

Abstract
The ubiquitin proteasome system (UPS) was first described as a mechanism for protein degradation more than three decades ago, but the critical roles of the UPS in regulating neuronal synapses have only recently begun to be revealed. Targeted ubiquitination of synaptic proteins affects multiple facets of the synapse throughout its life cycle; from synaptogenesis and synapse elimination to activity-dependent synaptic plasticity and remodeling. The recent identification of specific UPS molecular pathways that act locally at the synapse illustrates the exquisite specificity of ubiquitination in regulating synaptic protein trafficking and degradation events. Synaptic activity has also been shown to determine the subcellular distribution and composition of the proteasome, providing additional mechanisms for locally regulating synaptic protein degradation. Together these advances reveal that tight control of protein turnover plays a conserved, central role in establishing and modulating synapses in neural circuits.
Keywords: Ubiquitin, Proteasome, Synaptogenesis, Neurotransmission, Synaptic plasticity, Angelman syndrome
1. Introduction
Proteasome-mediated protein degradation has been known for 30 years [1,2], but the complexity, regulative capacity and importance of this system has only been widely appreciated in the past decade. Indeed, a high percentage of the human genome is devoted to encoding ubiquitin proteasome system (UPS) effector proteins, and a rapidly growing list of diseases are linked to mutations in these genes [3,4]. In particular, the UPS is closely implicated in many diseases that involve or are exclusive to the nervous system, including neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease [5], and neurodevelopmental diseases such as autism spectrum disorders and Angelman syndrome [6]. In all of these diseases, synaptic dysfunction is hypothesized to play a prominent role in pathogenesis.
The UPS functions locally at neuronal synapses [7,8], and protein degradation is now recognized alongside protein translation as a primary means for regulating the abundance and trafficking of proteins critical for neurotransmission. The multiple described UPS synaptic roles include feedback control of transcription, coordination of synapse development vs. elimination, local maintenance of neurotransmission strength, and mediating activity-dependent modulation of pre- and postsynaptic function [9–15]. Thus, elucidating UPS regulation at the synapse is now understood to be critical to understanding the physiological changes that determine neurotransmission strength during development and plasticity, as well as the pathological synaptic function changes that occur in a growing range of inherited neurological diseases.
There have been a number of relatively recent reviews of UPS regulation of neuronal and synaptic properties [7,15–18]. Therefore, in this review we focus primarily on the most recent published work that provides novel insights into UPS function at the synapse. In particular, we discuss newly-discovered UPS mechanisms that locally regulate protein degradation to control synaptic functions and new data revealing that proteasome trafficking dictates protein turnover locally at the synapse.
2. The ubiquitin proteasome system
2.1. Enzymatic cascade
Ubiquitin is a 76 amino acid protein conserved across eukaryotic cells. The covalent attachment of ubiquitin to a substrate protein tags that protein for intracellular trafficking or proteasomal degradation [19,20]. Tagging is a highly regulated process that can be controlled at multiple points. The first step is activation of ubiquitin by an activating enzyme (E1) that utilizes an ATP-requiring reaction to generate a high-energy thioester intermediate, E1-S~ubiquitin. The thioester attachment induces a conformational change in the E1 that promotes association with an ubiquitin carrier protein (E2). Next, activated ubiquitin is transferred to the E2 via formation of an additional high-energy thiol intermediate, E2-S~ubiquitin, leading to dissociation from the E1 [21]. In the third step, a substrate-specific ubiquitin E3 ligase interacts with the target protein-E2 ubiquitin complex to transfer ubiquitin to the target protein. In this process an isopeptide linkage is formed between the C-terminal carboxyl group of ubiquitin and the ε-amino group on a lysine of the target protein. Additional ubiquitin proteins may be attached to the initial ubiquitin via a lysine 48 (Lys48) linkage, or less commonly via one of the other six possible ubiquitin lysine linkages (Lys6, 11, 27, 29, 33, or 63), forming polyubiquitin chains that may be linear or branched [22,23]. In addition, ubiquitin attachment to additional substrate protein lysine residues may produce multi-ubiquitination.
Mono and multiple mono-ubiquitin attachments may serve as signals for endocytosis or endosomal trafficking of membrane proteins [19]. Polyubiquitin chains of at least four lysine 48 ubiquitins are generally thought to be necessary to target a protein to the proteasome for degradation [24,25]. However, in some cases, Lys29 and Lys63 linked polyubiquitin chains have also been shown to be proteasome degradation signals [22,26]. In contrast, forked chains with multiple linkages do not appear to signal proteasomal degradation [22]. Likewise, in other circumstances, Lys63 ubiquitin linkages allow for specific protein–protein interactions important in signal transduction [27], and in DNA repair [28]. In general, the effects of alternate lysine ubiquitin linkages are still poorly understood. The ubiquitination process is reversible through the action of deubiquitinating enzymes, cysteine proteases that remove the ubiquitin chains from substrate proteins to regenerate free ubiquitin [29].
2.2. E3 ubiquitin ligases
In mammals, there are only two E1 ligases [30], but dozens of E2 ligases, and hundreds of E3 ligases. Ubiquitination specificity is determined principally by this large variety of E3 ligases, which generate the vast number of E2/E3 combinations that each target specific groups of protein substrates. The E2/E3 combination has also recently been shown to determine the ubiquitin chain linkages that are possible [22], dictating the location and types of ubiquitin attachment. There are two primary domain classes of E3 ligases: HECT (homologous to E6-AP carboxy terminus) domain and RING (really interesting new gene)-finger domain. There are approximately 50 different HECT E3s in humans, of which the proto-typical member is E6AP (papillomavirus E6-associated protein), that has since been designated UBE3A. HECT-domain E3s participate directly in the catalytic transfer of ubiquitin to the substrate protein to form an E3-ubiquitin thioester complex at an active-site cysteine residue within the HECT domain [30]. RING-finger E3s number several hundred in humans, and subgroups include those that function as single proteins, such as parkin, and those that function in multi-subunit proteins. One example of a multi-subunit RING-finger E3 is the Skp1/Cullin/F-box (SCF), consisting of a core complex composed of the Skp1 linker, the Cdc43/Cul1 scaffold, the Rbx1/Roc1/Hrt1 RING-finger E3, and one of a suite of F-box proteins that serve as substrate recognition adaptors [32,33]. Another example is the anaphase promoting complex (APC), composed of at least twelve subunits, with the APC11 RING-finger E3, the APC2 Cul1-related scaffold, and either the Cdh1 or the Cdc20 activator protein, together forming the catalytic core of the complex [34,35]. There are also two related RING-finger like domains, the U-box and PHD/LAP (plant homeodomain and leukemia-associated protein) that confer E3 ligase activity [36]. Unlike HECT domain ligases, RING-finger E3s do not form ubiquitin thioester complexes, but instead serve primarily as scaffolds, orienting the E2-ubiquitin complex and the target protein for ubiquitin transfer. A more detailed characterization of E3 ligase structure and function can be found in several recent reviews [31–33,37].
2.3. Proteasome complex
Once committed to the degradation pathway, polyubiquitinated proteins are actively targeted to the proteasome, a proteolytic complex consisting of a 20S core catalytic component with two 19S regulatory subunits attached at either end [20]. The 20S proteasome is a 700 KDa complex composed of two each of α1–7 and β1–7 subunits. These 28 total subunits are arranged in four axially stacked heptameric rings with the successive layers consisting of α1–7, β1–7, β1–7, α1–7 [38]. The 19S regulatory complex, also known as PA700, contains 20 subunits, including six distinct AAA-family ATPases (Rpt1–Rpt6) and fourteen non-ATPase subunits (Rpn1–Rpn14), one of which (Rpn10/S5a) contains a ubiquitin interaction motif (UIM) that binds polyubiquitin [39]. Positioned on either end of the catalytic core, the 19S complexes serve as gatekeepers for substrate entry into the proteasome [38].
The 20S proteasome exists in at least two forms, with substitution of inducible subunits: β1i (LMP2), β2i (MECL-1), or β5i (LMP7) for the constitutive β1, β2, and β5 subunits under certain conditions such as after γ-interferon induction [40,41]. There are also multiple different 11S regulatory complexes that can replace the 19S regulator, designated P28α, P28β, and P28γ [42]. These alternate regulators do not have ATPase function and do not bind polyubiquitin chains. Proteasomes with inducible beta subunits or 11S substitutions for 19S regulators have higher levels of proteolytic activity [43,44].
