPA), chelidamic acid (CDA), and the various amino acids used in this study; l-proline (l-Pro), N-acetyl-l-proline (N-Ac-l-Pro), N-acetyl-l-prolinamide (N-Ac-l-Pro-NH2), l-arginine (l-Arg), l-asparagine (l-Asn), l-tyrosine (l-Tyr), and l-phenylalanine (l-Phe).
EXPERIMENTAL DETAILS
Starting materials consisting of lanthanide(III) chlorides, 2,6-pyridine-dicarboxylic acid (DPA), chelidamic acid (CDA), and the various amino acids were purchased from Aldrich, Bachem, or Acros, and used without further purification. The Ln content of solutions was determined by complexometric titrations with Titriplex III (Merck) in the presence of urotropine and xylene orange.
1H and 13C NMR spectra were performed on a Varian Inova 400 MHz NMR spectrometer. Chemical shifts are given with respect to TMS. Circularly polarized luminescence and total luminescence spectra were recorded on an instrument described previously.42 It is common to report the degree of CPL in terms of the luminescence dissymmetry factor, glum(λ), which is defined as
where IL and IR refer, respectively to the intensity of left and right circularly polarized emissions. The standard deviation, σd, in the measurement of the luminescence dissymmetry factor, glum, is defined as
where N is the total number of photon-pulses counted. The light source for excitation was a continuous wave 450 W xenon arc lamp from a Spex FluoroLog-2 spectrofluorometer, equipped with excitation and emission monochromators with dispersions of 4 nm/mm (SPEX, 1681B). 5D0 ← 7F0 excitation measurements for the Eu(III)-containing compounds were accomplished by using a Coherent-599 tunable dye laser (0.03 nm resolution) with a Coherent Innova Sabre TMS 15 or Innova-70 argon ion laser as a pump source. The laser dye used in all measurements was rhodamine 6G dissolved in ethylene glycol. Calibration of the emission monochromator (and subsequently the dye laser wavelength) was accomplished by passing scattered light from a low power He-Ne laser through the detection system. The error in the dye-laser wavelength is assumed to equal the resolution of the emission monochromator (0.1 nm). The optical detection system consisted of a focusing lens, long pass filter, and 0.22 m monochromator. The emitted light was detected by a cooled EMI-9558B photomultiplier tube operating in photon-counting mode. All measurements were performed in quartz cuvettes with a path length of 0.4 or 1.0 cm.
The measurements were performed in aqueous solutions at 295 K. Solutions for NMR, luminescence, and CPL measurements were prepared by mixing aqueous stock solutions of Eu(III) or Tb(III) chlorides and DPA or CDA in a 1:3.5 or 1:5.5 molar ratio, adding aqueous amino acid solutions, and waiting for ∼24 h before use in order to allow them to reach conformational equilibrium. The concentration of lanthanide(III) complexes and of amino acids were 0.04 mol/l and in the range of 0.6–1.2 mol/l. The final pH of solutions was approximately 7.0–7.2, 11.0–11.2, and 12.2–12.4.
“PFEIFFER EFFECT” THEORY
Consistent with previous work,34 it is assumed here that the effect of adding chiral amino acids (AA*) results in the preferential formation of one diastereomeric outer-sphere association complex. The three relevant equilibrium expressions are defined as follows
| (1) |
| (2) |
| (3) |
where the outer-sphere association complex is denoted by a colon (:). Previous studies have demonstrated that the addition of chiral amino acids to a racemic mixture of D3 lanthanide(III) complexes may lead to a perturbation of the ground state equilibrium without changing the local structure of the complexes involved.34 This perturbation is often referred to as the “Pfeiffer effect.” This latter leads to an enantiomeric excess in the ground state, η, defined as follows
| (4) |
where the square brackets in this equation denote ground state concentrations. As also shown in previous studies,37 in the limit of large concentrations of AA*, the concentration of free (i.e. unassociated) complex goes to zero, and η may be related to the diastereomeric association equilibrium constants as follows:
| (5) |
It should be noted that in the absence of other chiral effects such as enantioselective excited state quenching, it may be assumed that enantiomeric excesses in the excited and ground states are equal.
RESULTS AND DISCUSSION
We have chosen to study the 9-coordinate Eu(III) and Tb(III) complexes with the hydroxylated derivative of DPA, where a hydroxyl group is grafted onto the 4-position of the pyridine ring in lieu of a hydrogen atom. These compounds were selected due to the extensive research on these systems regarding stability, structural, luminescence, and CPL activity properties.34,43-49 The presence of a hydroxyl group in the CDA ligand greatly changes the environment where [Ln(CDA)3]6− exists compared to its analog compound, [Ln(DPA)3]3−. Although [Ln(DPA)3]3− occurs in aqueous solution as racemic mixtures of complexes with Δ and Λ helicity under physiological pH conditions (6–7), the tris complex with CDA is only present in a strong basic medium (pH > 12). The pH conditions were determined based on the identification of the various Eu-containing species using the 5D0 ← 7F0 excitation spectroscopy. At the lowest Eu(III):CDA ratio, the 1:1 complex is the major complex in solution (pH 3.2, Fig. 2). Increasing this metal to ligand ratio from 1:1 to 1:5.5 at a pH of 3.4 led to the observation that the 1:2 complex is the major complex in solution, while a third species corresponding to the tris complex is predominantly observed at higher pHs. The high resolution excitation spectra showed that the Eu(III) ions were in the form of the 1:3 species in a 0.01 M aqueous solution at a pH greater than 12. A Eu(III):CDA ratio of 1:5.5 was used to ensure complete formation of the tris complex. Eu(III) has in theory a simpler crystal field energy level pattern than Tb(III), but the latter has been chosen for the CPL and NMR studies due to its paramagnetic properties, and also because it was generally observed that the magnitude of the glum values was larger for the magnetic-allowed transitions of Tb(III).34 Knowing that the effect of adding chiral amino acids results in the preferential formation of one diastereomeric outer-sphere association complex without changing the local structure of the complexes involved, racemic complexes of, e.g., Eu(III) or Dy(III) could have been used too, as already demonstrated in previous studies.34 Moreover, the observation of identical 5D0 ← 7F0 excitation and 5D0 → 7F2 emission spectra for the aqueous solution of unassociated [Eu(CDA)3]6− and after addition of a 30- or 40-fold excess of l-Arg to [Eu(CDA)3]6− supported the assumption that the added amino acid does not significantly perturb the coordination of Eu(III) even at these high concentrations (Fig. 3).
