

Abstract
HIF-asparaginyl hydroxylase (FIH-1) normally couples O2-activation to hydroxylation of Asn803 on the α-subunit of the hypoxia-inducible factor (HIFα), a key step in pO2 sensing; in the absence of HIFα, O2-activation becomes uncoupled, leading to self-hydroxylation at Trp296 and a purple Fe(III)–O–Trp chromophore—this alternative reactivity may affect human hypoxia sensing.
Cellular responses to O2 levels are central to human health, controlling processes such as angiogenesis and basal metabolism. 1,2 Tight regulation of O2 is necessary, as while it is required for aerobic metabolism, an excess can lead to oxidative damage. In humans, the master regulator of O2-homeostasis is a transcription factor called the hypoxia-inducible factor (HIF), which is inactivated by HIF-asparaginyl hydroxylase (previously identified as FIH-1), a non-heme, Fe(II), α-ketoglutarate (αKG)-dependent dioxygenase.3–5 While tight coupling between O2-activation and HIF-hydroxylation would be expected for optimal O2 sensing, some αKG-dependent dioxygenases exhibit uncoupled O2-activation. We establish here that FIH-1 self-hydroxylates in the absence of HIF, suggesting that uncoupling between O2 and HIF may alter the activity levels of FIH-1.
HIF is a heterodimeric transcription factor, consisting of the HIFα monomer, which is regulated in response to pO2, and the ARNT monomer, which is insensitive with respect to pO2. Under the conditions of hypoxia (low pO2), HIF assembles the transcriptional co-activator, p300, upstream from HIF controlled genes, thereby promoting the expression of over 100 genes involved in iron transport, angiogenesis and basal metabolism.1,6 The key intermolecular interactions are found between transcriptional machinery and the C-terminal domain of HIFα.7,8 FIH-1 uses O2 to hydroxylate the β-carbon of HIFα-Asn803,9 preventing p300 from binding to HIFα, and thereby turning o. gene expression in response to increasing pO2.
Although the sensing of pO2 should require tight coupling between pO2 and the hydroxylation of HIFα, the chemical mechanism of FIH-1 activity suggests that O2 consumption could be uncoupled from HIF-hydroxylation. In the consensus mechanism, HIFα binding displaces the axial H2O from the Fe(II)– αKG center, which opens up a coordination site for O2 binding.10,11 This leads to the formation of the Fe(IV) = O intermediate, but only when the HIFα-Asn803 sidechain is nearby, ensuring that O2-activation only occurs when HIFα is present. However, spectroscopic studies suggest that the axial H2O in other members of the Fe(II), αKG-dependent dioxygenases can be released, even in the absence of a prime substrate;12,13 if this occurred on FIH-1, it would lead to uncoupling between O2 and HIFα. It is notable that in activity assays of FIH-1 and many other members of the non-heme, Fe(II), αKG-dependent dioxygenase superfamily, excess ascorbate was required for full activity, as this maintained the iron in the reduced state.10
In addition, a few members of this superfamily deactivate by auto-hydroxylation in the absence of a prime substrate. For example, aromatic residues within the active site of TauD (Tyr)14 and TfdA (Trp)15 are hydroxylated during uncoupling to generate an Fe(III) coordinated by an aromatic alcohol, suggesting that the electrophilic Fe(IV) = O species can oxidatively attack residues within ~10 Å of it, to create new metal-binding ligands. The active site of FIH-1 contains three aromatic residues that, based upon these chemical precedents in bacterial enzymes, could be susceptible to hydroxylation.
To determine whether FIH-1 can auto-hydroxylate, apo- FIH (100 μM in 50 mM HEPES, pH 7.50), FeSO4 (500 μM) and αKG (500 μM) were incubated anaerobically in a sealed cuvette for 1 h. The absorption spectrum was measured, and it showed a moderate-intensity absorption near 520 nm, which could be attributed to the Fe(II)–α KG charge transfer that would be expected for resting FIH-1.17 The cuvette was then opened to air to introduce O2, with the reaction being monitored by a UV-vis spectrophotometer over 10 000 seconds. A new absorption band appeared at λmax = 583 nm (ε583 = 3 × 103M−1 cm−1), which was consistent with the product of auto-hydroxylation: Fe(III) coordinated to an aromatic alcohol (Fig. 1). Negative controls, in which any individual component was omitted, showed that this chromophore required FIH-1, Fe(II), O2 and αKG for its formation.
Fig. 1.
