

Abstract
Background and Purpose
Animals subjected to an inflammatory insult at the time of stroke are predisposed to the development of an inflammatory autoimmune response to brain. This response is associated with worse neurological outcome. Because induction of immunologic tolerance to brain antigens before stroke onset is associated with improved outcome, we sought to determine whether this paradigm could prevent the deleterious autoimmune response to brain provoked by an inflammatory stimulus at the time of ischemia.
Methods
Male Lewis rats were tolerized to myelin basic protein (MBP) or ovalbumin by intranasal administration before middle cerebral artery occlusion. At the time of reperfusion, all animals received lipopolysaccharide (1 mg/kg intraperitoneal). Behavioral tests were performed at set time intervals.
Results
One month after middle cerebral artery occlusion, lymphocytes from the spleens of MBP-tolerized animals were less likely to evidence an autoimmune response and more likely to evidence a regulatory response (Treg) toward MBP than those from ovalbumin-tolerized animals. Animals that had an inflammatory response toward MBP (a Th1 response) performed worse on behavioral tests than those that did not. Fractalkine, a surrogate marker of inflammation, was elevated in animals with a Th1 response to MBP.
Conclusions
These data extend our previous findings and suggest that deleterious autoimmunity to brain antigens can be prevented by prophylactically inducing regulatory T-cell responses to those antigens.
Keywords: autoimmunity, mucosal tolerance, myelin basic protein, stroke, Th1, Treg
Recent data suggest that there is a systemic immunodepression after stroke.1 Teleologically, this immunodepression may be important for preventing autoimmune responses to brain given that the blood-brain barrier is compromised after stroke,2 which allows cells of the immune system to encounter novel brain antigens in the brain3,4 and the periphery.5-11 Another consequence of this immunodepression is enhanced susceptibility to infection,1,12 and studies show that patients in whom an infection develops in the immediate poststroke period have worse outcomes.13-15 Consistent with the premise of stroke-induced immunodepression, we previously showed that animals tend to evidence a regulatory immune response toward brain antigens after experimental ischemia and that development of a detrimental autoimmune response to brain is rare.16 When subjected to a systemic inflammatory insult with lipopolysaccharide (LPS) to mimic poststroke infection, however, a deleterious autoimmune response occurred in the majority of animals.16 This finding may help to explain why patients in whom an infection develops after stroke have worse outcomes.
Whereas an abundance of evidence suggests that inflammation contributes to postischemic brain injury, there are also data showing that induction of a regulatory immune response to brain antigens by mucosal administration of antigen before stroke can improve outcome.17-21 In the present study, we sought to determine whether induction of a regulatory immune response to the brain antigen myelin basic protein (MBP) before stroke onset could prevent the deleterious autoimmune response to MBP that follows a systemic inflammatory insult. We also assessed serological markers of the immune response, including antibodies to MBP and systemic levels of fractalkine, because the amount of this chemokine in circulation appears to correlate with disease activity in patients with autoimmune disorders.22-24
Materials and Methods
Animals
Experiments were approved by the Institution’s Animal Care and Use Committee. Male Lewis rats (250 to 300 grams; Charles River) were used for all studies. Rats were handled before tests/surgical procedures and housed 3 per cage to eliminate differences in socialization.
Tolerization Schedule
The experimental paradigm is outlined in Figure 1. Before sham surgery or stroke, either bovine MBP (100 μg/40 μL) or ovalbumin (OVA; 100 μg/40 μL) was instilled into each nostril every other day for a total of 5 doses; surgery was performed 1 to 2 days after the last instillation. Bovine MBP was chosen for the induction of tolerance because it can prevent experimental allergic encephalomyelitis25,26 and was previously shown to improve outcome in experimental models of stroke.20,21
Figure 1.
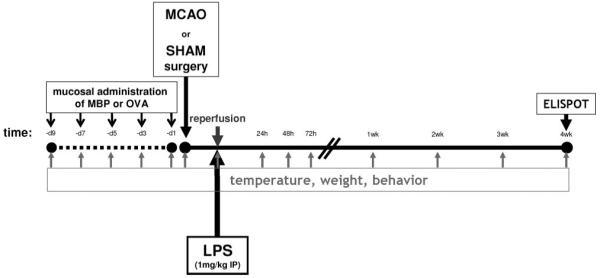
Experimental paradigm.