Fig. 2.
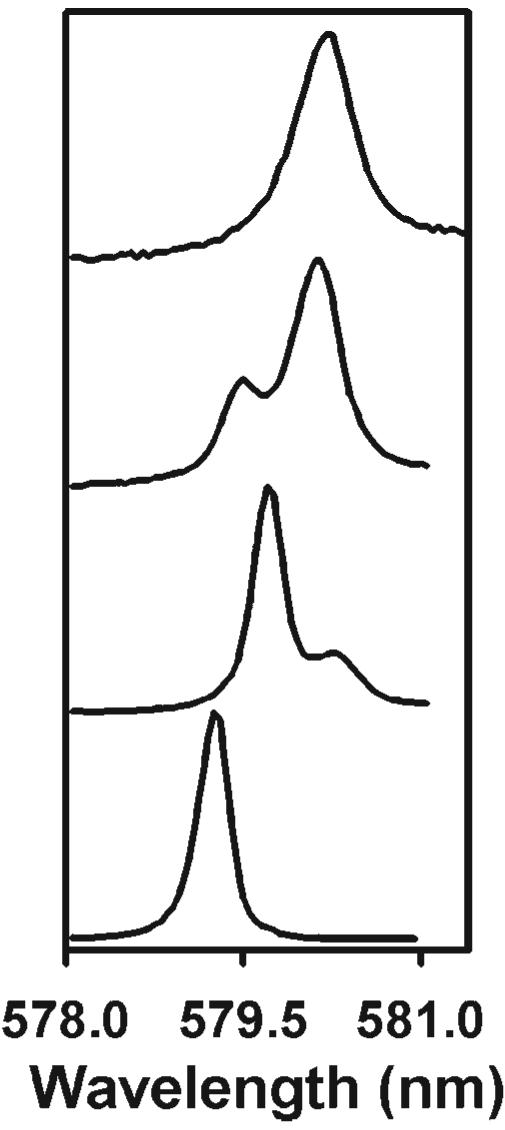
5D0 ← 7F0 excitation spectra for an aqueous solution of Eu(III) and CDA at a metal to ligand ratio of 1:1 (bottom) and 1:5.5 (three other spectra), and at different pHs (3.2, 3.4, 8.0, and 12.4 from bottom to top). The Eu(III) concentration was 0.01 mol/l and the luminescence was monitored at approximately 615 nm.
Fig. 3.
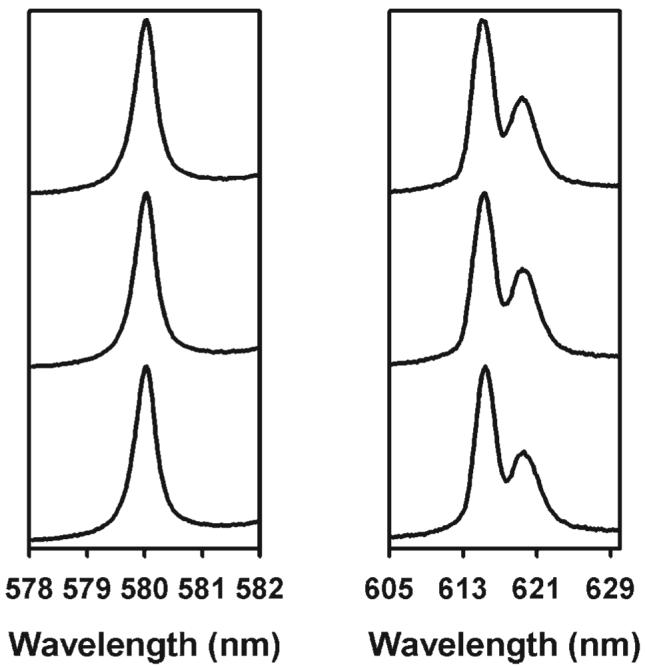
5D0 ← 7F0 excitation (left) and 5D0 → 7F2 emission (right) spectra for aqueous solutions of 0.040 mol/l [Eu(CDA)3]6−, and after addition of 1.2 and 1.6 mol/l of l-Arg at 295 K and at a pH of 12.2 (from bottom to top). The luminescence was monitored at approximately 615 nm for the excitation spectra, while the emission spectra were recorded following excitation at the maximum of the 5D0 ← 7F0 excitation spectra.
Although [Ln(DPA)3]3− and [Ln(CDA)3]6− are both negatively charged complexes, the “Pfeiffer effect” studies of the former system were mainly done under physiological pH conditions. In the case of [Tb(CDA)3]6−, the studies have been conducted at a pH above 12 and showed that the deprotonation of the hydroxyl group of CDA (pKa of 10.8)46,50 played a key role in the outer-sphere interactions responsible for the perturbation of the racemic equilibrium by l-amino acids (see later). It should be mentioned that the work discussed in this article represents the first example of using the racemic solution of the 9-coordinate terbium(III) complex with CDA. On the other hand, a basic pH also affected the form of the amino acids present in solution. Under physiological pH conditions (6–7) these amino acids are perhaps more correctly written as their zwitterionic form, whereas at strong basic conditions the amino acids have overall negative charges resulting from the deprotonation of the carboxylic group and loss of the positive charge on the amino part. It should be noted that we have focused on the glum values at the ∼543-nm peak of the total luminescence spectrum of the Tb(III) complexes in order to allow us to compare the sign and magnitude of the CPL signal for the different amino acids considered in the study. All the glum values reported in this study have been measured with a standard deviation, σd, of ±0.0003.