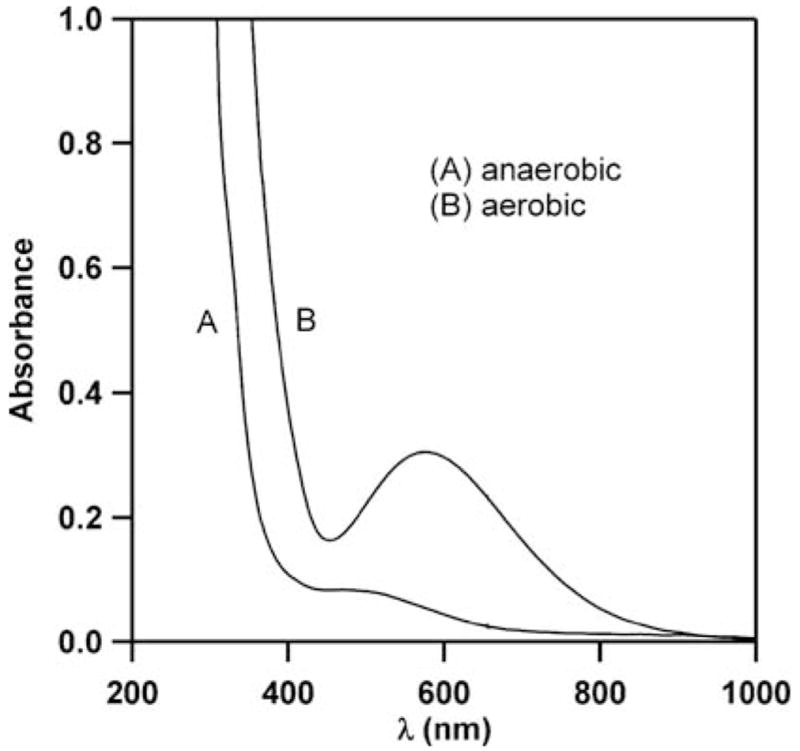
The UV-vis absorption spectrum of FIH-1 (100 μM in 50mM HEPES, pH 7.50), FeSO4 (500 μM) and αKG (500 μM). (A) Anaerobic conditions, (B) following exposure to air.
Auto-hydroxylation in TauD leads to the formation of a hydroxylated Tyr residue, resulting in an Fe(III)-catecholate with λmax = 550 nm (ε = 700 M−1 cm−1).14 A better spectroscopic match to the FIH-1 product is the Fe(III)-(hydroxy-Trp) formed by the auto-hydroxylation of TfdA, with λmax = 580 nm (ε = 1000 M−1 cm−1),15 suggesting that the purple chromophore that was formed by the auto-hydroxylation of FIH-1 contained a hydroxylated Trp residue, HO-Trp. Inspection of the FIH-1 sequence indicated that the Trp and Tyr residues within the active site (Fig. 2) would be located on separate tryptic fragments, suggesting that MS could be used to identify the hydroxylation site.
Fig. 2.
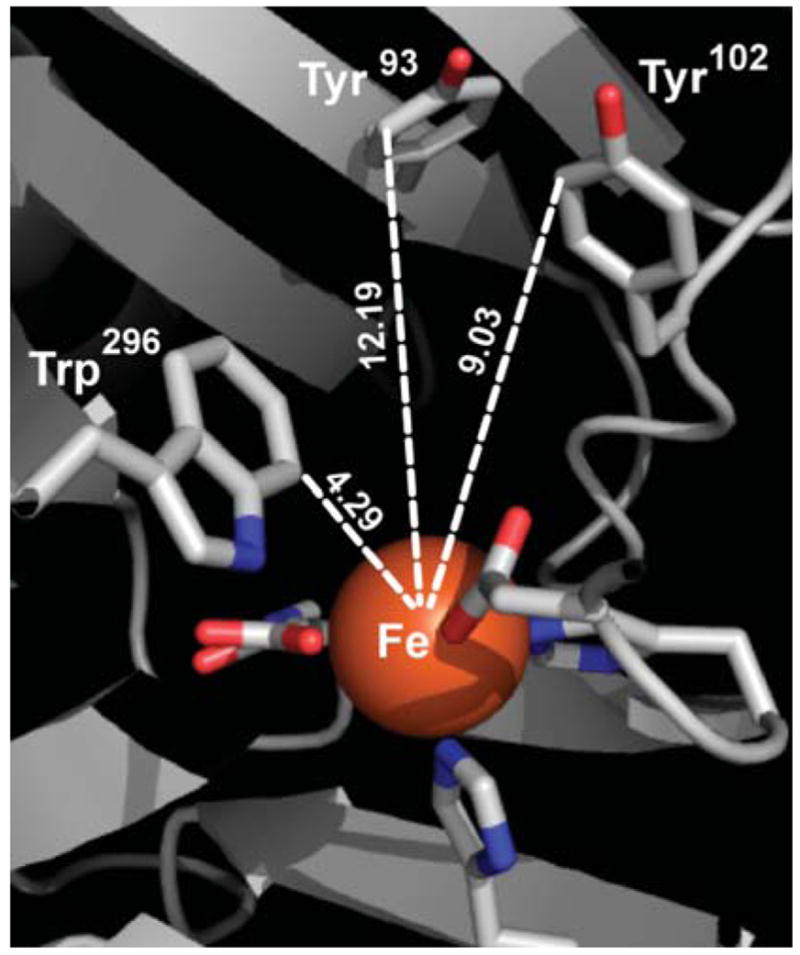
Aromatic residues within the active site of FIH-1 (PDB 1H2K).16
MS was used to identify the site of hydroxylation in a tryptic digest of FIH-1. The tryptic digest was separated by reverse-phase HPLC and detected via ESI-MS analysis performed on an ESI-Q-TOF instrument.‡ LC-MS/MS analysis of the tryptic digest showed that the tryptic fragments containing Tyr93 and Tyr102 were unmodified, while the fragment containing Trp296 (252–298, 5512.7 Da) appeared at m/z = 1379.2 (unmodified, MH44+) and 1383.2 (+16 Da, OMH44+) as z = 4+ ions. Co-elution of modified and unmodified Trp296 fragments revealed relative intensities of MH44+: (OMH44+) ~ 7:3 (Fig. 3).‡
Fig. 3.