3. UPS regulation of synaptogenesis
3.1. Axon growth and synapse formation
At the tip of outgrowing axons, motile growth cones sense guidance cues and translate this information into dynamic cytoskeletal reorganizations that orient growth in a specific direction [45]. UPS activity is stimulated by some of these guidance cues and, along with local protein synthesis, protein degradation is believed to play a dynamic role in the structural rearrangements that mediate axon outgrowth [46]. The anaphase promoting complex (APC), one of the first ubiquitin ligases to be identified in post-mitotic neurons [47], has been strongly implicated in coordinating axon growth programs. RNAi knockdown of the APC activator subunit, Cdh1, increased axon length and the rate of axon growth in cultured rat cerebellar granule neurons. Importantly, in cerebellar slice assays, Cdh1 knockdown also revealed axon tract patterning abnormalities, with defasciculation of fiber tracts and axons wandering across inappropriate layers, including across white matter tracts where myelin traditionally prevents axon growth [12,48]. In non-neuronal cells during cell division, the APC acts predominantly in the nucleus to regulate cell cycle transitions by coordinating the UPS-mediated degradation of transcription factors, such as the transcriptional co-repressor SnoN. Similarly, in post-mitotic neurons, the APC was found to predominantly localize to the nucleus where Cdh1-APC ubiquitinates and targets SnoN for degradation [12,48]. Preventing the APC interaction with SnoN completely eliminated the restraint on axon growth, but did not affect axon patterning in the cerebellum in vivo [48], suggesting the presence of additional APC targets.
Subsequently, Cdh1-APC was shown to ubiquitinate and downregulate another transcriptional repressor, Id2 (inhibitor of DNA binding 2) in cultured rat cerebellar neurons. This regulation followed a distinct developmental pattern with an increased rate of Id2 degradation during neuronal maturation. In a search for Id2 substrates, gene expression profiling identified the Nogo receptor, an inhibitor of axonal growth on which myelin inhibitory signals act. Intriguingly, expression of an Id2 mutant resistant to Cdh1-APC mediated degradation, allowed cerebellar neurons to extend axons in the presence of a Nogo receptor ligand, myelin-associated glycoprotein [49], supporting a prominent role for Id2 abundance in determining myelin inhibition of axonal growth. Moreover, the increased rate of Id2 degradation during development of neuronal connections suggests that Cdh1-APC may act as a “neuronal state switch” similar to its role in cell cycle transitions, coordinating the timing of transcriptional programs that regulate axon growth and patterning. Proteins involved in axon growth that are regulated downstream of Id2 include Semaphorin-3a, Jagged-2, Unc5a, and Notch1 [49]. Future work should address the upstream activators of Cdh1-APC in neurons to provide additional insight into the processes that coordinate the developmental timing of transcriptional regulation, and assess whether it has a similar role in the many other CNS regions where APC is expressed.
The critical role for the UPS in regulating terminal axon pathfinding and differentiation has been highlighted by exciting results from genetic screens in C. elegans and Drosophila. In C. elegans, screens for regulators of synaptic growth/differentiation of GABAergic and glutamatergic synapses identified the presynaptic E3 ligase, RPM-1 [50,51]. RPM-1 was found to act as a positive regulator of growth, as RPM-1 mutants have a reduced number of synapses. Another genetic screen identified FSN-1 as an RPM-1-interacting protein. FSN-1 contains an F-box domain and functions in a unique SCF ubiquitin ligase complex in which RPM-1 substitutes for the traditional Rbx1 E3 ligase [52]. Further evaluation showed RPM-1 to be a negative regulator of the p38 MAPK (mitogen-activated protein kinase) signaling pathway, mediating the ubiquitination and degradation of the MAPKKK (MAPK kinase kinase) DLK [53] (Fig. 1). In addition, RPM-1 was recently shown to function independent of its ubiquitin ligase activity in a pathway that signals axon termination [54].
Fig. 1.

UPS presynaptic pathways. UPS pathways regulate presynaptic differentiation and neurotransmission strength. In Drosophila and C. elegans, the large E3 ligase Highwire/Rpm-1 operates as part of a unique SCF complex that includes the F-box protein FSN-1/DsFn, which targets the MAPKKK Wallenda/DLK and regulates synaptic growth via JNK MAPK or p38 MAPK signaling, respectively. In C. elegans, Rpm-1 regulates axon termination through a separate incompletely characterized pathway. In Drosophila, Highwire also regulates presynaptic neurotransmitter release, as does targeted ubiquitination of dUNC-13, a synaptic vesicle priming protein. The APC E3 ligase complex operates presynaptically to regulate synaptic growth by targeting Liprin-α.
Independently, a Drosophila screen for neuromuscular junction (NMJ) synaptic overgrowth identified the RPM-1 homologue, Highwire. In contrast to the effects of RPM-1, highwire mutants showed a dramatic increase in the number of NMJ synaptic boutons and synaptic branches, and an increase in total synaptic area [55–57]. The signaling pathway driving this synaptic morphology regulation is complex (Fig. 1). Highwire negatively regulates the transforming growth factor β/bone morphogenetic protein (TGF-β/BMP) signaling cascade, by binding to and promoting the degradation of the SMAD transcriptional co-regulator protein, Medea, which acts as a downstream effector in the pathway [58]. Mutations that impair BMP signaling partially suppress the highwire synaptic overgrowth phenotype, whereas excess activation of the BMP pathway leads to synaptic overgrowth. The mechanisms through which BMP signaling tunes synaptic growth remain to be elucidated. More recently, Collins et al. used a forward genetic screen to show Highwire also targets Wallenda, a MAPKKK homologue to aforementioned C. elegans DLK [58], suggesting that UPS presynaptic regulation of a MAPK signaling pathway for regulating growth is highly conserved. Downstream of Wallenda, JNK MAPK and Fos transcription factor activity were necessary for expression of the synaptic overgrowth phenotype [59] (Fig. 1). Similar to RPM-1, Highwire has now been shown to participate in a SCF ubiquitin ligase complex with the F-box protein DSfn [60]. Thus, the RPM-1/Highwire E3 ligase plays an essential role in a conserved presynaptic signaling pathway targeting MAPKKK degradation and the encompassing MAPK pathways. Critical components of both the MAPK and TGF-β/BMP signaling cascades are present in the presynaptic terminal, but much work remains to determine whether the principle effects of RPM-1/Highwire mediated degradation of MAPKKK and Smad proteins are local and synapse-specific, or result from more global nuclear regulation of transcriptional programs that affect synaptic growth and patterning.
The work in C. elegans and Drosophila has led to the discovery that vertebrate orthologues of RPM-1/Highwire also affect axonal differentiation. In zebrafish, mutations in the RPM-1/Highwire orthologue, Esrom, were shown to disrupt fasciculation, targeting, and mapping retinal axons. This showed that this E3 ligase has additional roles in the CNS involved with axon patterning [61]. In mice, knockout of Phr1, a mammalian orthologue to Highwire/RPM-1/Esrom, led to impaired phrenic nerve development and severely disrupted phrenic nerve terminal morphology [62]. In the CNS, Phr1 knockout mice display striking defects in major axon tracts that include the internal capsule and anterior commissure. Mice with knockout of both Phr1 and the Wallenda/DLK MAPKKK homologue maintain the axon tract deficits, showing that this effect on axon patterning must be mediated through a separate signaling pathway than that regulating synaptic development in C. elegans and Drosophila [63]. These studies illustrate that the function of highwire/RPM-1/Esrom/Ph1 family of E3 ligases is highly conserved in presynaptic neurons, critically modulating regulation of axon growth and synapse differentiation. Despite this conservation, vertebrate studies demonstrate that these proteins have likely acquired additional functions in regulating axon tract patterning that utilize alternate signaling pathways. Further work on this exciting family of SCF E3 ligases is needed, particularly in mammalian systems, to more clearly define their mechanisms for determining axonal growth, differentiation, and patterning. It will be interesting to see if their effects overlap with those of Cdh-1 APC at the level of coordinating transcriptional programs.
Other neuronally expressed E3 ligases may be involved in overlapping mechanisms in growth cones and presynaptic terminals. APC was identified at the Drosophila NMJ through immunolabeling and found to negatively regulate synaptic size by initiating degradation of the scaffold protein Liprin-α [64]. It has not been determined whether APC is expressed in mammalian synapses. However, another RING-domain E3 ligase, Rnf6, was identified in cultured rat hippocampal neurons, where it is highly expressed in axonal projections and co-localizes with LIM kinase 1 (LIMK1) in the growth cone [65]. Rnf6 polyubiquitinates LIMK1, targeting it for proteasome-mediated degradation. Overexpression or knockdown of Rnf6 reduces or stimulates axon outgrowth, respectively [65].