l-Pro Derivative-Based Chiral Probes
The optical activity induced by the addition of a 30- or 15-fold excess of l-Pro to a racemic solution of [Tb(CDA)3]6− was the most pronounced amongst the various amino acids tested (glum = +0.0255 or +0.0102, Table 1). In addition, a ∼4-fold increase in the magnitude of the glum value has been observed for [Tb(CDA)3]6−: l-Pro compared to its analog system with [Tb(DPA)3]3− (glum = +0.0102 vs. +0.0024 at pH 12 and 7, respectively, Tables 1 and 2). Raising the pH of the [Tb(DPA)3]3−: l-Pro solution from 7 to ∼11 resulted in a significant decrease of glum from +0.0024 to +0.0006 (factor of 4), which was consistent with the fact that the added l-Pro did not perturb the racemic equilibrium of [Tb(DPA)3]3− anymore. An increase in the pH was accompanied by a decrease of the concentration of the zwitterionic form of l-Pro and, consequently, favored repulsive electrostatic forces between the negatively charged [Tb(DPA)3]3− complex and negatively charged l-Pro amino acid (pKa values of 2.0 and 10.8 for free l-Pro).51 As a result, the magnitude of glum decreased, indicating that the equilibrium shift became smaller with an increase of the pH towards basic conditions. The optimal interaction between [Tb(DPA)3]3− and l-Pro or other amino acids tested (i.e. l-Phe or l-Arg) was obtained under physiological pH conditions, where these amino acids are found in their zwitterionic form (Table 2). This finding led to the conclusion that the repulsive electrostatic forces in a basic medium offset the predominant intermolecular interactions (i.e. hydrogen bonding, coulombic forces, π-stacking, hydrophobic effects, or attractive electrostatic forces), responsible for the perturbation of the racemic [Tb(DPA)3]3− at pH ∼7.
TABLE 1.
Luminescence dissymmetry ratio values (glum) in the spectral range of the 5D4 → 7F5 transition for 0.040 mol/l [Tb(CDA)3]6− after the addition of 0.60–1.20 mol/l of various l-amino acids in deuterated aqueous solution at 295 K
| [AA] | ||
|---|---|---|
| Amino acid |
[Tb(CDA) 3]6− |
glum (542.5 nm) σd = ±0.0003 |
| l-proline | 30 (pH 12.2) | +0.0255 |
| 15 (pH 12.2) | +0.0102 | |
| N-Ac-l-proline | 15 (pH 12.1) | −0.0002 |
| N-Ac-l-proline-NH2 | 15 (pH 12.2) | −0.0083 |
| l-asparagine | 30 (pH 12.2) | +0.0034 |
| l-arginine | 30 (pH 12.2) | +0.0036 |
| l-tyrosine | 15 (pH 12.3) | +0.0001 |
| l-phenylalanine | 15 (pH 12.2) | +0.0020 |
Excitation wavelengths were in the 318–323 nm range.
TABLE 2.
Luminescence dissymmetry ratio values (glum) in the spectral range of the 5D4 → 7F5 transition for 0.010 mol/l [Tb(DPA)3]3− after addition of 0.15 mol/l of various l-amino acids in aqueous solution at 295 K
| [AA] | ||
|---|---|---|
| Amino acid |
[Tb(DPA)3]3− |
glum (543.0 nm), σd = ±0.0003 |
| l-proline | 15 (pH 7.0) | +0.0024 |
| 15 (pH 11.0) | +0.0006 | |
| l-tyrosine | 15 (pH 10.0) | +0.0081 |
| l-phenylalanine | 15 (pH 7.0) | −0.0077 |
| 15 (pH 10.0) | −0.0012 |
Excitation wavelengths were in the 318–323 nm range.
Knowing that [Tb(CDA)3]6− is present at a pH above 12, one may expect that other intermolecular forces may contribute to the recognition between [Tb(CDA)3]6− and the added chiral probes. As shown in Table 1, the determination of glum values with different orders of magnitude and opposite signs (− to +) confirmed the role of a multitude of factors contributing to the detection of an induced CPL signal. In particular, hydrogen-bonding has been found to be a strong dictator of these intermolecular interactions responsible in the induced optical activities detected at pH ∼12.2. Deprotonation of the hydroxyl group in CDA allowed effective hydrogen bonding between l-Pro (through its NH group) and the ligand interface of [Tb(CDA)3]6−. The use of 13C-NMR proved the occurrence of hydrogen-bonding, where the largest chemical-shift differences (Δδ) between the free l-Pro (i.e. unassociated) and associated l-Pro in the Pfeiffer-perturbed system occurred in the surrounding carbon atoms to the hydrogen-bonding site on the nitrogen atom of l-Pro. The Δδ values for the carbons of the pyrrolidine ring of l-Pro amounted between 0.8 and 1.1 ppm, while a chemical shift of only 0.1 ppm was observed for the carbon of the carboxylate group (Table 3), suggesting that this group remained practically unaffected by the presence of [Tb(CDA)3]6− (terbium is well known to induce chemical shifts and, therefore, the carbon atoms closest to the paramagnetic [Tb(CDA)3]6− would shift to the greatest degree).52 This latter observation indicated that the carboxylate substituent was not actively participating in the outer-sphere interactions. This result is in agreement with previous studies done by Brittain in which he suggested that the association of [Tb(DPA)3]3− and l-Pro happened through a possible hydrogen-bonding mechanism involving the amino group of l-Pro and the DPA π-orbitals.28 It should be noted that our study using [Tb(CDA)3]6− (pH ∼12) in lieu of [Tb(DPA)3]3− (pH ∼11) with l-Pro also resulted in a positive CPL signal.
TABLE 3.
