

Abstract
Toll-like receptors (TLRs), which are activated by invading microorganisms or endogenous molecules, evoke immune and inflammatory responses. TLR activation is closely linked to the development of many chronic inflammatory diseases including rheumatoid arthritis. Auranofin, an Au(I) compound, is a well-known and long-used anti-rheumatic drug. However, the mechanism as to how auranofin relieves the symptom of rheumatoid arthritis has not been fully clarified. Our results demonstrated that auranofin suppressed TLR4-mediated activation of transcription factors, NF-κB and IRF3 and expression of COX-2, a pro-inflammatory enzyme. This suppression was well correlated with the inhibitory effect of auranofin on the homodimerization of TLR4 induced by an agonist. Furthermore, auranofin inhibited NF-κ activation induced by MyD88-dependent downstream signaling components of TLR4, MyD88, IKKβ, and p65. IRF3 activation induced by MyD88-independent signaling components, TRIF and TBK1, was also downregulated by auranofin. Our results first demonstrate that auranofin suppresses the multiple steps in TLR4 signaling, especially the homodimerization of TLR4. The results suggest that the suppression of TLR4 activity by auranofin may be the molecular mechanism through which auranofin exerts anti-rheumatic activity.
Keywords: auranofin, gold, Toll-like receptor, LPS, dimerization, NF-κB, COX-2, MyD88, TRIF, IKKβ
1. Introduction
Toll-like receptors (TLRs) play an important role in innate immune responses that are essential for host defense against invading microbial pathogens [1-4]. TLRs interact with different combinations of adapter proteins. TLR signaling pathways can trigger the activation of NF-κ through the MyD88 dependent pathway, but they also induce type I IFN expression through the MyD88-dependent and -independent pathways. MyD88 is the immediate adaptor molecule which is common to all mammalian TLRs [1]. MyD88 recruits IL-1 receptor-associated kinase-4 (IRAK-4) and induces phosphorylation of IRAK-4 leading to the phosphorylation of IRAK-1. The phosphorylated IRAK-1 associates with tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) leading to the activation of the canonical IKK complex resulting in the activation of NF-κ transcription factor. The activation of NF-κ leads to the induction of inflammatory gene products such as cytokines and cyclooxygenase-2 [5].
TLR3 and TLR4 activate MyD88-independent signaling pathway mediated through TIR domain-containing adapter inducing IFNβ (TRIF). TRIF-dependent pathway triggers the expression of type I IFN mediated through TBK1 and IKKε [6, 7]. The C-terminal portion of TRIF also was shown to be associated with RIP1 leading to the delayed activation of NF-κ [8]. Thus, TRIF is likely to use TBK1 for IRF3- and RIP1 for NF-κ activation [9].
Deregulated activation of TLRs can lead to severe systemic inflammatory and joint destructive process in rheumatoid arthritis (RA) [10]. RA is an autoimmune disease characterized by chronic inflammation in most major joints [10]. There are several anti-rheumatic drugs that can be divided into two groups. First, the non-steroidal anti-inflammatory drugs (NSAIDs), such as aspirin, are used to suppress pain and inflammation. Second, disease-modifying anti-rheumatic drugs (DMARDS) are used to limit joint damage and to suppress the underlying autoimmune dysfunction of RA [11]. The latter class of drugs includes methotrexate, chloroquine, sulphasalazine, D-penicillamine, and gold compounds such as auranofin. The mechanism for the efficiency of these drugs on RA has not been fully understood. Recent strategies for drug development have focused on cytokines that are detected in the synovium of RA. Many cytokines such as TNF, IL-1, IL-6, IL-8, IL-10, IL-12, IL-15 and IL-18 are detected in the RA except for IL-4 [12, 13]. The expression of these inflammatory cytokines can be regulated by TLR-NF-κ signaling pathway [10].