The HECT domain E3 ligase, Ube3a, has also recently been shown to be present in the growth cone [66]. A maternally inherited deficiency for UBE3A causes Angelman syndrome (AS), a neurodevelopment disease characterized by mental retardation, impaired expressive language development, frequent jerky limb movements and difficult to control seizures [67]. At the Drosophila NMJ, overexpression of the Drosophila homologue (Dube3a) causes an increase in the number of synaptic boutons when expressed presynaptically, and a decrease in bouton number when expressed postsynaptically (Lawrence Reiter, personal communication). These changes may be mediated through the Rho-GEF (Rho-guanine exchange factor) Pebble, and its regulation of the actin-binding GTPase Rac2, which function in a pathway that regulates dynamic reorganization of the actin cytoskeleton. Mutations in pebble have been shown to disrupt neuronal outgrowth at the NMJ [68], and the Pebble protein has been identified as a putative Dube3a target in a Drosophila proteomic screen. The involvement of the Pebble orthologue, ECT2, in the pathogenesis of AS was validated in Ube3a deficient mice where ECT2 is mislocalized in hippocampus and cerebellum [69]. While this is an intriguing starting point for future work, many questions about the function of Ube3a at the synapse remain, as no definitive synaptic substrate proteins for Ube3a have yet been identified. In particular, it will be interesting to see a detailed analysis of synaptic changes in both Drosophila and mouse AS disease models.
The process of synaptogenesis involves both structural and functional specialization of presynaptic and postsynaptic specializations. The sequence of synaptogenic events has been best characterized at the NMJ synapse, in both invertebrates and vertebrates [45,70]. At the Drosophila NMJ, Commissureless (Comm) is a postsynaptic membrane protein that must undergo endocytosis for normal synaptogenesis to occur [71]. Recently, Ing et al. reported that the UPS regulates Comm endocytosis through the action of the HECT-domain E3 ligase Nedd4 [11]. Nedd4 ubiquitinates Comm to trigger its endocytosis. Multiple conditions that prevent Comm ubiquitination or the Nedd4-Comm interaction lead to accumulation of Comm on the surface membrane and consequent aberrant innervation [11]. Use of a monobubiquitin-Comm construct that did not allow longer ubiquitin chains to form was sufficient to promote Comm endocytosis during synaptogenesis [11], suggesting that polyubiquitination is not required.
At the mammalian NMJ, the postsynaptic receptor tyrosine kinase MuSK (muscle specific kinase) and the presynaptically secreted proteoglycan Agrin serve as critical mediators of the signaling cascade that regulates juxtaposed clustering of pre-synaptic and postsynaptic elements [45]. Lu et al. identified a RING-domain E3 ligase, PDZRN3 (PDZ domain containing RING finger 3), that is concentrated postsynaptically at the NMJ, interacts with MuSK and promotes MuSK ubiquitination [13]. Using siRNA knockdown or transgenic overexpression of PDZRN3 in cultured myotubes, Lu et al. showed that PDZRN3 regulates MuSK surface expression, and provided multiple lines of evidence that this occurs by MuSK ubiquitination-dependent endocytosis. The process was largely blocked by lysosomal inhibitors, but proteasome inhibitors had no effect [13], suggesting that ubiquitin is involved in protein targeting, not turn-over.
The similar UPS-mediated endocytosis of Comm and MuSK demonstrate how specific synaptic E3 ligases locally regulate the surface expression of critical effectors of synaptogenesis. As the timing of events is paramount during synapse development, the temporal expression of these E3 ligases must presumably be tightly regulated. Future work will need to address how specific UPS pathways are turned on and off to mediate their precisely ordered effects.
3.2. Pruning and synapse elimination
The remodeling and elimination of synaptic connections is an ongoing process that occurs during normal development and neural circuit refinement, and also aberrantly in progressive neurodegenerative disorders. UPS mechanisms are involved in preventing the dying back of an axon distal to the site of injury, in a process known as Wallerian degeneration [72,73]. Similarly, the UPS plays a critical role in initial stages of normal axon pruning in the Drosophila Mushroom Body, a primary brain learning and memory center; in a process that closely resembles Wallerian degeneration [74]. Recently, a Drosophila screen for loss of dendritic pruning in sensory neurons identified ubcD1 as an E2 conjugating enzyme involved in the degradation of DIAP1, a caspase-antagonizing E3 ligase. This UPS pathway regulates local caspase activation leading to severing of dendrites, with preservation of the sensory neurons [75,76]. Thus, UPS pathways play a prominent role in both axonal and dendritic pruning during Drosophila development. Hopefully, these exciting findings will stimulate further investigation of conserved pathways in mammalian systems.
Synapse elimination also involves local control of targeted protein degradation. Recently, Ding et al. elegantly showed that local control of synapse elimination in C. elegans occurs via a UPS-mediated mechanism [9]. An egg laying motor neuron (HSNL) connects to its target vulval muscles via a cluster of finely mapped synapses in the Primary Synapse Region (PSR). At early stages of development, synapses also form in an adjacent vulvar region, termed the Secondary Synapse Region (SSR), but these synapses are subsequently eliminated to give rise to the adult pattern. Ding et al. have shown that local control of synapse elimination is dependent on the interaction of an immuno-adhesion protein, Syg1, which interferes with the binding of the F-box protein SEL-1 with the SCF complex [9]. If this Syg1-SEL1 interaction is prevented, or if SEL-1 is over-expressed, the SCF complex forms a competent ubiquitin ligase complex that targets synaptic organizing proteins for degradation, leading to synapse elimination. These new data demonstrate that through tight spatial and temporal regulation of E3 ligase activity either synapse stabilization or elimination can be favored. It remains to be tested whether similar pathways are involved in mammalian pruning and synapse elimination.
4. UPS regulation of presynaptic neurotransmission
Following UPS regulation of synapse formation, the UPS has a maintained role in regulating neurotransmission strength. We have identified high levels of proteasome expression in Drosophila NMJ presynaptic boutons, and found that both pharmacological and genetic inhibition of the proteasome causes rapid strengthening of neurotransmission via a presynaptic mechanism [8]. Blocking the proteasome resulted in a significant increase in transmission strength within minutes. UNC-13 proteins are known to regulate the efficacy of presynaptic neurotransmitter release by mediating a critical step of synaptic vesicle priming [77]. Drosophila UNC-13 is ubiquitinated and accumulates in the presynaptic terminal after proteasome inhibition over the same time course as presynaptic strengthening occurs [8,78]. Thus, UPS-mediated turnover of synaptic proteins occurs on a time scale that is rapid enough to drive acute forms of synaptic plasticity, and UNC-13 is one primary target for this UPS-mediated regulation.
Willeumier et al. have recently shown that acute proteasome inhibition increases the size of the recycling pool of synaptic vesicles in cultured hippocampal neurons [14]. Using optical dye imaging to track synaptic vesicle release and uptake, they found a dramatic increase in the size of the recycling vesicle pool after 2 h of proteasome inhibition. This effect was increased by heightened synaptic activity, and decreased by reduced synaptic activity [14], suggesting an important role for the UPS in activity-mediated regulation of presynaptic transmitter release. A similar increase in the size of the recycling pool was produced by forskolin activation of PKA pathways, but there was no additive effect of PKA and proteasome inhibition, suggesting a shared or intersecting pathway [14]. A cAMP-mediated recruitment of reserve pool vesicles to the recycling pool had been previously demonstrated at the Drosophila NMJ [79]. The proteasome inhibition-induced rapid increase in levels of dUNC-13 is abolished by PKA antagonists [78], suggesting one attractive mechanism for this convergence.
Of the presynaptic E3 ligases that have been identified, the best characterized is the membrane-localized SCF E3 ligase SCRAPPER. Yao et al. identified SCRAPPER and demonstrated that it ubiquitinates and mediates the proteasomal degradation of RIM-1 (rab3-interacting molecule 1), a presynaptic active zone protein involved in priming of synaptic vesicles for release. In a SCRAPPER knock-out mouse, RIM-1 expression was increased together with a 3-fold increase in miniature EPSC frequency [80], supporting the prediction that SCRAPPER regulates presynaptic transmitter release. The SCRAPPER gene also contains a cyclic-AMP response element, further implicating the presynaptic PKA signaling cascade in UPS regulation of presynaptic release.
Several additional E3 ligases have been identified at the presynaptic terminal. The aforementioned Highwire has been shown to facilitate presynaptic neurotransmission at the Drosophila NMJ, through a yet unidentified pathway that is separate from its effects on synaptic growth regulation [56,59]. Other presynaptic E3 ligases include the ZNRF family of RING-finger ligases [81], the RING-finger E3 ligase Staring, shown to ubiquitinate and regulate t-SNARE Syntaxin-1 levels [82], and the RING-finger domain ligases, Siah-1A and Siah-2A, that bind to the integral synaptic vesicle protein Synaptophysin and promote its ubiquitination and degradation [83]. The identification of these presynaptic E3 ligases and several target proteins involved in synaptic vesicle trafficking and release mechanisms provide a starting point, but more work needs to be focused on how UPS regulation of these proteins affects presynaptic neurotransmission.