Carbon chemical shift values for deuterated aqueous solutions of 1.20 mol/l of free amino acids and for 0.040 mol/l racemic solutions of [Tb(CDA)3]6− after addition of 1.20 mol/l of various amino acids at 295 K and at a pH of 12.2
| Amino acid | Free amino acid |
Associated amino acid |
Δδ (ppm)a |
|---|---|---|---|
| l-proline | 181.9 | 182.0 | −0.1 |
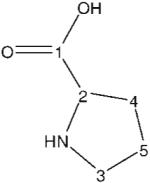 |
60.9 | 61.8 | −0.9 |
| 45.4 | 46.5 | −1.1 | |
| 30.2 | 31.0 | −0.8 | |
| 24.7 | 25.7 | −1.0 | |
| l-arginine | 182.7 | 182.9 | −0.2 |
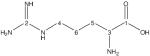 |
156.3 | 155.0 | 1.3 |
| 55.1 | 55.5 | −0.4 | |
| 40.5 | 40.7 | −0.2 | |
| 31.3 | 31.7 | −0.4 | |
| 24.1 | 24.4 | −0.3 | |
| l-asparagine | 181.2 | 181.7 | −0.5 |
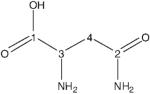 |
176.6 | 177.0 | −0.4 |
| 53.2 | 53.6 | −0.4 | |
| 40.4 | 41.0 | −0.6 |
Obtained by subtracting the associated amino acid value from the free amino acid value.
The importance of the amino group of l-Pro in the induced optical activity of [Tb(CDA)3]6− has been confirmed by testing a derivative of l-Pro, N-Ac-l-Pro, where the hydrogen of the NH group has been substituted by a bulkier acetyl group. The capping of this amino acid at its hydrogen-bonding site resulted in a considerable decrease of magnitude in the perturbation of the racemic [Tb(CDA)3]6− upon addition of a 15-fold excess of N-Ac-l-Pro (glum values went from +0.0102 to almost zero, see Fig. 4 and Table 1). This result suggested that the presence of the NH group is critical for effective intermolecular interactions between l-Pro and [Tb(CDA)3]6−. The formation of hydrogen-bonding serves as the basis for further interactions (i.e. planar stacking interactions) which probably explain the strength of l-Pro to effectively perturb the racemic solution of [Tb(CDA)3]6−. A study by Rudolph and Crowe has shown that pyrrolidone rings of l-Pro form planar stacking arrangements of aromatic groups upon one another.53 These planar stacking interactions are thought to allow l-Pro to closely interact with [Tb(CDA)3]6−, where the pyrrolidone rings of l-Pro and the ligand interface of [Tb(CDA)3]6− are thought to stack directly upon one another. On the other hand, the presence of the acetyl group in N-Ac-l-Pro prevents the formation of hydrogen-bonding interactions and, thus, diminishes the possibility of forming a planar stacking arrangement due to (i) steric hindrances resulting from the addition of the bulky acetyl substituent, and/or (ii) the absence of an efficient recognition mechanism between [Tb(CDA)3]6− and the chiral probe. As a result, the driving force in the weak perturbation of the racemic solution of [Tb(CDA)3]6− by N-Ac-l-Pro is probably electrostatic and/or hydrophobic in nature.
Fig. 4.
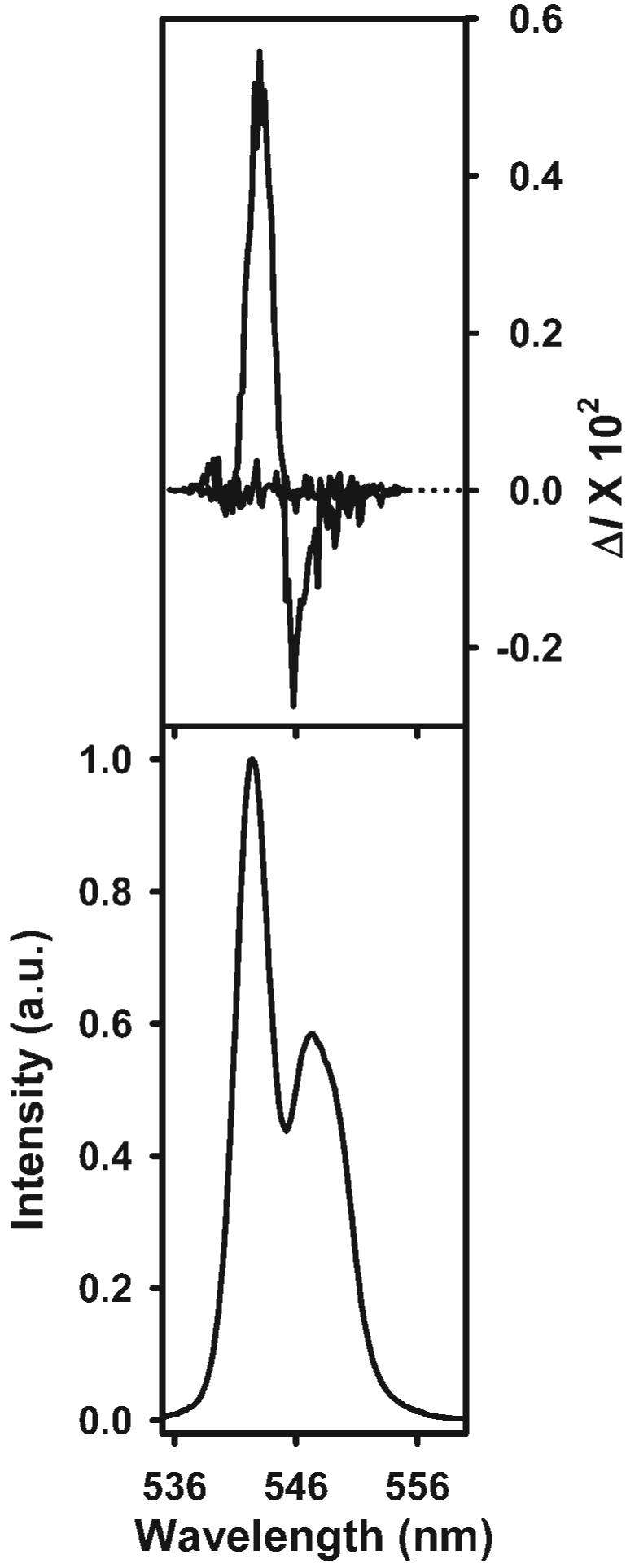
Circularly polarized luminescence (upper curves) and total luminescence (lower curves) spectra for the 5D4 → 7F5 transition of 0.040 mol/l [Tb(CDA)3]6− after addition of 0.60 mol/l of l-proline (solid line) and N-acetyl-l-proline (bold line) in aqueous solution at 295 K. Excitation wavelengths were in the 318–323 nm range.