Gold compounds have been used for the treatment of RA for more than 100 years [14]. The two most common gold compounds used as anti-rheumatic drugs are sodium aurothiomalate, which is water-soluble and administered by the intramuscular route, and auranofin, which is a hydrophobic compound and can be taken orally [14]. Both compounds are Au(I) compounds with sulfur-linked organic ligands. These gold compounds inhibited endotoxin-induced IL-1 and TNF production in the inflammatory monocytes and macrophages in the RA synovial membranes [15, 16]. Gold compounds have also been shown to inhibit the endotoxin-induced activation of NF-κB and AP-1 dependent transfected reporter genes [17, 18].
Identifying the direct targets of gold compounds in TLR pathways would be important because the activation of TLRs by agonists can induce inflammatory responses and consequently increases the risk of the development of chronic inflammatory diseases. Therefore, we attempted to identify the molecular target of gold compounds in TLR signaling pathways.
2. Materials and Methods
2.1. Reagents
Auranofin was purchased from Biomol (Plymouth Meeting, PA). Sodium tetrachloroaurate(III) dihydrate was purchased from Sigma-Aldrich (St. Louis, MO). Purified LPS was obtained from List Biological Lab. Inc. All other reagents were purchased from Sigma unless otherwise described.
2.2. Cell culture
Ba/F3 cells, an IL-3-dependent murine pro-B cell line, expressing TLR4 (Flag or GFP-tagged), CD14, MD2 (Flag-tagged), and NF-κB luciferase reporter gene were described previously [19]. Cells were cultured in RPMI1640 medium containing recombinant murine IL-3 (70 U/ml), 10% (v/v) heat-inactivated fetal bovine serum (FBS, Invitrogen), 100 units/ml Penicillin, and 100 μg/ml Streptomycin (GIBCOBRL). RAW264.7 cells (a murine monocytic cell line, ATCC TIB-71) and 293T cells (human embryonic kidney) were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% (v/v) FBS, 100 units/ml Penicillin, and 100 μg/ml Streptomycin. Cells were maintained at 37°C in a 5% CO2/air environment.
2.3. Transfection and reporter gene luciferase assay
NF-κB(2x)-luciferase reporter gene assay were performed as described previously [20, 21]. Cells were co-transfected with a luciferase plasmid and HSP70-β-galactosidase plasmid as an internal control using SuperFect transfection reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions. Various expression plasmids or equal amounts of empty vector for signaling components were co-transfected. Luciferase enzyme activities were determined using the Luciferase Assay System (Promega, Madison, WI) according to the manufacturer's instructions. Luciferase activity was normalized by β-galactosidase activity.
2.4. Immunoblotting and immunoprecipitation
These were performed the same as previously described [20]. Protein extracts from Ba/F3 cells expressing TLR4 (Flag or GFP-tagged), CD14, MD2 (Flag-tagged), and NF-κB luciferase reporter gene for immunoprecipitation were prepared as described [19]. The samples were immunoprecipitated with mouse-GFP antibody (Molecular Probes Inc., Eugene, OR) for overnight. The solubilized immune complex was resolved on 8% SDS-PAGE and electrotransferred to polyvinylidene difluoride membrane. The membrane was blocked with phosphate-buffered saline containing 0.1% Tween 20 and 5% nonfat dry milk and was blotted with the Flag antibodies for overnight. Thereafter, the blot was exposed to horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences, Arlington Heights, IL) for 1 hr, and detected with ECL Western blot detection reagents (Amersham Biosciences, Arlington Heights, IL). The blot was reprobed with rabbit GFP antibodies.
3. Results
3.1. Auranofin inhibited LPS-induced NF-κB and IRF3 activation and target gene (COX-2) expression
Auranofin, an anti-rheumatic gold compound, inhibited LPS-induced activation of NF-κB and IRF3 as determined by luciferase reporter gene assays in RAW264.7 cells (Fig. 1A,B). The decrease in the activation of these transcription factors resulted in the reduction of the expression of the target gene, COX-2 (Fig. 1C).
Figure 1. Auranofin inhibits LPS-induced NF-κB and IRF3 activation.