Many of these recent studies highlight a potential overlap of UPS-mediated degradation pathways and PKA signaling pathways in presynaptic modulation. This connection is not unexpected given that phosphorylation can serve as a trigger for subsequent ubiquitination [84,85]. Conversely, there is a well-characterized role for UPS-mediated protein degradation of the PKA regulatory subunit that enables PKA activation during Aplysia long-term facilitation [86]. The bidirectional interaction between protein phosphorylation and ubiquitination pathways provides an attractive mechanism for temporally linking changes in synaptic activity to protein degradation.
5. UPS regulation of postsynaptic neurotransmission
A fundamental mechanism for adjusting postsynaptic strength is regulation of the number of neurotransmitter-gated ion channel receptors. The UPS regulates postsynaptic receptor numbers through multiple mechanisms, including controlling forward receptor trafficking, the number of postsynaptic density (PSD) insertion slots, endocytosis, and trafficking through recycling and lysosomal pathways (Fig. 2). In C. elegans, direct ubiquitination and proteasomal degradation of glutamate receptor GluR-1 was demonstrated in a process requiring clathrin-mediated endocytosis [87]. A subsequent study identified Kel-8, a SCF E3 ligase substrate adaptor protein, as being instrumental in this process [88], although direct association with GluR1 was not demonstrated. The UPS also indirectly regulates postsynaptic GluR1 abundance through pathways involving APC [89] and LIN-23 F-box SCF [90] E3 ligases. In cultured rat hippocampal neurons, UPS function was similarly shown to regulate AMPA-type glutamate receptor internalization, as both AMPA-induced and NMDA-induced endocytosis of AMPA receptors was blocked by brief pretreatments with proteasome inhibitors and was dependent on polyubiquitination [91]. The ubiquitin-like modifier, SUMO, has been shown to target postsynaptic GluR6-containing kainate receptors [92]. While structurally similar to ubiquitin, SUMO does not form chains. Treatment of rat hippocampal neurons with kainate increased GluR6 SUMOylation and deSUMOylation prevented kainate-mediated GluR6 endocytosis. In contrast, SUMOylation did not affect NMDA-induced GluR6 endocytosis [92], suggesting that it is not involved in all endocytic mechanisms. These results imply that ubiquitination and SUMOylation-induced regulation of endocytosis are likely to operate through separate mechanisms. Other potential synaptic ubiquitination targets include long splice variants of type I metabotropic glutamate receptors (mGluR1 and mGluR5) that bind to the E3 ligase Siah (Seven in absentia homologue). When expressed in a heterologous system, mGluR1 and mGluR5 were directly polyubiquitinated and rapidly degraded [93]. If these findings can be extended to neurons, regulation of downstream signaling from type 1 mGluRs provides another potential indirect UPS mechanism for regulating ionotropic glutamate receptor abundance.
Fig. 2.

UPS postsynaptic mechanisms regulating neurotransmitter receptors. There are multiple modes of UPS regulation of receptors in the postsynaptic density. 1. ERAD: Receptor subunits are targeted for degradation by the proteasome after ubiquitination and translocation from the ER. 2. Forward trafficking: Ubiquitination regulates the rate and efficiency of forward trafficking from the ER to the plasma membrane. 3. Scaffold control: Polyubiquitination and targeted degradation of postsynaptic density scaffolding proteins determines the number of receptor slots. 4. Membrane recycling: Mono- or polyubiquitination of receptors and/or endocytic machinery triggers endocytosis. 5. Vesicle trafficking: Monoubiquitination serves as a trafficking signal, directing receptors to the late endosome for lysosomal degradation. 6. Retrotranslocation: Reverse trafficking of surface membrane receptors back to the ER can lead to extracellular domain polyubiquitination and translocation from the membrane for proteasome degradation.
There are a growing number of examples showing UPS control of the assembly and forward trafficking of postsynaptic receptors. For nicotinic acetylcholine receptors, surface expression was shown to be regulated by UPS-mediated endoplasmic reticulum associated degradation (ERAD) of unassembled receptor subunits [94]. Moreover, binding of the ubiquitin-like protein, PLIC-1/ubiquilin-1, limited the availability of unassembled nicotinic acetylcholine receptor subunits by targeting them to the proteasome [95]. PLIC-1/ubiquilin-1 has also been shown to bind to an intracellular domain of GABAA receptor α and β subunits [96], where it seems to have the opposing effect of preventing receptor degradation. In hippocampal neurons, activity-dependent ubiquitination of β3 subunit-containing GABAA receptors negatively regulates forward trafficking through the ER [97]. In addition, recent work has shown that GABAA α1 subunits are ubiquitinated. An autosomal dominant form of juvenile myoclonic epilepsy is caused by a missense α1 A322D mutation, and this mutant α1 subunit protein was shown to be misfolded in the ER leading to rapid UPS-mediated ERAD and reduced surface GABAA receptor expression [98]. This mechanism can also be reversed: NMDA receptors have been shown to be retrotranslocated from the plasma membrane to the ER [99], where the NR1 subunit is ubiquitinated and targeted for degradation (Fig. 2).
Ubiquitination also targets postsynaptic scaffold proteins for degradation, suggesting a coordinate mechanism for regulating linked proteins in the PSD. In a rat hippocampal synaptosomal preparation, Ehlers demonstrated that proteasome inhibition modulated activity-dependent changes in the PSD over a period of 48 h, and that the multivalent scaffold proteins Shank, GKAP and AKAP79/150 were ubiquitination targets. These UPS-mediated changes were accompanied by changes in the signaling properties of the PSD as measured by assays of the phosphorylation states of CREB and ERK-MAPK [100]. NMDA-mediated internalization of GluR2-containing AMPA receptors is UPS-mediated through a pathway involving direct ubiquitination of the PSD-95 scaffold protein by the E3 ligase MDM2 [101], although an independent study was not able to demonstrate direct PSD-95 ubiquitination [102]. In cultured hippocampal neurons, SPAR (Spine-associated guanosine triphosphatase activating protein), an actin regulatory protein, becomes an ubiquitination target after phosphorylation. Over 24 h, UPS-mediated SPAR degradation was accompanied by a decrease in the abundance of PSD-95 and a loss of dendritic spines [85]. Recently, CaMKII (calcium/calmodulin-dependent protein kinase II) activation was shown to promote degradation of the synaptic scaffold protein Liprin-α1, through both proteasome dependent and independent mechanisms [103]. CaMKII function itself is regulated by the HECT E3 ligase Ube3a. In the chronic absence of Ube3a, CaMKII inhibitory autophosphorylation is increased, resulting in reduced CaMKII activity and reduced PSD localization [104]. Strikingly, learning and behavioral deficits in the AS mouse model were rescued by introducing a mutant CaMKII that cannot undergo inhibitory autophosphorylation [105]. These studies show that over the longer time scale of 24–48 h, UPS-regulated turnover of postsynaptic scaffold proteins is a key element for the morphological and functional remodeling of the PSD.
Recently, we showed that the UPS differentially regulates two ionotropic glutamate receptor classes present in the Drosophila NMJ postsynaptic density. These two spatially and functionally distinct GluR classes are composed of common GluRIII (IIC), IID and IIE subunits, and variant GluRIIA (A-class) or GluRIIB (B-class) subunits [106–108]. Genetic postsynaptically-targeted proteasome inhibition caused a rapid increase in B-class receptors over the course of a few hours, with little effect on A-class receptor levels (Fig. 3) [109]. The mechanism likely involves ubiquitin-mediated turnover of Discs Large (DLG), a PSD-95 (postsynaptic density-95) scaffold homolog. DLG was shown to be ubiquitinated and accumulated after genetic proteasome inhibition [109] and is a known determinant of B-class receptor localization/maintenance at the Drosophila NMJ [110]. Direct ubiquitination of B-class receptors also remains as a possibility that needs to be explored. These results show that UPS regulation is critically important in determining not only the number of postsynaptic receptors, but also the subclass specification of these receptors, thereby providing for fine-tuning of postsynaptic responsiveness.
Fig. 3.