The importance of hydrogen-bonding in the recognition mechanism between [Tb(CDA)3]6− and l-Pro derivative-based chiral probes has further been investigated with the use of N-Ac-l-Pro-NH2, a derivative of N-Ac-l-Pro. The hydrogen-donating hydroxyl group of the carboxylate function in N-Ac-l-Pro has been converted into a hydrogen-accepting amino substituent, more favorable to hydrogen-bonding interactions with negatively charged complexes such as [Tb(CDA)3]6−. Addition of a 15-fold excess of N-Ac-l-Pro-NH2 to a racemic solution of [Tb(CDA)3]6− led to the observation of a relatively strong induced CPL signal compared to l-Pro and N-Ac-l-Pro (glum = −0.0083 vs. +0.0102 and +0.0002, Table 1). It is interesting to note that the glum values obtained for [Tb(CDA)3]6−:l-Pro and [Tb(CDA)3]6−:N-Ac-l-Pro-NH2 adduct systems have opposite signs, suggesting that these chiral probes interact preferentially with one of the two enantiomeric forms (Λ or Δ) of [Tb(CDA)3]6−, but opposite of each other (influence of the bulky acetyl group). However, the derivatized amino acid N-Ac-l-Pro-NH2 also corroborated that the driving force in the recognition mechanism is largely influenced by the hydrogen-bonding forces. N-Ac-l-Pro-NH2 most probably interacts in a similar fashion to l-Pro (via a hydrogen-bonding-based mechanism through the amino function). This is favored due to the removal of the negative charge that was located on the carboxylate function in N-Ac-l-Pro when its hydroxyl group was converted into an amino group in N-Ac-l-Pro-NH2.
Amino Acids with Additional Hydrogen Bonding Side Chain Sites
To further examine the importance of the hydrogen-bonding forces in the chiral-induced equilibrium shift of [Tb(CDA)3]6−, we have tested l-Arg, an amino acid that contains a guanidino group in its side chain. The study with l-Pro derivatives indicated that the primary amino acid site of recognition contained an NH or NH2 group, favorable for hydrogen-bonding interactions with the ligand interface of [Tb(CDA)3]6−. Knowing that the guanidino group of l-Arg has a pKa of 12.48, l-Arg is positively charged in neutral, acidic, and most basic environments. The delocalization of the positive charge because of the conjugation between the nitrogen lone pairs and the double bond enables the guanidino group to form hydrogen-bonds, principally with negatively charged groups. Addition of a 30-fold excess of l-Arg to [Tb(CDA)3]6− yielded hydrogen-bonding interactions which seemed to dictate the nature of this amino acid's induction of optical activity. The data obtained through 13C NMR strongly indicated that the site of interaction occurred at the carbon position within the guanidino side chain of l-Arg, supporting the notion that hydrogen-bonding is indeed the driving force. The largest chemical shift observed between the unassociated l-Arg and associated l-Arg to [Tb(CDA)3]6− was for the carbon atom of the guanidino substituent (Δδ = 1.3 ppm), while the other carbons of l-Arg were moderately shifted with Δδ values ranging between 0.2 and 0.4 ppm (Table 3). This result implies that the hydrogen-bonding character of the negatively charged hydroxyl group in CDA plays an important role in the recognition mechanism between [Tb(CDA)3]6− and added amino acid-based chiral probes such as l-Arg or l-Pro, susceptible to form hydrogen bonds. These hydrogen-bonding networks serve as the basis for effective noncovalent chiral discriminatory interactions thereafter. However, the induced CPL signal for [Tb(CDA)3]6−:l-Arg was ∼7-fold weaker than for [Tb(CDA)3]6−:l-Pro (glum = +0.0036 vs. +0.0255, Table 1), and this may be due to a lesser degree of complementation between the interacting surfaces of the l-Arg side chain moiety and the ligand interface of [Tb(CDA)3]6−.
A similar result has been found when a 30-fold excess of l-Asn (pKa values of 2.2 and 8.8 for free l-Asn)51 was added to the racemic solution of [Tb(CDA)3]6− at pH 12.2. The magnitude of the induced CPL signal is also smaller than for l-Pro (factor of ∼7), but of opposite sign (glum = −0.0034 vs. +0.0255, Table 1). Knowing that the side chain of l-Asn can make efficient hydrogen bond interactions, this amino acid is another example where the chiral induced equilibrium shift of [Tb(CDA)3]6− is driven by the formation of hydrogen bonds between the complex and l-Asn. It is interesting to note that all carbon atoms in l-Asn are shifted by approximately 0.4–0.6 ppm upon addition of [Tb(CDA)3]6− (Table 3), suggesting that most of the hydrogen bond donor and acceptor groups of l-Asn participate in an energetically favorable hydrogen-bonding network. Because amino acids are rather flexible structures, the active sites of l-Asn with hydrogen-bonding characters (i.e. amide, amine, and carboxylate functions) reshape until a point where a complementary fit is reached between the ligand interface of [Tb(CDA)3]6− and the structure of l-Asn. This rearrangement in highly complementary structures is probably similar to the one found in protein–protein interactions, which is often referred as the induced fit model.54,55 The active sites reshape and continue to change until an optimal arrangement has been found, at which point the noncovalent discriminatory interactions are maximal. However, the complementary shape, charge, steric, and hydrophobic/hydrophilic characteristics of the systems of interest will dictate the stability of the adduct formed, resulting from the association of the amino acid to the Pfeiffer-perturbed system.