Middle Cerebral Artery Occlusion
Anesthesia was induced with 5% and maintained with 1.5% halothane. After midline neck incision, the right common carotid, internal carotid, and pterygopalatine arteries were ligated. A monofilament suture (4.0) was inserted into the common carotid artery and advanced into the internal carotid artery;27 38 OVA-tolerized and 28 MBP-tolerized animals were subjected to middle cerebral artery occlusion (MCAO). Animals were maintained at normothermia during surgery. Reperfusion was performed 3 hours after MCAO. In sham-operated animals (27 OVA-tolerized and 15 MBP-tolerized), the suture was inserted into the carotid but not advanced. Rectal temperature and body weight were assessed at set time intervals (Figure 1). Animals were euthanized 1 month after MCAO or sham surgery. All animals received LPS (serotype 026:B6; 1 mg/kg intraperitoneally) 3 hours after MCAO (at reperfusion) or sham surgery.
Neurological Outcome
Neurological status was assessed at set time points after MCAO; tests included a modification of the Bederson scale and an adaptation of the “sticky tape test.”28,29 Sensorimotor performance was assessed using a rotating drum or “rotarod.” Average time to fall (100 sec maximum) from a rod rotating at 5 rpm for 3 trials were recorded.
ELISPOT Assay
Animals were euthanized 1 month after MCAO/sham surgery and mononuclear cells were isolated from the brain and spleen using previously described methods.16,20 Mononuclear cells were cultured (1×105 cells/well) for 48 hours in 96-well plates (MultiScreen-IP; Millipore) in media alone or in media supplemented with MBP (25 μg/mL), OVA (25 μg/mL), or LPS (5 μg/mL). Lymphocyte reaction to whole MBP was used in these assays because the relevant epitope of MBP in this experimental scenario is unknown and unlikely to be that associated with encephalomyelitis. Furthermore, MBP is highly conserved among mammals and an autoimmune response to bovine MBP was previously shown to be associated with worse outcome in experimental stroke.16,30 All experiments were performed in triplicate. Antigen-specific secretion of interferon (IFN) γ (in comparison to unstimulated cells) was used as an indicator of the Th1 response; antigen-specific secretion of transforming growth factor (TGF) β1 was used as an indicator of the Treg response. Spots were counted under a dissecting microscope by 2 independent investigators blinded to treatment status. Results are expressed as the ratio of the increase in MBP-specific IFN-γ-secreting cells to the ratio of the increase in MBP-specific TGF-β1-secreting cells. Based on previous ELISPOT data from our laboratory, the lower interquartile range for this ratio in animals sensitized to MBP (by injection in complete Freund’s adjuvant) was 1.48. We thus considered having a Th1+ response to MBP if the ratio of the IFN:TGF response was ≥1.48; conversely, a Treg was considered to be induced if this ratio was ≤0.68 (ie, 1.00÷1.48).
Among animals undergoing MCAO, ELISPOT assay was performed on the brains of 17 OVA-tolerized and 16 MBP-tolerized animals. Among sham-operated animals, ELISPOT assay was performed on the brains of 16 OVA-tolerized and 15 MBP-tolerized animals. In some animals the brains were frozen at euthanization but ELISPOT analyses were still performed on spleen.
Enzyme-Linked Immunosorbent Assays
Blood was collected by cardiac puncture at the time of euthanization. Serum was stored at -80° until use. Fractalkine levels were assessed using a commercially available kit (R&D Systems). Titers of IgG antibodies specific for MBP were measured using indirect enzyme-linked immunosorbent assay. Data are presented as relative absorption units.
Statistics
Categorical data were evaluated using the χ2 test statistic. Parametric data were compared using the t test, paired t test, or ANOVA, and expressed as mean±SEM. Nonparametric data were compared using the Mann-Whitney U test. Significance was set at P ≤0.05.
Results
OVA-Tolerized Versus MBP-Tolerized Animals
Overall mortality after MCAO and LPS administration was 34.2% (13/38) in OVA-tolerized and 17.9% (5/28) in MBP-tolerized rats (not significant). Mortality in sham-operated animals that received LPS was 1 of 15 (6.7%) in MBP-tolerized animals and 0 of 27 OVA-tolerized animals (not significant).
Temperatures were similar in OVA-tolerized and MBP-tolerized animals at all time points after MCAO and sham surgery (data not shown).
Animals treated with MBP regained more weight over the course of the month after MCAO than animals treated with OVA (Figure 2a). Neurological scores tended to be lower (better) (supplemental Figure I, available online at http://stroke.ahajournals.org) and the time to notice adhesive tape on the paw was shorter in MBP-tolerized animals. MBP-tolerized animals also spent longer times on the rotarod before falling (Figure 2b). Sham-operated animals performed consistently better on all tests and there were no differences in the neurological performance of OVA-treated and MBP-treated sham-operated rats (data not shown).