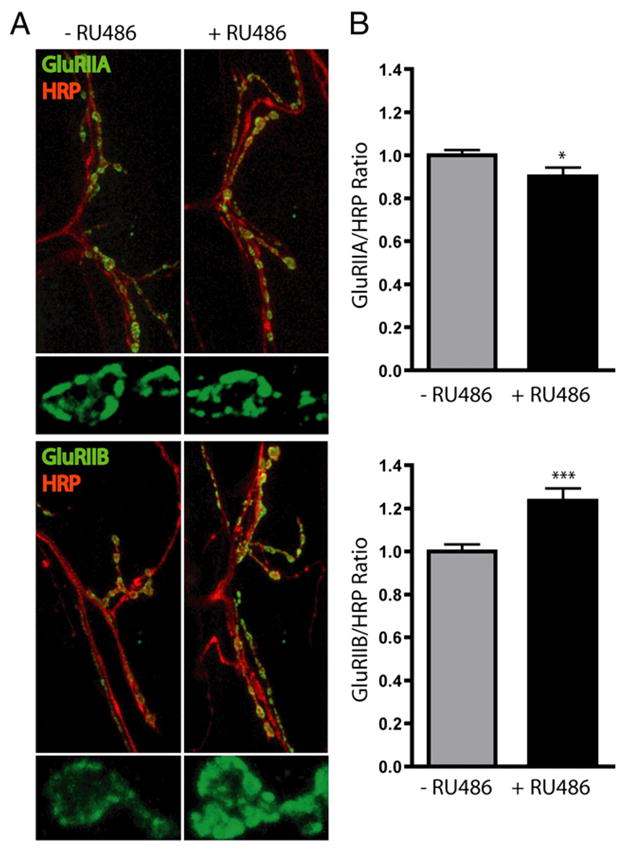
Genetically targeted postsynaptic proteasome inhibition acutely regulates class-specific glutamate receptor expression. Drosophila genetic targeting of postsynaptic proteasome inhibition was induced by feeding third instar larvae RU486-containing food for a brief interval (6 h). Confocal imaging of a NMJ synapse was performed in control (−RU486) and proteasome-blocked (+RU486) animals. A, Representative images from synapses co-labeled for A-class (GluRIIA) or B-class (GluRIIB) receptors and synaptic membrane marker (HRP). Smaller images show higher magnification of individual synaptic boutons labeled for GluRIIA or GluRIIB. B, GluR expression intensity was quantified for GluRIIA and GluRIIB relative to HRP. GluRIIA was decreased and GluRIIB was increased by proteasome blockade. Bars represent mean± SEM, *p<0.05, ***p<0.001.
Much remains to be learned about the specific E2/E3 combinations that are active in the postsynaptic compartment, their localization signals and the time course over which they function. In the ER, there are transmembrane E3 ligases that work in concert with AAA-ATPases to translocate membrane proteins into the cytoplasm, where they are targeted through ERAD to the proteasome for degradation [111]. Are similar local mechanisms available for transmembrane protein degradation at the plasma membrane, or is the primary mechanism for their degradation monobubiquitin tagging, that can serve as both an endocytosis and lysosomal sorting signal? It is clear that for each postsynaptic receptor type, UPS-mediated regulation at all points in the receptor life-cycle must be understood. For different receptor types there may well be multiple points of UPS regulation, with individual mechanisms more or less active depending on the postsynaptic environment.
6. UPS role in activity-dependent synaptic modulation
6.1. UPS regulation of LTP and LTD
Recent work suggests that the UPS modulates rapidly-induced types of plasticity, including both long-term potentiation (LTP) and depression (LTD). Earlier study had shown that proteasome inhibition markedly reduces hippocampal LTD mediated by degradation of the postsynaptic PSD-95 scaffold protein, regulating the number of glutamate receptor slots [101]. More recently, Fonseca et al. examined the effects of pharmacological proteasome inhibition on LTP, showing no effect on early LTP but a strong impairment of late-phase LTP (L-LTP) [10]. When proteasome inhibition was combined with protein synthesis inhibition, the effect was reversed and L-LTP restored. The authors suggest a model in which an activity-induced increase in proteasome activity leads to the degradation of proteins that are negative regulators of L-LTP. When combined with activity-dependent protein synthesis of positive L-LTP regulators, L-LTP is favored. In the setting of both protein synthesis and proteasome inhibition, the existing balance of positive and negative regulators must favor L-LTP, but if either proteasome activity or protein synthesis is absent, the balance shifts to favor negative regulators, and L-LTP does not occur [10]. Thus, in this model, protein synthesis and degradation mechanisms work in harmony to mediate activity-dependent changes in synaptic efficacy.
6.2. UPS trafficking in synapse regulation
Recent work suggests that gross movement of proteasomes may be an important mechanism for the activity-dependent regulation of protein composition within individual synapses. Exciting new results have shown that proteasomes are transported in and out of synapses in response to synaptic activity [112]. Bingol et al. used a GFP-labeled proteasome subunit in conjunction with restricted photobleaching of individual dendritic spines and shafts to follow proteasome movements. Increasing synaptic activity led to a 1.5-fold increase in the GFP proteasome signal within dendritic spines. Furthermore, with activity, the immobile fraction of spine proteasomes increased significantly, reflected by a 6-fold decrease in their exit rate from the synapse [112]. This observation suggests that proteasomes are anchored or tethered in the postsynaptic compartment in an activity-dependent manner, likely due to proteasome association with the actin cytoskeleton. As polyribosomes are also transported into dendritic spines during heightened synaptic activity [113], this new study reinforces the idea that local protein synthesis and degradation mechanisms are likely working in concert to fine-tune synaptic plasticity.
Shen et al. have recently demonstrated that proteasome inhibition or bicuculline disinhibition in cultured hippocampal neurons caused translocation of the proteasome from nucleus to cytoplasm [114]. We have discovered a similar shift of UPS components from nucleus to cytoplasm upon transgenic proteasome inhibition in Drosophila, where ubiquitinated proteins no longer accumulated in the nucleus after acute proteasome inhibition (Fig. 1) [115]. Shen et al. show that a 19S ATPase regulatory proteasome subunit, Mov34, complexes with a subunit of the SCF E3 ligase complex, Cul3, and an immediate early gene, NAC1, that is upregulated after psychostimulant-induced behavioral plasticity. Heightened synaptic activity promotes rapid and persistent NAC1-dependent translocation of the proteasome and Cul3 to the region of postsynaptic dendritic spines [114]. The activity-dependent trafficking of proteasomes and associated UPS machinery provides an exciting mechanism for tuning local protein degradation by coupling synaptic activity levels with proteasome availability. Identifying the upstream signaling pathways that mediate this change will be an important area for future research.
The composition of the proteasome may also change in an activity-dependent manner. Zif268 is an immediate early gene that is upregulated by heightened synaptic activity, and has been shown to be critical for LTP. A recent study by James et al. showed that the potential transcriptional targets of Zif268 include a large number of UPS related genes [116]. Of the genes identified, 4 (psmb9, psme2, SGK, and TAP1) were evaluated further using RT-PCR and their transcription was confirmed to be downregulated by Zif268. The psmb9 gene encodes the inducible proteasome beta subunit, β2i, and psme2 encodes for an inducible regulatory 11S proteasome complex that can replace the 19S regulator complex. Thus, the predicted change in proteasome composition with Zif268 activation would be to decrease proteolytic activity and also potentially increase the specificity of the proteasome for binding ubiquitinated substrates. In a follow-up study, Zif268 activation was also shown to decrease the abundance of the APC cdc20 subunit, reducing APC activity [117]. Taken together, these recent studies suggest that synaptic activity regulates proteasome localization and composition, providing mechanisms for very specific temporal and spatial regulation of the turnover of synaptic proteins.
7. Summary and conclusions
Recent work shows that the UPS plays a critical role in both acute and chronic changes in protein abundance throughout the life-cycle of the synapse; ranging from early axonal pathfinding and target recognition, through synaptogenesis and synapse elimination, and continuing to regulate activity-dependent plasticity in both presynaptic and postsynaptic compartments. The recent identification of many E3 ligases that are active at the synapse should open new frontiers into understanding specific UPS pathways. The identification of neuronal APC and SCF E3 ligase complexes is striking, as these ligases were originally characterized in regulation of cell cycle transitions in development. This has led to speculation that neurons undergoing active axonal growth may be in a relatively undifferentiated state compared with neurons whose axons have already reached their targets [49]. As neurogenesis has been shown to persist in some regions in the adult brain, and as neurons frequently undergo sprouting following injury, it will be important to determine whether APC and SCF ligase mediate “neuronal state switches” in these settings. Moreover, localization of many E3 ligases to both nucleus and synapse suggests that there may be interplay between UPS regulation of transcriptional programs that function in synaptic modulation and local synaptic regulation of protein degradation. Further underscoring this potential association is the finding that neuronal activity mediates transport of UPS elements between nucleus and synapse.
The many recent discoveries of UPS regulation of synaptic function have revealed exquisite fine-tuning of protein levels and localization at the synapse. Many major advances have come from elegant studies utilizing the powerful genetic tools in C. elegans and Drosophila coupled to increasingly sophisticate synaptic assays in these systems. Remarkable progress has also been made in unraveling UPS regulation in mammalian synapses, primarily through utilizing neuronal culture systems. However, it is clear that we are just now beginning to appreciate the wide-ranging mechanisms by which the UPS controls synapse assembly, maintenance, and function. Given the vast number of E3 ligases, there are likely a great many disparate pathways to be discovered that tune synaptic function. There is also a great need to understand the neuronal and synaptic targets of these ligases and how their effects interrelate at synapses in vivo. These advances are of paramount importance to elucidating the mechanisms of and developing treatments for the many neurodevelopmental and neurodegenerative diseases that are characterized by UPS dysfunction.