Amino Acids with Aromatic Rings
The importance of the factors discussed earlier can be evaluated in the magnitude of the induced CPL signal determined when chiral amino acids are added to racemic solutions of labile D3 terbium(III) complexes with achiral ligands such as CDA or DPA. For instance, we have found from our studies that working with amino acids with aromatic rings such as l-Phe or l-Tyr led to little induced optical activity when added to a racemic solution of [Tb(CDA)3]6− (Table 1). This is an effective example of the importance of the steric and electrostatic properties brought about by the addition of a hydroxyl group in the para-position of the pyridine ring in CDA. The addition of l-Phe or l-Tyr to a racemic solution of [Tb(DPA)3]3− resulted in a relatively strong induced CPL signal at pH 7 or 10 (glum = −0.0077 or +0.0081, Table 2), whereas the magnitude of the induced CPL signal dropped significantly in the case of a racemic solution of [Tb(CDA)3]6− at pH 12.2 (glum = +0.0020 or +0.0001, Table 1). This is thought to be due to the bulky nature of these amino acids with aromatic rings, the deprotonation of the hydroxyl group in the l-Tyr side chain (pKa of 10.6),51 and the resultant electrostatic repulsions between l-Tyr and [Tb(CDA)3]6−.
CONCLUSION
This work showed that the “Pfeiffer effect” induced in a 9-coordinate terbium(III) complex with CDA, the hydroxylated derivative of DPA, was influenced by the nature of the l-amino acids, and was also different from its analog compound, [Tb(DPA)3]3−. The ligand interface of the racemic complex plays a key role in the nature of the outer-sphere interactions responsible for the perturbation of the racemic equilibrium by l-amino acids. In particular, the hydrogen-bonding character of the negatively charged hydroxyl group in CDA leads to a larger “Pfeiffer effect” with l-amino acids susceptible to form hydrogen bonds with negatively charged groups, while these hydrogen-bonding effects are less important with DPA. However, the hydrogen-bonding networks formed between the ligand interface of the racemic [Tb(CDA)3]6− and the l-amino acids such as l-Pro or l-Arg serve as the basis for further outer-sphere interactions (i.e. electrostatics, ring stacking, coulombic forces, etc).
This study confirms that the chiral recognition of l-amino acids can be modulated by the nature of the ligand interface of the racemic 9-coordinate lanthanide(III) complexes and, in particular, by varying the substituent in the para-position of the pyridine ring of DPA. The combination of these factors opens new opportunities for the design of lanthanide triple helical complexes acting as probes for chiral recognition. Research in this direction is currently underway.
These results also suggested that CPL spectroscopy is a convenient and useful tool for evaluating the chiral recognition properties of promising functionalized luminescent chiral lanthanide complexes used as probes for specific target molecules due to (i) its high degree of sensitivity and reliability, (ii) its ease of use, and (iii) its minimal sample preparation. This work is intended to serve as a starting point aimed at demonstrating that CPL spectroscopy would be an attractive complementary method to current techniques used as specific structural probes of chiral molecules such as circular dichroism.
ACKNOWLEDGMENTS
We are also grateful to San José State University and San José State University Research Foundation for an Award for Research, Scholarship or Creativity Activity for G.M.
Contract grant sponsor: National Institute of Health Minority Biomedical Research Support; Contract grant number: 2 S06 GM008192-24A1.
Contract grant sponsor: Research Corporation Cottrell Science Award;
Contract grant number: CC6624.
LITERATURE CITED
- 1.Nassar AEF, Kamel AM, Clarimont C. Improving the decision-making process in the structural modification of drug candidates: enhancing metabolic stability. Drug Disc Today. 2004;9:1020–1028. doi: 10.1016/S1359-6446(04)03280-5. [DOI] [PubMed] [Google Scholar]
- 2.Di L, Kerns EH. Application of pharmaceutical profiling assays for optimization of drug-like properties. Curr Opin Drug Disc Develop. 2005;8:495–504. [PubMed] [Google Scholar]
- 3.Rouhi AM. Chiral chemistry. Chem Eng News. 2004;82(24):47–62. [Google Scholar]
- 4.Thayer AM. Centering on chirality. Chem Eng News. 2007;85(32):11–19. [Google Scholar]
- 5.Zhang Y, Wu DR, Wang-Iverson DB, Tymiak A. Enantioselective chromatography in drug discovery. Drug Discov Today. 2005;10:571–577. doi: 10.1016/S1359-6446(05)03407-0. [DOI] [PubMed] [Google Scholar]
- 6.Welch CJ, DaSilva J, Bilba M, Albaneze-Walker J, Henderson D, Laing B, Mathre D. Preparative chiral SFC as a green technology for rapid access to enantiopurity in pharmaceutical process research. LC-GC. 2005;23:16–29. [Google Scholar]
- 7.Zhai ZD, Shi YP, Wang T. Development and validation of HPLC methods for enantioseparation of mirtazapine enantiomers at analytical and semipreparative scale using polysaccharide chiral stationary phases. Anal Chim Acta. 2005;550:123–129. [Google Scholar]