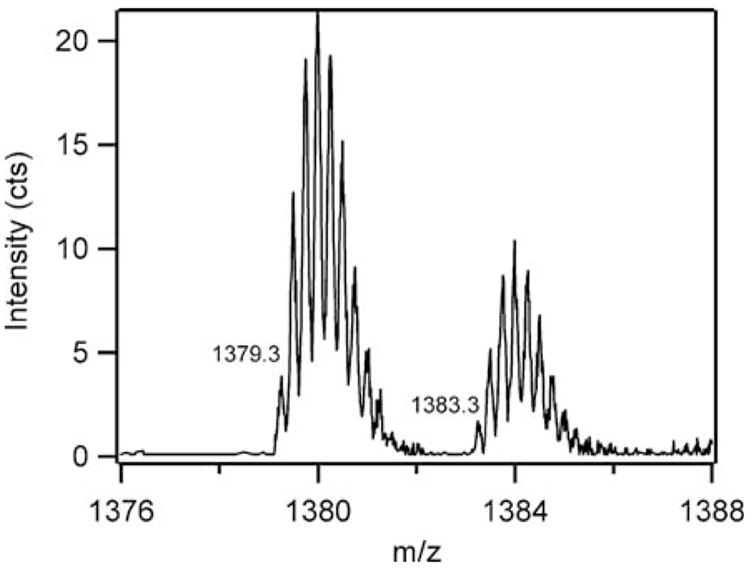
ESI-TOF spectrum of the tryptic digest fragment 252–298 from purple FIH-1.
The hydroxylated residue was identified by LC purification of the Trp296 peptides, followed by direct infusion into the ESI-TOF spectrometer. ESI-MS/MS analysis was performed on the z = 4+ ions for both peptide peaks. The b- and y-ion sequence of the MH44+ ion corresponded to the genetically encoded sequence. In contrast, the b- and y-ion sequence of the OMH44+ ion revealed that the additional O-atom was located on Trp296 (Table 1). This positively identified the active-site Trp residue as the sole site of O-atom insertion.
Table 1.
Observed y-ions from ESI-MS/MS analysis of the Trp296 peptide (252–298) by direct infusion and fragmentation of the z=4+ ion at m/z = 1383.2
| Amino acid | y-ion number | Unmodified m/z observed (calc.) | Modified m/z observed (calc.) |
|---|---|---|---|
| T | 7 | 973.48 (973.48) | |
| V | 6 | 872.41 (872.44) | |
| N | 5 | 773.39 (773.37) | |
| F | 4 | 659.35 (659.32) | |
| W | 3 | 512.28 (512.26) | |
| Y | 2 | 310.19 (310.18) | |
| K | 1 | 147.11 (147.11) |
The purple chromophore that formed on FIH-1 was identified as an Fe(III)–O–Trp296 species by the identification of O-atom addition to Trp296 and its similarity to the blue Fe(III)–O–Trp chromophore found in TfdA. While the Trp296 of FIH-1 is separated in a 1° sequence from the Fe ligands, it is positioned very close (<5 Å) to the Fe center. As C7 of the indole ring of Trp296 is located closest to the Fe, this may be the site of hydroxylation.
By analogy to the consensus chemical mechanism for normal turnover,10 we propose that the Fe(III)–O–Trp center in FIH-1 is formed by the uncoupled activation of O2, leading to the formation of an Fe(IV)=O intermediate in the absence of HIFα. H-atom abstraction, followed by rebound, led to HO-Trp296 and Fe(II), which, through subsequent oxidation and coordination, formed the observed Fe(III)–O–Trp species. As the normal rate of turnover for FIH-1 to hydroxylate HIFα substrates is ca. 0.24 s−1,18,19 and the auto-hydroxylation occurred on the timescale of hours, auto-hydroxylation would only be kinetically relevant under conditions of low HIFα concentration.