Acknowledgments
This work was supported by NIH (National Institute of Health) grants NS048882 to K.F.H. and NS41740 to K.B.
References
- 1.Etlinger JD, Goldberg AL. A soluble ATP-Dependent proteolytic system responsible for the degradation of abnormal proteins in reticulocytes. PNAS. 1977;74:54–58. doi: 10.1073/pnas.74.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hershko A, Ciechanover A, Heller H, Haas AL, Rose IA. Proposed role of ATP in protein breakdown — conjugation of proteins with multiple chains of the polypeptide of ATP-Dependent proteolysis. PNAS. 1980;77:1783–1786. doi: 10.1073/pnas.77.4.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang YH, Beaudet AL. Human disorders of ubiquitination and proteasomal degradation. Curr Opin Pediatr. 2004;16:419–426. doi: 10.1097/01.mop.0000133634.79661.cd. [DOI] [PubMed] [Google Scholar]
- 4.Reinstein E, Clechanover A. Protein degradation and human diseases: the ubiquitin connection. Ann Intern Med. 2006;145:676–684. doi: 10.7326/0003-4819-145-9-200611070-00010. [DOI] [PubMed] [Google Scholar]
- 5.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 6.Jiang Y, Sahoo T, Michaelis RC, Bercovich D, Bressler J, Kashork CD, Liu Q, Shaffer LG, Schroer RJ, Stockton DW, Spielman RS, Stevenson RE, Beaudet AL. A mixed epigenetic/genetic model of autism with a limited role for UBE3A. Amer J Med Genet. 2004;131A:1–10. doi: 10.1002/ajmg.a.30297. [DOI] [PubMed] [Google Scholar]
- 7.Hegde AN, DiAntonio A. Ubiquitin and the synapse. Nat Rev Neurosci. 2002;3(11):854–861. doi: 10.1038/nrn961. [DOI] [PubMed] [Google Scholar]
- 8.Speese SD, Trotta N, Rodesch CK, Aravamudan B, Broadie K. The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr Biol. 2003;13(11):899–910. doi: 10.1016/s0960-9822(03)00338-5. [DOI] [PubMed] [Google Scholar]
- 9.Ding M, Chao D, Wang G, Shen K. Spatial regulation of an E3 ubiquitin ligase directs selective synapse elimination. Science. 2007;317:947–951. doi: 10.1126/science.1145727. [DOI] [PubMed] [Google Scholar]
- 10.Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, Nagerl UV. A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron. 2006;52:239–245. doi: 10.1016/j.neuron.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 11.Ing B, Shteiman-Kotler A, Castelli M, Henry P, Pak Y, Stewart B, Boulianne GL, Rotin D. Regulation of Commissureless by the ubiquitin ligase DNedd4 is required for neuromuscular synaptogenesis in Drosophila melanogaster. Mol Cell Biol. 2007;27:481–496. doi: 10.1128/MCB.00463-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konishi Y, Stegmuller J, Matsuda T, Bonni S, Bonni A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 2004;303:1026–1030. doi: 10.1126/science.1093712. [DOI] [PubMed] [Google Scholar]
- 13.Lu Z, Je HS, Young P, Gross J, Lu B, Feng G. Regulation of synaptic growth and maturation by a synapse-associated E3 ubiquitin ligase at the neuromuscular junction. J Cell Biol. 2007;177:1077–1089. doi: 10.1083/jcb.200610060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willeumier K, Pulst SM, Schweizer FE. Proteasome inhibition triggers activity-dependent increase in the size of the recycling vesicle pool in cultured hippocampal neurons. J Neurosci. 2006;26:11333–11341. doi: 10.1523/JNEUROSCI.1684-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yi JJ, Ehlers MD. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]
- 16.Bingol B, Schuman EM. Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol. 2005;15:536–541. doi: 10.1016/j.conb.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 17.DiAntonio A, Hicke L. Ubiquitin-dependent regulation of the synapse. Ann Rev Neurosci. 2004;27:223–246. doi: 10.1146/annurev.neuro.27.070203.144317. [DOI] [PubMed] [Google Scholar]
- 18.Patrick GN. Synapse formation and plasticity: recent insights from the perspective of the ubiquitin proteasome system. Curr Opin Neurobiol. 2006;16:90–94. doi: 10.1016/j.conb.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 19.Hicke L. Protein regulation by monoubiquitin. Nat Rev, Mol Cell Biol. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- 20.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 21.Huang DT, Hunt HW, Zhuang M, Ohi MD, Holten JM, Schulman BA. Basis for a ubiquitin-like protein thioester switch toggling E1–E2 affinity. Nature. 2007;445:394–398. doi: 10.1038/nature05490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HT, Kim KP, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, Gygi SP, Goldberg AL. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem. 2007;282:17375–17386. doi: 10.1074/jbc.M609659200. [DOI] [PubMed] [Google Scholar]
- 23.Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 24.Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain Is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 25.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson ES, Ma PCM, Ota IM, Varshavsky A. A proteolytic pathway that recognizes ubiquitin as a degradation signal. J Biol Chem. 1995;270:17442–17456. doi: 10.1074/jbc.270.29.17442. [DOI] [PubMed] [Google Scholar]
- 27.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the I[kappa]B kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 28.Spence J, Sadis S, Haas AL, Finley D. A ubiquitin mutant with specific defects in DNA-repair and multiubiquitination. Mol Cell Biol. 1995;15:1265–1273. doi: 10.1128/mcb.15.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischer JA. Deubiquitinating enzymes: their roles in development, differentiation, and disease. Int Rev Cytol. 2003;229:43–72. doi: 10.1016/s0074-7696(03)29002-1. [DOI] [PubMed] [Google Scholar]
- 30.Jin J, Li X, Gygi SP, Harper JW. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature. 2007;447:1135–1138. doi: 10.1038/nature05902. [DOI] [PubMed] [Google Scholar]
- 31.Kee Y, Huibregtse JM. Regulation of catalytic activities of HECT ubiquitin ligases. Biochem Biophys Res Comm. 2007;354:329–333. doi: 10.1016/j.bbrc.2007.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deshaies RJ. SCF and cullin/RING H2-based ubiquitin ligases. Ann Rev Cell Dev Biol. 1999;15:435–467. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- 33.Willems AR, Schwab M, Tyers M. A hitchhiker’s guide to the cullin ubiquitin ligases: SCF and its kin. Biochimica et Biophysica Acta (BBA) Mol Cell Res. 2004;1695:133–170. doi: 10.1016/j.bbamcr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 34.Kim AH, Bonni A. Thinking within the D box: Initial identification of Cdh1-APC substrates in the nervous system. Mol Cell Neurosci. 2007;34:281–287. doi: 10.1016/j.mcn.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 35.Visintin R, Prinz S, Amon A. CDC20 and CDH1: A family of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- 36.Fang S, Lorick KL, Jensen JP, Weissman AM. RING finger ubiquitin protein ligases: implications for tumorigenesis, metastasis and for molecular targets in cancer. Sem Can Biol. 2003;13:5–14. doi: 10.1016/s1044-579x(02)00095-0. [DOI] [PubMed] [Google Scholar]
- 37.Jackson PK, Eldridge AG, Freed E, Furstenthal L, Hsu JY, Kaiser BK, Reimann JDR. The lore of the RINGs: substrate recognition and catalysis by ubiquitin ligases. Trends Cell Biol. 2000;10:429–439. doi: 10.1016/s0962-8924(00)01834-1. [DOI] [PubMed] [Google Scholar]
- 38.DeMartino GN, Gillette TG. Proteasomes: machines for all reasons. Cell. 2007;129:659–662. doi: 10.1016/j.cell.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 39.Pickart CM, Cohen RE. Proteasomes and their kin: proteases in the machine age. Nat Rev, Mol Cell Biol. 2004;5:177–187. doi: 10.1038/nrm1336. [DOI] [PubMed] [Google Scholar]
- 40.Groettrup M, Ruppert T, Kuehn L, Seeger M, Standera S, Koszinowski U, Kloetzel PM. The interferon-[IMAGE]-inducible 11S regulator (PA28) and the LMP2/LMP7 subunits govern the peptide production by the 20S proteasome in vitro. J Biol Chem. 1995;270:23808–23815. doi: 10.1074/jbc.270.40.23808. [DOI] [PubMed] [Google Scholar]
- 41.Eleuteri AM, Kohanski RA, Cardozo C, Orlowski M. Bovine spleen multicatalytic proteinase complex (proteasome) — replacement of X, Y, and Z subunits by LMP7, LMP2, and MECL1 and changes in properties and specificity. J Biol Chem. 1997;272:11824–11831. doi: 10.1074/jbc.272.18.11824. [DOI] [PubMed] [Google Scholar]
- 42.Hill CP, Masters EI, Whitby FG. The 11S regulators of 20S proteasome activity. Curr Top Microbiol Immunol. 2002;268:73–89. doi: 10.1007/978-3-642-59414-4_4. [DOI] [PubMed] [Google Scholar]