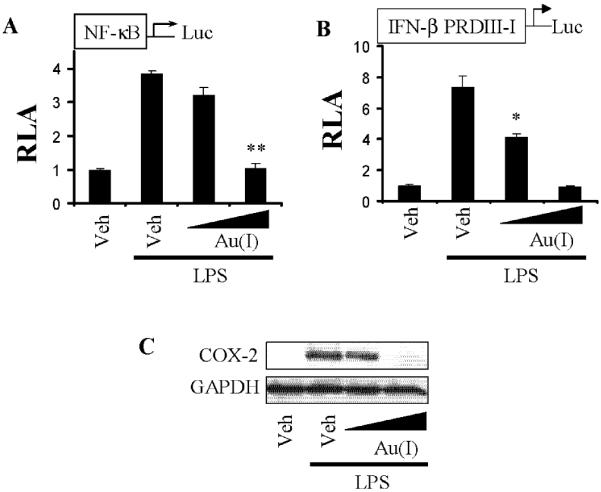
A,B) RAW264.7 cells were transfected with (A) NF-κB or (B) IFNβ promoter with specific IRF3 binding site (IFNβ PRDIII-I) luciferase reporter plasmid and pre-treated with auranofin (5, 10 μmM) for 1 h, and then treated with LPS (5 ng/ml) for an additional 6 hrs. Cell lysates were prepared and luciferase and β-galactosidase enzyme activities were measured as described in “Materials and Methods”. Relative luciferase activity (RLA) was normalized with β-galactosidase activity. Values are mean±SEM (n=3). *, Significantly different from LPS alone, p<0.05. **, Significantly different from LPS alone, p<0.01. C) RAW264.7 cells were pretreated with auranofin (5, 10 μM) for 1 hr and then further stimulated with LPS (5 ng/ml) for 6 hrs. Cell lysates were analyzed for COX-2 and GAPDH protein by immunoblots. The panels are representative data from more than three independent experiments. Veh, vehicle; Au(I), auranofin.
3.2. LPS-induced dimerization of TLR4 was inhibited by gold compounds
We next determined whether auranofin inhibits LPS-induced dimerization of TLR4 using IL-3-dependent Ba/F3 cells stably transfected with murine TLR4-Flag and TLR4-GFP, MD2, CD14, and NF-κB-luciferase reporter gene as described previously [19, 22]. Auranofin inhibited the dimerization of TLR4 (Fig. 2A) induced by LPS in a dose dependent manner. The inhibitory potency of auranofin [Au(I)] is greater than that of another gold compound, tetrachloroauric (III) acid [Au(III)]. The inhibition of TLR4 dimerization by the gold compounds was well correlated the suppressive effects on NF-κB activation (Fig. 2B).
Figure 2. Gold compounds inhibit LPS-induced homodimerization of TLR4.

A) Ba/F3 cells expressing TLR4-Flag (TLR4F), TLR4-GFP (TLR4G), MD2-Flag (MD2F), CD14 and NF-κB luciferase were pre-treated with auranofin (5, 10 μM) or sodium tetrachloroaurate (50 μM) for 1 h and then treated with LPS (50 ng/ml) for 20 mins. Cells were then subjected to immunoprecipitation with anti-mouse GFP antibody and immunoblotted with anti-Flag (upper) or anti-rabbit GFP (lower) antibody. B) The same Ba/F3 cells in Fig. 2A were pre-treated with auranofin (5, 10 μM) or sodium tetrachloroaurate (20, 50 μM) for 1 h and then treated with LPS (5 ng/ml) for an additional 6 hrs. Cell lysates were prepared and luciferase enzyme activities were measured as described in the legend of Fig. 1. Values are mean±SEM (n=3). **, Significantly different from LPS alone, p<0.01. The panels are representative data from more than three independent experiments. Veh, vehicle; Au(I), auranofin; Au(III), sodium tetrachloroaurate dihydrate.
3.3. Gold compounds inhibited both MyD88-dependent and TRIF-dependent signaling pathways of TLR4
TLR4 activates both MyD88-dependent and independent pathways. Since the gold compounds inhibit the LPS-induced dimerization of TLR4. They should inhibit both MyD88-dependent and -independent pathways. TLR4 activation induced by LPS leads to the recruitment of MyD88. MyD88 in turn recruits IRAK-4 which phosphorylates IRAK-1 leading to its degradation. IRAK-1 associates with TRAF6 leading to the activation of IKKβ complex resulting in the activation of NF-κB transcription factor. Both auranofin and Au (III) acid inhibited NF-κB activation induced by the overexpression of MyD88, IKKβ, or p65 in 293T cells (Fig. 3A-C). LPS-induced IRAK-1 degradation was also inhibited by gold compounds (Fig. 3D).