- 8.Pirkle WH, Pochapsky TC. Considerations of chiral recognition relevant to the liquid chromatographic separation of enantiomers. Chem Rev. 1989;89:347–362. [Google Scholar]
- 9.Schlingmann G, Roll DM. Absolute stereochemistry of unusual biopolymers from Ascomycete culture LL-W1278: examples that derivatives of (S)-6-hydroxymellein are also natural fungal metabolites. Chirality. 2005;17:S48–S51. doi: 10.1002/chir.20105. [DOI] [PubMed] [Google Scholar]
- 10.Jaki B, Franzblau S, Pauli GF. An NMR method towards the routine chiral determination of natural products. Phytochem Anal. 2004;15:213–219. doi: 10.1002/pca.760. [DOI] [PubMed] [Google Scholar]
- 11.Maas G. Determination of absolute and relative configuration by X-ray and neutron diffraction methods. In: Houben-Weyl, editor. Methoden der Organischen Chemie, and E21a. Thieme Verlag; Stuttgart: 1995. p. 1170. [Google Scholar]
- 12.Stout G, Jensen L. X-ray structure determination: a practical guide. 2nd ed. Wiley; New York: 1989. p. 480. [Google Scholar]
- 13.Batra D, Shea KJ. Combinatorial methods in molecular imprinting. Curr Opin Chem Biol. 2003;7:434–442. doi: 10.1016/s1367-5931(03)00060-7. [DOI] [PubMed] [Google Scholar]
- 14.Lin JM, Nakagama T, Wu XZ, Uchiyama K, Hobo T. Capillary electrochromatographic separation of amino acid enantiomers with molecularly imprinted polymers as chiral recognition agents. Fresenius J Anal Chem. 1997;357:130–132. [Google Scholar]
- 15.Kusumi T, Ooi T, Ohkubo Y, Yabuuchi T. The modified mosher's method and the sulfoximine method. Bull Chem Soc Jpn. 2006;79:965–980. [Google Scholar]
- 16.Dale JA, Mosher HS. Nuclear magnetic resonance enantiomer reagents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and α-methoxy-α-trifluoromethylphenylacetate (MTPA) Esters. J Am Chem Soc. 1973;95:512–519. [Google Scholar]
- 17.Seco JM, Quiñoá E, Riguera R. The assignment of absolute configuration by NMR. Chem Rev. 2004;104:17–117. doi: 10.1021/cr2003344. [DOI] [PubMed] [Google Scholar]
- 18.Seco JM, Quiñoá E, Riguera R. A practical guide for the assignment of the absolute configuration of alcohols, amines and carboxylic acids by NMR. Tetrahedron: Asymmetry. 2001;12:2915–2925. [Google Scholar]
- 19.Marathias VM, Tawa GJ, Goljer I, Bach AC., II Stereochemical identification of (R)- and (S)-ibuprofen using residual dipolar couplings, NMR, and modeling. Chirality. 2007;19:741–750. doi: 10.1002/chir.20338. [DOI] [PubMed] [Google Scholar]
- 20.McConnell O, He Y, Nogle L, Sarkahian A. Application of chiral technology in a pharmaceutical company. Enantiomeric separation and spectroscopic studies of key asymmetric intermediates using a combination of techniques. Phenylglycidols. Chirality. 2007;19:716–730. doi: 10.1002/chir.20368. [DOI] [PubMed] [Google Scholar]
- 21.Bringmann G, Maksimenka K, Mutanyatta-Comar J, Knauer M, Bruhn T. The absolute axial configurations of knipholone and knipholone anthrone by TDDFT and DFT/MRCI CD calculations: a revision. Tetrahedron. 2007;63:9810–9824. [Google Scholar]
- 22.Sforza S, Haaima G, Marchelli R, Nielsen PE. Chiral peptide nucleic acids (PNAs): helix handedness and DNA recognition. Eur J Org Chem. 1999:197–204. [Google Scholar]
- 23.Wittung P, Nielsen PE, Buchardt O, Egholm M, Nordén B. DNA-like double helix formed by peptide nucleic acid. Nature. 1994;368:561–563. doi: 10.1038/368561a0. [DOI] [PubMed] [Google Scholar]
- 24.Corradini R, Sforza S, Tedeschi T, Marchelli R. Chirality as a tool in nucleic acid recognition: principles and relevance in biotechnology and in medicinal chemistry. Chirality. 2007;19:269–294. doi: 10.1002/chir.20372. [DOI] [PubMed] [Google Scholar]
- 25.Brittain HG. Circularly polarized luminescence studies of chiral lanthanides complexes. Pract Spectrosc. 1991;12:179–200. [Google Scholar]
- 26.Brittain HG. Circularly polarized luminescence studies of chiral lanthanide complexes. J Coord Chem. 1989;20:331–347. [Google Scholar]
- 27.Brittain HG. Studies of the pfeiffer effect induced in tris(pyridine-2,6-dicarboxylato)terbate(III) by monosaccharide aldose sugars. J Chem Soc Dalton Trans. 1984;7:1367–1370. [Google Scholar]
- 28.Brittain HG. Optical activity induced in terbium(III) tris(pyridine-2,6-dicarboxylate) through association with certain chiral amino acids. Inorg Chem. 1981;20:3007–3013. [Google Scholar]
- 29.Pfeiffer P, Nakasuka Y. The activation of complex salts in aqueous solutions III. Chem Ber. 1933;66B:415–418. [Google Scholar]
- 30.Pfeiffer P, Quehl K. Activation of complex salts in aqueous solution. Chem Ber. 1932;65:560–565. [Google Scholar]
- 31.Pfeiffer P, Quehl K. A new effect in solutions of optically active substances. Chem Ber. 1931;64:2667–2671. [Google Scholar]
- 32.Kirschner S. The pfeiffer effect, outer-sphere complexation, and the absolute configuration of coordination compounds. J Indian Chem Soc. 1974;LI:28–31. [Google Scholar]
- 33.Muller G, Riehl JP. Use of induced circularly polarized luminescence (CPL) from racemic D3 lanthanide complexes to determine the absolute configuration of amino acids. J Fluorescence. 2005;15:553–558. doi: 10.1007/s10895-005-2828-4. [DOI] [PubMed] [Google Scholar]