- 43.Cascio P, Call M, Petre BM, Walz T, Goldberg AL. Properties of the hybrid form of the 26S proteasome containing both 19S and PA28 complexes. EMBO Journal. 2002;21:2636–2645. doi: 10.1093/emboj/21.11.2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fruh K, Gossen M, Wang KN, Bujard H, Peterson PA, Yang Y. Displacement of housekeeping proteasome subunits by MHC-encoded LMPs — a newly discovered mechanism for modulating the multi-catalytic proteinase complex. EMBO J. 1994;13:3236–3244. doi: 10.1002/j.1460-2075.1994.tb06625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Ann Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- 46.Campbell DS, Holt CE. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron. 2001;32:1013–1026. doi: 10.1016/s0896-6273(01)00551-7. [DOI] [PubMed] [Google Scholar]
- 47.Gieffers C, Peters BH, Kramer ER, Dotti CG, Peters JM. Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. PNAS. 1999;96:11317–11322. doi: 10.1073/pnas.96.20.11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stegmuller J, Konishi Y, Huynh MA, Yuan ZQ, DiBacco S, Bonni A. Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron. 2006;50:389–400. doi: 10.1016/j.neuron.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 49.Lasorella A, Stegmuller J, Guardavaccaro D, Liu GC, Carro MS, Rothschild G, Torre-Ubieta L, Pagano M, Bonni A, Lavarone A. Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature. 2006;442:471–474. doi: 10.1038/nature04895. [DOI] [PubMed] [Google Scholar]
- 50.Zhen M, Huang X, Bamber B, Jin Yishi. Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron. 2000;26:331–343. doi: 10.1016/s0896-6273(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 51.Schaefer AM, Hadwiger GD, Nonet ML. RPM-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron. 2000;26:345–356. doi: 10.1016/s0896-6273(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 52.Liao EH, Hung W, Abrams B, Zhen M. An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature. 2004;430:345–350. doi: 10.1038/nature02647. [DOI] [PubMed] [Google Scholar]
- 53.Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, Jin Y. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell. 2005;120:407–420. doi: 10.1016/j.cell.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 54.Grill B, Bienvenut WV, Brown HM, Ackley BD, Quadroni M, Jin Y. C. elegans RPM-1 regulates axon termination and synaptogenesis through the Rab GEF GLO-4 and the Rab GTPase GLO-1. Neuron. 2007;55:587–601. doi: 10.1016/j.neuron.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 55.DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS. Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature. 2001;412:449–452. doi: 10.1038/35086595. [DOI] [PubMed] [Google Scholar]
- 56.Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS. Highwire regulates synaptic growth in Drosophila. Neuron. 2000;26:313–329. doi: 10.1016/s0896-6273(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 57.Wu C, Wairkar YP, Collins CA, DiAntonio A. Highwire function at the Drosophila neuromuscular junction: spatial, structural, and temporal requirements. J Neurosci. 2005;25:9557–9566. doi: 10.1523/JNEUROSCI.2532-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mccabe BD, Hom S, Aberle H, Fetter RD, Marques G, Haerry TE, Wan H, O’Connor MB, Goodman CS, Haghighi AP. Highwire regulates presynaptic BMP signaling essential for synaptic growth. Neuron. 2004;41:891–905. doi: 10.1016/s0896-6273(04)00073-x. [DOI] [PubMed] [Google Scholar]
- 59.Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 60.Wu C, Daniels RW, DiAntonio A. DFsn collaborates with Highwire to downregulate the Wallenda/DLK kinase to restrain synaptic terminal growth. Neural Develop. 2007;2:16. doi: 10.1186/1749-8104-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burgess RW, Peterson KA, Johnson MJ, Roix JJ, Welsh IC, O’Brien TP. Evidence for a conserved function in synapse formation reveals Phr1 as a candidate gene for respiratory failure in newborn mice. Mol Cell Biol. 2004;24:1096–1105. doi: 10.1128/MCB.24.3.1096-1105.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.D’Souza J, Hendricks M, Le Guyader S, Subburaju S, Grunewald B, Scholich K, Jesuthasan S. Formation of the retinotectal projection requires Esrom, an ortholog of PAM (protein associated with Myc) Development. 2004;132:247–256. doi: 10.1242/dev.01578. [DOI] [PubMed] [Google Scholar]
- 63.Bloom AJ, Miller BR, Sanes JR, DiAntonio A. The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev. 2007;21:2593–2606. doi: 10.1101/gad.1592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van Roessel P, Elliott DA, Robinson LM, Prokop A, Brand AH. Independent regulation of synaptic size and activity by the anaphase-promoting complex. Cell. 2004;119:707–718. doi: 10.1016/j.cell.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 65.Tursun B, Schluter A, Peters MA, Viehweger B, Ostendorff HP, Soosairajah J, Drung A, Bossenz M, Johnsen SA, Schweizer M, Bernard O, Bac L. The ubiquitin ligase Rnf6 regulates local LIM kinase 1 levels in axonal growth cones. Genes Dev. 2005;19:2307–2319. doi: 10.1101/gad.1340605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17:111–1118. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- 67.Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, Wagstaff J. Angelman syndrome 2005: updated consensus for diagnostic criteria. Amer J Med Genet Part A. 2006;140A:413–418. doi: 10.1002/ajmg.a.31074. [DOI] [PubMed] [Google Scholar]
- 68.Prokopenko SN, He Y, Lu Y, Bellen HJ. Mutations affecting the development of the peripheral nervous system in Drosophila: a molecular screen for novel proteins. Genetics. 2000:1691–1715. doi: 10.1093/genetics/156.4.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reiter LT, Seagroves TN, Bowers M, Bier E. Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum Mol Genet. 2006;15:2825–2835. doi: 10.1093/hmg/ddl225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Broadie KS, Richmond JE. Establishing and sculpting the synapse in Drosophila and C. elegans. Curr Opin Neurobiol. 2002;12:491–498. doi: 10.1016/s0959-4388(02)00359-8. [DOI] [PubMed] [Google Scholar]
- 71.Wolf B, Seeger MA, Chiba A. Commissureless endocytosis is correlated with initiation of neuromuscular synaptogenesis. Development. 1998;125:3853–3863. doi: 10.1242/dev.125.19.3853. [DOI] [PubMed] [Google Scholar]
- 72.Zhai Q, Wang J, Kim A, Liu Q, Watts R, Hoopfer E, Mitchison T, Luo L, He Z. Involvement of the ubiquitin-proteasome system in the early stages of Wallerian degeneration. Neuron. 2003;39:217–225. doi: 10.1016/s0896-6273(03)00429-x. [DOI] [PubMed] [Google Scholar]
- 73.Mack TGA, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Hugh V, Michael P, Coleman P. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 74.Watts RJ, Hoopfer ED, Luo LQ. Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron. 2003;38:871–885. doi: 10.1016/s0896-6273(03)00295-2. [DOI] [PubMed] [Google Scholar]
- 75.Kuo CT, Jan LY, Ja YN. Dendrite-specific remodeling of Drosophila sensory neurons requires matrix metalloproteases, ubiquitin-proteasome, and ecdysone signaling. PNAS. 2005;102:15230–15235. doi: 10.1073/pnas.0507393102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuo CT, Zhu S, Younger S, Jan LY, Jan YN. Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron. 2006;51:283–290. doi: 10.1016/j.neuron.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 77.Aravamudan B, Fergestad T, Davis WS, Rodesch CK, Broadie K. Drosophila Unc-13 is essential for synaptic transmission. Nat Neurosci. 1999;2:965–971. doi: 10.1038/14764. [DOI] [PubMed] [Google Scholar]
- 78.Aravamudan B, Broadie K. Synaptic Drosophila UNC-13 is regulated by antagonistic G-protein pathways via a proteasome-dependent degradation mechanism. J Neurobiol. 2003;54(3):417–438. doi: 10.1002/neu.10142. [DOI] [PubMed] [Google Scholar]
- 79.Kuromi H, Kidokoro Y. Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron. 2000;27:133–143. doi: 10.1016/s0896-6273(00)00015-5. [DOI] [PubMed] [Google Scholar]
- 80.Yao I, Takagi H, Ageta H, Kahyo T, Sato S, Hatanaka K, Fukuda Y, Chiba T, Morone N, Yuasa S, Inokuchi K, Ohtsuka T, MacGregor GR, Tanaka K, Setou M. SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell. 2007;130:943–957. doi: 10.1016/j.cell.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Araki T, Milbrandt J. ZNRF proteins constitute a family of presynaptic E3 ubiquitin ligases. J Neurosci. 2003;23:9385–9394. doi: 10.1523/JNEUROSCI.23-28-09385.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chin LS, Vavalle JP, Li L. Staring, a novel E3 ubiquitin-protein ligase that targets Syntaxin 1 for degradation. J Biol Chem. 2002;277:35071–35079. doi: 10.1074/jbc.M203300200. [DOI] [PubMed] [Google Scholar]