Figure 3. Gold compounds inhibit MyD88-dependent signaling pathways.

A-C) 293T cells were transfected with NF-κB binding site(2x)-luciferase reporter plasmid and the expression plasmid of (A) MyD88, (B) IKKβ, or (C) p65. After 24 hrs, cells were further treated with auranofin (5, 10 μM) or sodium tetrachloroaurate (20, 50 μM) for 6 hrs. Relative luciferase activity (RLA) was determined as described in the legend of Fig. 1. Values are mean±SEM (n=3). (A) **, Significantly different from MyD88 plus vehicle, p<0.01. (B) ++, Significantly different from IKκB plus vehicle, p<0.01. +, Significantly different from IKKβ plus vehicle, p<0.05. (C) ##, Significantly different from p65 plus vehicle, p<0.01. D) RAW264.7 cells were pretreated with auranofin (5, 10 μM) for 1 h and then stimulated with LPS (50 ng/ml) for 30 mins. Cell lysates were subjected to SDS-PAGE and probed with anti-IRAK-1 (upper) or anti-β-actin (lower) antibody. The panels are representative data from more than three independent experiments. Veh, vehicle; Au(I), auranofin; Au(III), sodium tetrachloroaurate dihydrate.
The results suggest that gold compounds inhibit NF-κB activation induced through MyD88-dependent pathway of TLR4. The TRIF-dependent pathway of TLR4 leads to the activation of IRF3 transcription factor mediated through TBK1 [23]. Gold compounds also inhibited TRIF or TBK1 (downstream kinase of TRIF-dependent pathway)-induced IRF3 activation as demonstrated by the transcription activation and the phosphorylation of IRF3 (Fig. 4A-C). These results showed that Gold compounds inhibit TRIF/TBK1/IRF3 signaling pathway of TLR4.
Figure 4. Gold compounds inhibit TRIF or TBK1-induced IRF3 activation.

A,B) 293T cells were co-transfected with IFNβ promoter (IFNβ PRDIII-I)-luciferase reporter plasmid and an expression plasmid for TRIF or TBK1. pCMV for TRIF and pcDNA for TBK1were used as a vector control. After 24 hrs, cells were treated with auranofin (5, 10 μM) or sodium tetrachloroaurate (20, 50 μM) for 6 hrs. Relative luciferase activity (RLA) was determined by normalization with β-galactosidase activity. Values are mean±SEM (n=3). (A) **, Significantly different from TRIF plus vehicle, p<0.01. (B) ++, Significantly different from TBK1 plus vehicle, p<0.01. C) RAW264.7 cells were treated with auranofin (5, 10 μM) for 1 h and further stimulated with LPS (50 ng/ml) for 1.5 h. Cell lysates were analyzed for phospho-IRF3 (S396) and IRF3 immunoblots. The panels are representative data from more than three independent experiments. Veh, vehicle; Au(I), auranofin; Au(III), sodium tetrachloroaurate dihydrate.
Together, our results demonstrate that gold compounds have the multiple targets in TLR4 signaling pathways to inhibit the NF-κB and IRF3 activation.