Figure 2.
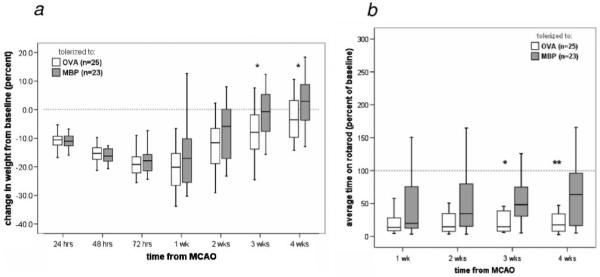
Effect of mucosal administration of OVA and MBP on outcome. Animals tolerized to MBP gained more weight over the month after MCAO than animals tolerized to OVA (a). MBP-treated animals performed better on the rotarod than OVA-tolerized animals (b). *P≤0.05, **P ≤0.01.
Difference Between the Immune Response in Brain and Spleen
A differential was seen in the ability of lymphocytes from brain and spleen to respond to mitogenic stimuli (ie, LPS) in vitro. Among a group of naive unmanipulated animals (n=5), fewer cells from brain than spleen responded to LPS by the secreting IFN-γ (15.7±3.7 per 1×105 vs 129.3±17.6 per 1×105; P=0.004). To account for differences in spontaneous secretion of IFN-γ by brain-derived lymphocytes and by splenocytes, the relative increase in the number of IFN-γ-secreting cells after LPS stimulation was assessed. Again, brain-derived lymphocytes seemed to respond less well than splenocytes to this mitogen (118%±20% vs 177%±22%; P=0.056). After sham-surgery, there was similar a differential in the percentage of brain-derived lymphocytes and splenocytes responding to LPS (86%±9% vs 131%±17%; P=0.040). After MCAO the response to LPS in spleen is enhanced. The difference in the percentage of cells from brain and spleen responding to LPS is thus more marked (101%±6% vs 214%±32%; P=0.001; Figure 3).
Figure 3.
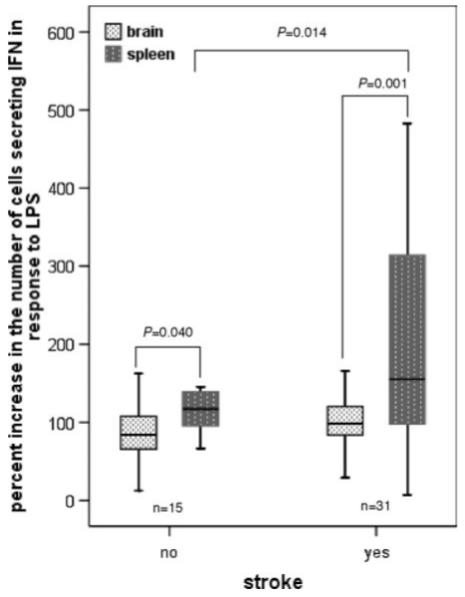
Mononuclear cells from brain are less able to respond to LPS. One month after surgery, the percentage increase in the number of cells responding to LPS by secreting IFN-γ (compared to the number of cells spontaneously secreting IFN-γ) is less in brain than spleen. This observation is true in both shamoperated and ischemic animals. Ischemia appears to enhance the ability of splenocytes to respond to LPS.
Cellular Immune Responses in OVA-Tolerized and MBP-Tolerized Animals
Immune responses could only be assessed in animals that survived. Sham-operated animals did not evidence an immune response to MBP. After MCAO, the ratio of IFN:TGF secretion by splenocytes in response to MBP was greater in OVA-tolerized than in MBP-tolerized animals (1.55±0.19 vs 0.83±0.07; P=0.001). The ratio of MBP-stimulated IFN:TGF secretion by brain-derived lymphocytes was similar (1.26±0.20 vs 0.86±0.08; P=0.076; Figure 4a,b). Animals with a Th1+ response to MBP in spleen also evidenced an enhanced humoral immune response to MBP with higher antibody titers to MBP than animals without a Th1+ response (P=0.001). Among animals with a Th1+ response to MBP in spleen, antibody titers were highly correlated with the intensity of the cellular immune response (Figure 4c).
Figure 4.