- 83.Wheeler TC, Chin LS, Li Y, Roudabush FL, Li L. Regulation of Synaptophysin degradation by mammalian homologues of Seven in Absentia. J Biol Chem. 2002;277:10273–10282. doi: 10.1074/jbc.M107857200. [DOI] [PubMed] [Google Scholar]
- 84.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Ann Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 85.Pak DTS, Yang S, Rudolph-Correia S, Kim E, Sheng M. Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron. 2001;31:289–303. doi: 10.1016/s0896-6273(01)00355-5. [DOI] [PubMed] [Google Scholar]
- 86.Hedge AN, Goldberg AL, Schwartz JH. Regulatory subunits of cAMP-dependent protein kinases are degraded after conjugation to ubiquitin: a molecular mechanism underlying long-term synaptic plasticity. PNAS. 1993;90:7436–7440. doi: 10.1073/pnas.90.16.7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM. Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron. 2002;35(1):107–120. doi: 10.1016/s0896-6273(02)00749-3. [DOI] [PubMed] [Google Scholar]
- 88.Schaefer H, Rongo C. KEL-8 is a substrate receptor for CUL3-dependent ubiquitin ligase that regulates synaptic glutamate receptor turnover. Mol Biol Cell. 2006;17:1250–1260. doi: 10.1091/mbc.E05-08-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Juo P, Kaplan JM. The anaphase-promoting the abundance of GLR-1 complex regulates glutamate receptors in the ventral nerve cord of C. elegans. Curr Biol. 2004;14:2057–2062. doi: 10.1016/j.cub.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 90.Dreier L, Burbea M, Kaplan JM. LIN-23-mediated degradation of [beta]-catenin regulates the abundance of GLR-1 glutamate receptors in the ventral nerve cord of C. elegans. Neuron. 2005;46:51–64. doi: 10.1016/j.neuron.2004.12.058. [DOI] [PubMed] [Google Scholar]
- 91.Patrick GN, Bingol B, Weld HA, Schuman EM. Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr Biol. 2003;13(23):2073–2081. doi: 10.1016/j.cub.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 92.Martin S, Nishimune A, Mellor JR, Henley JM. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature. 2007;447:321–326. doi: 10.1038/nature05736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moriyoshi K, Iijima K, Fujii H, Ito H, Cho Y, Nakanishi S. Seven in absentia homolog 1A mediates ubiquitination and degradation of group 1 metabotropic glutamate receptors. PNAS. 2004;101:8614–8619. doi: 10.1073/pnas.0403042101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Christianson JC, Green WN. Regulation of nicotinic receptor expression by the ubiquitin-proteasome system. EMBO J. 2004;23:4156–4165. doi: 10.1038/sj.emboj.7600436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ficklin B, Zhao SL, Feng GP. Ubiquilin-1 regulates nicotine-induced up-regulation of neuronal nicotinic acetylcholine receptors. J Biol Chem. 2005;280:34088–34095. doi: 10.1074/jbc.M506781200. [DOI] [PubMed] [Google Scholar]
- 96.Bedford FK, Kittler JT, Muller E, Thomas P, Uren JM, Merlo D, Wisden W, Triller A, Smart TG, Moss SJ. GABAA receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein Plic-1. Nat Neurosci. 2001;4:908–916. doi: 10.1038/nn0901-908. [DOI] [PubMed] [Google Scholar]
- 97.Saliba RS, Michels G, Jacob TC, Pangalos MN, Moss SJ. Activity-dependent ubiquitination of GABAA receptors regulates their accumulation at synaptic sites. J Neurosci. 2007;27:13341–13351. doi: 10.1523/JNEUROSCI.3277-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gallagher MJ, Ding L, Maheshwari A, Macdonald RL. The GABAA receptor {alpha}1 subunit epilepsy mutation A322D inhibits transmembrane helix formation and causes proteasomal degradation. PNAS. 2007;104:12999–13004. doi: 10.1073/pnas.0700163104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kato A, Rouach N, Nicoll RA, Bredt DS. Activity-dependent NMDA receptor degradation mediated by retrotranslocation and ubiquitination. PNAS. 2005;102:5600–5605. doi: 10.1073/pnas.0501769102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6(3):231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- 101.Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40(3):595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bingol B, Schuman EM. A proteasome-sensitive connection between PSD-95 and GluR1 endocytosis. Neuropharmacology. 2004;47:755–763. doi: 10.1016/j.neuropharm.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 103.Hoogenraad CC, Feliu-Mojer MI, Spangler SA, Milstein AD, Dunah AW, Hung AY, Sheng M. Liprin alpha 1 degradation by calcium/calmodulin-dependent protein kinase II regulates LAR receptor tyrosine phosphatase distribution and dendrite development. Dev Cell. 2007;12:587–602. doi: 10.1016/j.devcel.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 104.Weeber EJ, Jiang YH, Elgersma Y, Varga AW, Carrasquillo Y, Brown SE, Christian JM, Mirnikjoo B, Silva A, Beaudet AL, Sweatt JD. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman Mental Retardation Syndrome. J Neurosci. 2003;23:2634–2644. doi: 10.1523/JNEUROSCI.23-07-02634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qui S, de Avila Freire R, Jiang Y, Elgersma Y, Weeber EJ. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of αCaMKII inhibitory phosphorylation. Nat Neurosci. 2007;10:280–282. doi: 10.1038/nn1845. [DOI] [PubMed] [Google Scholar]
- 106.Featherstone DE, Rushton E, Rohrbough J, Liebl F, Karr J, Sheng Q, Rodesch CK, Broadie K. An essential Drosophila glutamate receptor subunit that functions in both central neuropil and neuromuscular junction. J Neurosci. 2005;25:3199–3208. doi: 10.1523/JNEUROSCI.4201-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marrus SB, Portman SL, Allen MJ, Moffat KG, DiAntonio A. Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junction. J Neurosci. 2004;24:1406–1415. doi: 10.1523/JNEUROSCI.1575-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Qin G, Schwarz T, Kittel RJ, Schmid A, Rasse TM, Kappei D, Ponimaskin E, Heckmann M, Sigrist SJ. Four different subunits are essential for expressing the synaptic glutamate receptor at neuromuscular junctions of Drosophila. J Neurosci. 2005;25:3209–3218. doi: 10.1523/JNEUROSCI.4194-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Haas KF, Miller SLH, Friedman DB, Broadie K. The ubiquitin-proteasome system postsynaptically regulates glutamatergic synaptic function. Mol Cell Neurosci. 2007;35:64–75. doi: 10.1016/j.mcn.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen K, Featherstone DE. Discs-large (DLG) is clustered by presynaptic innervation and regulates postsynaptic glutamate receptor subunit composition in Drosophila. BMC Biol. 2005;3 doi: 10.1186/1741-7007-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Carvalho P, Goder V, Rapoport TA. Distinct ubiquitinligase complexes define convergent pathways for the degradation of ER proteins. Cell. 2006;126:361–373. doi: 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- 112.Bingol B, Schuman EM. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441:1144–1148. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- 113.Ostroff LE, Fiala JC, Allwardt B, Harris KM. Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron. 2002;35:535–545. doi: 10.1016/s0896-6273(02)00785-7. [DOI] [PubMed] [Google Scholar]
- 114.Shen H, Korutla L, Champtiaux N, Toda S, LaLumiere R, Vallone J, Klugmann M, Blendy JA, Mackler SA, Kalivas PW. NAC1 regulates the recruitment of the proteasome complex into dendritic spines. J Neurosci. 2007;27:8903–8913. doi: 10.1523/JNEUROSCI.1571-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Haas KF, Woodruff E, III, Broadie K. Proteasome function is required to maintain muscle cellular architecture. Biol Cell. 2007;99:615–626. doi: 10.1042/BC20070019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.James AB, Conway AM, Morris BJ. Regulation of the neuronal proteasome by Zif268 (Egr1) J Neurosci. 2006;26:1624–1634. doi: 10.1523/JNEUROSCI.4199-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Conway AM, James AB, Zang J, Morris BJ. Regulation of neuronal cdc20 (p55cdc) expression by the plasticity-related transcription factor Zif268. Synapse. 2007;61:463–468. doi: 10.1002/syn.20387. [DOI] [PubMed] [Google Scholar]