4. Discussion
Several lines of evidence have shown the significant role of TLR4 in the development and progress of rheumatoid arthritis. Repeated exposure to LPS, a TLR4 agonist, generates a mouse model of reactive arthritis [24]. The endogenous TLR4 agonists such as fibrinogen and hyaluronan are found in arthritis joints. Small heat shock protein B8 (HSP22) abundantly expressed in synovial tissue from patients with rheumatoid arthritis was identified as a TLR4 agonist [25]. Mice deficient of MyD88, an adaptor molecule of TLR, did not develop streptococcal cell wall-induced arthritis [26]. Some anti-rheumatic gold compounds have been shown to suppress LPS activity. Auranofin and gold sodium thiomalate inhibited LPS-induced production of IL-1 or TNF in peripheral blood mononuclear cells or murine macrophages [15, 16]. Therefore, the anti-rheumatic activity of gold compounds has been partly explained by the downregulation of the expression of inflammatory mediators. However, the mechanism through which gold compounds suppress LPS activity has not been fully understood. The homodimer formation of TLR4 in response to an agonist is considered as a critical step to recruit the adaptor molecules and to trigger the activation of downstream signaling pathways [19]. Our study demonstrated that auranofin, an anti-rheumatic gold compound, and tetrachloroauric (III) acid to a lesser extent, suppress LPS-induced formation of TLR4 homodimers. The suppression resulted in the decrease of the activation of downstream transcription factors, IRF3 and NF-κB. Therefore, our results suggest a novel mechanism as to how gold compounds suppress LPS activity and exert anti-rheumatic activity.
The biochemical mechanism as to how auranofin interacts with TLR4 to exert the inhibitory effects is not understood. Cysteine residues have been implicated as potential targets for gold compounds. Gold(I) salts have a high thiol binding affinity. Auranofin inhibits IβB kinase but the cysteine mutant of IKKβ (C179A) is resistant to inhibition [27]. This suggests that auranofin inhibits IKK by modifying Cys179 of IKKβ. In addition, gold salts react with Cys272 and Cys154 in the DNA-binding domains of Jun and Fos, respectively, thereby inhibiting DNA binding and transcription of AP-1 transcription factor [28]. TLRs have several cysteine residues, which form disulfide bonds for the dimerization of the receptors, in both cytoplasmic and extracellular domains [29]. Therefore, it can be speculated that auranofin may bind to cysteine residues of TLR4 to interfere with the activation.
The engagement of TLR4 by LPS establishes stably sustained phase of NF-κB activation composed of the early phase that is dependent on MyD88 and the TRIF-dependent delayed phase [30]. The activation of IRF3 through TRIF and TBK1 is responsible for the late phase of NF-κB activation. IRF3 activation leads to the expression of TNFα which in turn acts on TNFα receptor to trigger the delayed activation of NF-κB. Therefore, TRIF/TBK1/IRF3 pathway is now recognized to play an important role in TLR-mediated inflammation. Our results showed that auranofin suppressed the functional activity of TRIF and TBK1 as demonstrated by the phosphorylation and the transcriptional activation of IRF3.
Several gold compounds such as auranofin, aurothiomalate, aurothioglucose, and AuCl3 were shown to inhibit kinase activity of IKK immune complex prepared by the immunoprecipitation with IKKα antibody [31]. This was suggested as the mechanism through which gold compounds suppress NF-κB activation. Consistently, auranofin, aurathioglucose and aurothiomalate inhibited 12-O-tetradecanoylphorbol 13-acetate (TPA)-induced nuclear translocation of NF-κB in rat peritoneal macrophages when cells were treated with each of the gold compounds [32]. However, the contradicting results on the effect of each gold compound on NF-κB activation were also reported. Aurothiomalate did not suppress TNFα-induced NF-κB activation in HUVEC as determined by EMSA while auranofin inhibited the NF-κB activation [33]. Our study demonstrated that auranofin inhibited IKKβ-induced NF-κB reporter activity. In addition, auranofin and tetrachloroauric (III) acid suppressed p65-induced NF-κB reporter activity suggesting the direct inhibition of p65 transcriptional activity by gold compounds.
Collectively, our study for the first time demonstrated that auranofin inhibits the dimerization of TLR4 and the activation of TRIF-dependent signaling pathways. Auranofin and other gold compound suppress TLR signaling at multiple steps. These results provide an important clue to understand the mechanism for gold compounds to exert anti-rheumatic activity.