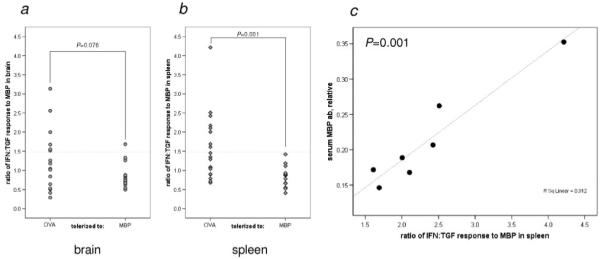
Immunologic response to MBP. Data represent individual animals and compare the ratio of the MBP-induced secretion of IFN:TGF in brain cells (a) and splenocytes (b) in OVA-tolerized and MBP-tolerized animals. Among animals with a Th1 response to MBP in spleen, there was a significant correlation between the degree of that response and the titer of anti-MBP antibodies (c). Antibody measurements were not available for 1 of the Th1+ animals.
Based on lymphocyte responses in spleen, 38% of animals treated with intranasal MBP before MCAO developed a Treg response to MBP, whereas 38% of OVA-tolerized animals had a Th1+ response to MBP (P<0.001). The percentage of MBP-tolerized and OVA-tolerized animals with Treg and Th1+ responses to MBP in brain were similar (Table). That the paradigm of mucosal tolerance can be used to create a Treg response to a given antigen is further illustrated by the fact that among sham-operated animals, 9 of 20 (45%) of animals treated with OVA had a Treg response to OVA in spleen whereas 0 of 9 MBP-treated sham-operated animals had a Treg response to OVA (P=0.015). After MCAO, the percentages of OVA-treated and MBP-treated animals with a Treg response to OVA in spleen were similar: 9 of 20 (45%) versus 1 of 15 (6.7%) (P=0.030). A Treg response to OVA was also seen in the brains of sham-operated OVA-treated but not MBP-treated animals: 6 of 16 (37.5%) versus 0 of 9 (P=0.013). After MCAO, the percentages of OVA-treated and MBP-treated animals with a Treg response to OVA in brain were 8 of 16 (50%) and 1 of 15 (6.7%), respectively (P=0.008).
Table. Immunologic Response to MBP in Brain Lymphocytes and Splenocytes 1 Month After MCAO.
| Brain-Derived Lymphocytes | OVA (N=17) | MBP (N=16) | P |
|---|---|---|---|
| No response | 6 (35%) | 10 (62%) | 0.084 |
| Th1+* | 6 (35%) | 1 (6%) | |
| Treg† | 5 (29%) | 5 (31%) | |
| Splenocytes | OVA (N=21) | MBP (N=16) | P |
| No response | 13 (62%) | 10 (62%) | <0.001 |
| Th1(+)* | 8 (38%) | 0 | |
| Treg† | 0 | 6 (38%) | |
Animals were considered to have a Th1+ response if the ratio of the number of MBP-reactive lymphocytes secreting IFN to those secreting TGF was ≥1.48.
Animals were considered to have a Treg response if the ratio of the number of MBP-reactive lymphocytes secreting IFN to those secreting TGF was ≤0.68.
The number of animals in which the spleen was assessed is greater than the number of animals in which the brain was assessed because the brains of some animals were frozen for immunohistochemistry.
Animals demonstrating a Th1+ response to MBP in brain had higher body temperatures 72 hours after MCAO than animals without a Th1+ response to MBP (37.8±0.1°C vs 37.0±0.1°C; P=0.009); animals with a Th1+ response to MBP in brain also regained weight more slowly than animals without (Figure 5a). Development of a Treg response did not influence weight gain (Figure 5b). Neurological scores after MCAO were similar in animals with and without a Th1+ response to MBP in brain (supplemental Figure I). Animals with a Th1+ response to MBP in brain, however, performed consistently worse on the rotarod (shorter times to fall) after MCAO (Figure 5c), whereas animals with a Treg response performed better on this task (Figure 5d). Furthermore, the degree of the immune response to MBP in brain correlated with rotarod performance: the more robust the response to MBP, the worse the performance (P=0.020). The effect of a Th1+ response to MBP in spleen was not predictive of outcome (supplemental Figure II).
Figure 5.