We speculate that the auto-hydroxylation of FIH-1 may comprise an important regulatory feature of physiological hypoxia sensing. Under normoxic conditions, HIFα is rapidly degraded within cells, thereby depleting FIH-1 of its physiological substrate—such conditions would favor uncoupled O2-activation. As suggested within the context of TauD and TfdA function,10 FIH-1 auto-hydroxylation may protect the cell from deleterious effects of reactive oxygen species by serving in a sacrificial role. Alternatively, auto-hydroxylation may serve as a negative feedback mechanism to regulate hypoxia sensing by reducing the activity of FIH-1.
Supplementary Material
Acknowledgments
Acknowledgement is made to the University of Massachusetts, the American Cancer Society (#IRG 93-033) and the NIH (GM077413) for funding.
Footnotes
Electronic supplementary information (ESI) available: ESI-MS/MS spectra of trypsin-digested purple FIH-1.
Experimental details: FIH-1 (10 μM) was digested by trypsin overnight (37 °C, 50 mM Tris, pH 8.00, 1 mM CaCl2). The tryptic digest (30 μl injection) was separated by reverse-phase HPLC, using a CH3CN/H2O (0.1% formic acid) gradient, over a 150 × 2.1 mm PLRP-S column with a 300 Å pore size and a 3 μm particle size (Polymer Laboratories, Amherst, MA). High resolution MS utilized a Qstar XL hybrid ESI-TOF spectrometer (Applied Biosystems, Inc.) that was set to detect positive ions over the 500–2000 m/z range, and fragment the 3+, 4+ and 5+ ions of the unmodified peptide (1838.6, 1379.2 and 1103.5 m/z) and hydroxylated peptide (1843.9, 1383.2 and 1106.7 m/z). The positive ions of the 252–298 peptide fragment were found at both unmodified (5512.7 Da) and O-atom added (5528.7 Da) masses.
Notes and references
- 1.Schofield CJ, Ratcliffe PJ. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 2.Guillemin K, Krasnow MA. Cell. 1997;89:9–12. doi: 10.1016/s0092-8674(00)80176-2. [DOI] [PubMed] [Google Scholar]
- 3.Mahon PC, Hirota K, Semenza GL. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ. J Biol Chem. 2002;277:26351–26355. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 5.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semenza GL. Science. 2007;318:62–64. doi: 10.1126/science.1147949. [DOI] [PubMed] [Google Scholar]
- 7.Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE. Proc Natl Acad Sci U S A. 2002;99:5271–5276. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freedman SJ, Sun ZYJ, Poy F, Kung AL, Livingston DM, Wagner G, Eck MJ. Proc Natl Acad Sci U S A. 2002;99:5367–5372. doi: 10.1073/pnas.082117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McNeill LA, Hewitson KS, Claridge TD, Seibel JF, Horsfall LE, Schofield CJ. Biochem J. 2002367:571–575. doi: 10.1042/BJ20021162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 11.Zhou J, Gunsior M, Bachmann BO, Townsend CA, Solomon EI. J Am Chem Soc. 1998;120:13539–13540. doi: 10.1021/ja004025+. [DOI] [PubMed] [Google Scholar]
- 12.Zhou J, Kelly WL, Bachmann BO, Gunsior M, Townsend CA, Solomon EI. J Am Chem Soc. 2001;123:7388–7398. doi: 10.1021/ja004025+. [DOI] [PubMed] [Google Scholar]
- 13.Neidig ML, Brown CD, Light KM, Fujimori DG, Nolan EM, Price JC, Barr EW, Bollinger JM, Krebs C, Walsh CT, Solomon EI. J Am Chem Soc. 2007;129:14224–14231. doi: 10.1021/ja074557r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryle MJ, Liu A, Muthukumaran RB, Ho RYN, Koehntop KD, McCracken J, Que L, Hausinger RP. Biochemistry. 2003;42:1854–1862. doi: 10.1021/bi026832m. [DOI] [PubMed] [Google Scholar]
- 15.Liu A, Ho RYN, Que LJ, Ryle MJ, Phenney BS, Hausinger RP. J Am Chem Soc. 2001;123:5126–5127. doi: 10.1021/ja005879x. [DOI] [PubMed] [Google Scholar]
- 16.Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. J Biol Chem. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- 17.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 18.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ. J Biol Chem. 2002;277:26351–26355. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 19.Ehrismann D, Flashman E, Genn DN, Mathioudakis N, Hewitson KS, Ratcliffe PJ, Schofield CJ. Biochem J. 2007;401:227–234. doi: 10.1042/BJ20061151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.