Acknowledgement
This work was supported by grants DK064007, DK41868 and CA75613 from the National Institutes of Heath, grant (2001-35200-10721) from the United States Department of Agriculture (USDA), grant (01A095Rev) from the American Institute for Cancer Research, and program funds from the Western Human Nutrition Research Center/ARS/USDA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
A list of non-standard abbreviations: TLR, Toll-like receptors; LPS, lipopolysaccharide; COX-2, cyclooxygenase-2; NFκB, nuclear factor κB; IRF-3, IFN-regulatory factor-3; MyD88, myeloid differential factor 88; TRIF, TIR domain-containing adapter inducing IKK, IκB kinase; TBK1, TANK-binding kinase 1; IRAK1, interleukin-1 receptor-associated kinase 1.
References
- [1].Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- [2].Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- [3].O'Neill LA. TLRs: Professor Mechnikov, sit on your hat. Trends Immunol. 2004;25:687–693. doi: 10.1016/j.it.2004.10.005. [DOI] [PubMed] [Google Scholar]
- [4].Vogel SN, Fitzgerald KA, Fenton MJ. TLRs: differential adapter utilization by toll-like receptors mediates TLR-specific patterns of gene expression. Mol Interv. 2003;3:466–477. doi: 10.1124/mi.3.8.466. [DOI] [PubMed] [Google Scholar]
- [5].Rhee SH, Hwang D. Murine TOLL-like receptor 4 confers lipopolysaccharide responsiveness as determined by activation of NF kappa B and expression of the inducible cyclooxygenase. J Biol Chem. 2000;275:34035–34040. doi: 10.1074/jbc.M007386200. [DOI] [PubMed] [Google Scholar]
- [6].Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- [7].Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- [8].Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, Tschopp J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. 2004;5:503–507. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- [9].Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. 2005;17:338–344. doi: 10.1016/j.coi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- [10].Andreakos E, Sacre S, Foxwell BM, Feldmann M. The toll-like receptor-nuclear factor kappaB pathway in rheumatoid arthritis. Front Biosci. 2005;10:2478–2488. doi: 10.2741/1712. [DOI] [PubMed] [Google Scholar]
- [11].Simon LS. DMARDs in the treatment of rheumatoid arthritis: current agents and future developments. Int J Clin Pract. 2000;54:243–249. [PubMed] [Google Scholar]
- [12].Andreakos ET, Foxwell BM, Brennan FM, Maini RN, Feldmann M. Cytokines and anti-cytokine biologicals in autoimmunity: present and future. Cytokine Growth Factor Rev. 2002;13:299–313. doi: 10.1016/s1359-6101(02)00018-7. [DOI] [PubMed] [Google Scholar]
- [13].Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- [14].Stern I, Wataha JC, Lewis JB, Messer RL, Lockwood PE, Tseng WY. Anti-rheumatic gold compounds as sublethal modulators of monocytic LPS-induced cytokine secretion. Toxicol In Vitro. 2005;19:365–371. doi: 10.1016/j.tiv.2004.11.001. [DOI] [PubMed] [Google Scholar]
- [15].Yanni G, Nabil M, Farahat MR, Poston RN, Panayi GS. Intramuscular gold decreases cytokine expression and macrophage numbers in the rheumatoid synovial membrane. Ann Rheum Dis. 1994;53:315–322. doi: 10.1136/ard.53.5.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bondeson J, Sundler R. Auranofin inhibits the induction of interleukin 1 beta and tumor necrosis factor alpha mRNA in macrophages. Biochem Pharmacol. 1995;50:1753–1759. doi: 10.1016/0006-2952(95)02030-6. [DOI] [PubMed] [Google Scholar]
- [17].Williams DH, Jeffery LJ, Murray EJ. Aurothioglucose inhibits induced NF-kB and AP-1 activity by acting as an IL-1 functional antagonist. Biochim Biophys Acta. 1992;1180:9–14. doi: 10.1016/0925-4439(92)90020-n. [DOI] [PubMed] [Google Scholar]