- 34.Riehl JP, Muller G. Circularly polarized luminescence spectroscopy from lanthanide systems. In: Gschneidner KA Jr, Bünzli JCG, Pecharsky VK, editors. Handbook on the physics and chemistry of rare earths. Vol. 34. North-Holland Publishing Company; Amsterdam: 2005. pp. 289–357. Chapter 220, and references therein. [Google Scholar]
- 35.Yan F, Copeland RA, Brittain HG. Optical activity induced in the terbium(III) and europium(III) tris complexes of pyridine-2,6-dicarboxylate through association with monoamino- and diaminocarboxylic acids. Inorg Chem. 1982;21:1180–1185. [Google Scholar]
- 36.Wu S, Hilmes GL, Riehl JP. Nature of chiral-induced equilibrium shifts in racemic labile lanthanide complexes. J Phys Chem. 1989;93:2307–2310. [Google Scholar]
- 37.Huskowska E, Riehl JP. Perturbation of the racemic equilibrium between D3 lanthanide complexes through the addition of sugars. Inorg Chem. 1995;34:5615–5621. [Google Scholar]
- 38.Brittain HG. Luminescence studies of the stereoselectivity in the mixed-ligand complexes formed by terbium(III) with enantiomerically resolved 1,2-propanediaminetetraacetic acid and α-hydroxyphenylcarboxylic acids. Chirality. 1997;9:583–592. [Google Scholar]
- 39.Hilmes GL, Riehl JP. Circularly polarized luminescence spectroscopy of mixed-ligand complexes of europium(III) with 2,6-pyridine-dicarboxylic acid and L-malic acid. Inorg Chim Acta. 1987;129:123–125. [Google Scholar]
- 40.Parac-Vogt TN, Binnemans K, Görller-Walrand C. Nature of equilibrium shifts in racemic praseodymium(III) tris(2,2′-oxydiacetate) induced by interaction with chiral probes. J Chem Soc Dalton Trans. 2002:1602–1606. [Google Scholar]
- 41.Parac-Vogt TN, Binnemans K, Görller-Walrand C. Absolute configuration assignment of D3-symmetric lanthanide complexes based on circular dichroism induced by interaction with a chiral probe. Chem Phys Chem. 2001;12:767–769. doi: 10.1002/1439-7641(20011217)2:12<767::AID-CPHC767>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 42.Bonsall SD, Houcheime M, Straus DA, Muller G. Optical isomers of N,N′-bis(1-phenylethyl)-2,6-pyridinedicarboxamide coordinated to europium(III) ions as reliable circularly polarized luminescence calibration standards. Chem Commun. 2007;35:3676–3678. doi: 10.1039/b704346e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brayshaw PA, Bünzli JCG, Froidevaux P, Harrowfield JM, Kim Y, Sobolev AN. Synthetic, structural, and spectroscopic studies on solids containing tris(dipicolinato) rare earth anions and transition or main group metal cations. Inorg Chem. 1995;34:2068–2076. [Google Scholar]
- 44.Grenthe I. Stability relationships among the rare earth dipicolinates. J Am Chem Soc. 1961;83:360–364. [Google Scholar]
- 45.Latva M, Takalo H, Mukkala VM, Matachescu C, Rodriguez-Ubis JC, Kankare J. Correlation between the lowest triplet state energy level of the ligand and lanthanide(III) luminescence quantum yield. J Luminesc. 1997;75:149–169. [Google Scholar]
- 46.Choppin GR, Fugate GA. Applications of the Judd-Ofelt theory in lanthanide-chelidamic acid complexation. Mol Phys. 2003;101:935–939. [Google Scholar]
- 47.Lima PP, Malta OL, Alves Júnior S. Spectroscopic study of the Eu3+, Tb3+ and Gd3+ complexes with ligands derived from dicarboxylic acids. Quim Nova. 2005;28:805–808. [Google Scholar]
- 48.Pike MM, Yarmush DM, Balschi JA, Lenkinski RE, Springer CSJ. Aqueous shift reagents for high-resolution cationic nuclear magnetic resonance. II. 25Mg, 39K, and 23Na resonances shifted by chelidamate complexes of dysprosium(III) and thulium(III) Inorg Chem. 1983;22:2388–2392. [Google Scholar]
- 49.Devlin MT, Stephens EM, Richardson FS. Comparison of electric-dipole intensity parameters for a series of structurally related neodymium, holmium, and erbium complexes in aqueous solution. Theory and experiment. Inorg Chem. 1988;27:1517–1524. [Google Scholar]
- 50.Bag SP, Fernando Q, Freiser H. The influence of metal chelation on the structure of chelidamic acid. Inorg Chem. 1962;1:887–890. [Google Scholar]
- 51.Henchoz Y, Schappler J, Geiser L, Prat J, Carrupt PA, Veuthey JL. Rapid determination of pKa values of 20 amino acids by CZE with UV and capacitively coupled contactless conductivity detections. Anal Bioanal Chem. 2007;389:1869–1878. doi: 10.1007/s00216-007-1568-5. [DOI] [PubMed] [Google Scholar]
- 52.Di Bari L, Salvadori P. Solution structure of chiral lanthanide complexes. Coord Chem Rev. 2005;249:2854–2879. [Google Scholar]
- 53.Rudolph AS, Crowe JH. A calorimetric and infrared spectroscopic study of the stabilizing solute proline. Biophys J. 1986;50:423–430. doi: 10.1016/S0006-3495(86)83478-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Longo A, Guanga GP, Rose RB. Structural basis for induced fit mechanisms in DNA recognition by the Pdx1 homeodomain. Biochemistry. 2007;46:2948–2957. doi: 10.1021/bi060969l. [DOI] [PubMed] [Google Scholar]
- 55.Sandak B, Wolfson HJ, Nussinov R. Flexible docking allowing induced fit in proteins: insights from an open to closed conformational isomers. Proteins. 1998;32:159–174. [PubMed] [Google Scholar]