- [18].Yang JP, Merin JP, Nakano T, Kato T, Kitade Y, Okamoto T. Inhibition of the DNA-binding activity of NF-kappa B by gold compounds in vitro. FEBS Lett. 1995;361:89–96. doi: 10.1016/0014-5793(95)00157-5. [DOI] [PubMed] [Google Scholar]
- [19].Saitoh S, Akashi S, Yamada T, Tanimura N, Kobayashi M, Konno K, Matsumoto F, Fukase K, Kusumoto S, Nagai Y, Kusumoto Y, Kosugi A, Miyake K. Lipid A antagonist, lipid IVa, is distinct from lipid A in interaction with Toll-like receptor 4 (TLR4)-MD-2 and ligand-induced TLR4 oligomerization. Int Immunol. 2004;16:961–969. doi: 10.1093/intimm/dxh097. [DOI] [PubMed] [Google Scholar]
- [20].Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, Lee WH, Fitzgerald KA, Hwang DH. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem. 2004;279:16971–16979. doi: 10.1074/jbc.M312990200. [DOI] [PubMed] [Google Scholar]
- [21].Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278:37041–37051. doi: 10.1074/jbc.M305213200. [DOI] [PubMed] [Google Scholar]
- [22].Youn HS, Saitoh SI, Miyake K, Hwang DH. Inhibition of homodimerization of Toll-like receptor 4 by curcumin. Biochem Pharmacol. 2006;72:62–69. doi: 10.1016/j.bcp.2006.03.022. [DOI] [PubMed] [Google Scholar]
- [23].Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- [24].Kyo F, Futani H, Matsui K, Terada M, Adachi K, Nagata K, Sano H, Tateishi H, Tsutsui H, Nakanishi K. Endogenous interleukin-6, but not tumor necrosis factor alpha, contributes to the development of toll-like receptor 4/myeloid differentiation factor 88-mediated acute arthritis in mice. Arthritis Rheum. 2005;52:2530–2540. doi: 10.1002/art.21213. [DOI] [PubMed] [Google Scholar]
- [25].Roelofs MF, Boelens WC, Joosten LA, Abdollahi-Roodsaz S, Geurts J, Wunderink LU, Schreurs BW, van den Berg WB, Radstake TR. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J Immunol. 2006;176:7021–7027. doi: 10.4049/jimmunol.176.11.7021. [DOI] [PubMed] [Google Scholar]
- [26].Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, Joosten LA, Akira S, Netea MG, Adema GJ. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–494. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jeon KI, Byun MS, Jue DM. Gold compound auranofin inhibits IkappaB kinase (IKK) by modifying Cys-179 of IKKbeta subunit. Exp Mol Med. 2003;35:61–66. doi: 10.1038/emm.2003.9. [DOI] [PubMed] [Google Scholar]
- [28].Handel ML, Watts CK, deFazio A, Day RO, Sutherland RL. Inhibition of AP-1 binding and transcription by gold and selenium involving conserved cysteine residues in Jun and Fos. Proc Natl Acad Sci U S A. 1995;92:4497–4501. doi: 10.1073/pnas.92.10.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tao X, Xu Y, Zheng Y, Beg AA, Tong L. An extensively associated dimer in the structure of the C713S mutant of the TIR domain of human TLR2. Biochem Biophys Res Commun. 2002;299:216–221. doi: 10.1016/s0006-291x(02)02581-0. [DOI] [PubMed] [Google Scholar]
- [30].Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- [31].Jeon KI, Jeong JY, Jue DM. Thiol-reactive metal compounds inhibit NF-kappa B activation by blocking I kappa B kinase. J Immunol. 2000;164:5981–5989. doi: 10.4049/jimmunol.164.11.5981. [DOI] [PubMed] [Google Scholar]
- [32].Yamashita M, Ashino S, Oshima Y, Kawamura S, Ohuchi K, Takayanagi M. Inhibition of TPA-induced NF-kappaB nuclear translocation and production of NO and PGE2 by the anti-rheumatic gold compounds. J Pharm Pharmacol. 2003;55:245–251. doi: 10.1211/002235702513. [DOI] [PubMed] [Google Scholar]
- [33].Bratt J, Belcher J, Vercellotti GM, Palmblad J. Effects of anti-rheumatic gold salts on NF-kappa B mobilization and tumour necrosis factor-alpha (TNF-alpha)-induced neutrophil-dependent cytotoxicity for human endothelial cells. Clin Exp Immunol. 2000;120:79–84. doi: 10.1046/j.1365-2249.2000.01190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]