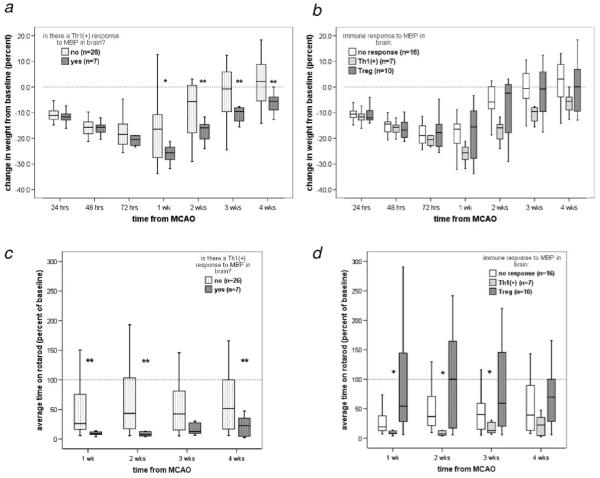
Immune response to MBP in brain and outcome. Animals that had a Th1+ response to MBP in brain regained weight more slowly than animals without a Th1+ response (a). Development of a Treg response to MBP did not affect the rate of weight gain (b). Animals with a Th1+ response to MBP in brain fell more quickly from the rotarod than those without (c); animals with a Treg response to MBP performed better than those with both a Th1+ response and no response (d). *P≤0.05, **P≤0.01.
Peripheral Markers of the Immune Response in Ischemic Animals
Fractalkine levels were higher in animals with a Th1+ response to MBP in brain than in those with no response to MBP (471.9±96.3 pg/mL vs 274.2±25.4 pg/mL; P=0.019). Fractalkine levels were similar in animals with a Th1+ response to MBP in spleen and those without (429.9±83.1 pg/mL vs 306.4±31.5 pg/mL), but among animals with a Th1+ response, serum fractalkine levels correlated to the degree of the cellular immune response to MBP (P=0.038).
Discussion
In previous studies we showed that mucosal administration of MBP before MCAO decreased infarct size and was associated with induction of a Treg response to MBP.20,21 The primary aim of the present study was to determine if the paradigm of mucosal tolerance could also be used to prevent a Th1 response to MBP in a model of stroke associated with a high prevalence of such an autoimmune response, specifically, MCAO with LPS administration.16 Mortality was high in OVA-tolerized animals, likely reflecting the severity of the stroke model (3 hours of MCAO and the concomitant dose of LPS). Because ELISPOT data are available only for surviving animals, the animals with the most severe strokes (and likely the most robust inflammatory responses) were eliminated from analysis by death. The high mortality in OVA-treated animals thus dilutes the effect of MBP treatment on the immunologic outcome. Nonetheless, we found that pre-ischemic induction of mucosal tolerance to MBP suppresses the Th1 response to MBP and increases the probability of a Th3, or Treg, response to this antigen. The present study also extends our previous findings that demonstrated that the paradigm of mucosal tolerance improves early neurological outcome after stroke by now showing that this improvement is sustained over the period of a month. Given that the neurological scores of OVA-tolerized and MBP-tolerized animals were similar immediately after MCAO, the difference in neurological outcome is likely attributable to difference in the immunologic status of these animals. The possibility that the pre-ischemic induction of a Treg response to MBP led to smaller infarcts, which in turn reduced the chances of a Th1 response to MBP must also be considered. The potential benefit to postischemic induction of a Treg response to MBP therefore needs to be addressed.
That the relationship between neurological outcome and the immunologic response in brain was more robust than in spleen is not surprising considering studies in animals with experimental allergic encephalomyelitis (EAE). In EAE, MBP reactive T cells are found in the central nervous system (CNS) of animals with clinical disease but are absent from the CNS during periods of recovery.31 Our findings similarly suggest that there is selective retention of MBP-reactive cells in the brains of animals with active CNS inflammation leading to worse behavioral outcomes in these animals. Unfortunately, the procedures for isolating lymphocytes from brain for the ELISPOT assay do not allow for simultaneous histological analyses. The presence of a Th1 response to MBP in spleen but not in brain in some animals suggests that an autoimmune response to brain occurred but that the autoreactive cells are not pathological in their present state of activation; however, it is possible that reactivation of these cells, either by another inflammatory stimulus or by ischemic insult, could lead to inflammation within the CNS.
A difference in the ability of lymphocytes isolated from the brain and spleen to respond to mitogenic, and presumably antigenic, stimuli in vitro was detected in this study. It is now widely appreciated that the supposed “immune privilege” of the brain is only relative and that the response of immune cells is regulated within this organ by a number of different mechanisms.32-36 One consequence of this endogenous immunoregulation is that lymphocytes in brain have a reduced capacity to produce IFN-γ and other proinflammatory cytokines.32,34,35 Teleologically, the CNS-mediated immunosuppression serves to prevent the development of CNS autoimmunity. As we have shown in previous studies, however, inflammation induced by systemic administration of LPS in the setting of cerebral ischemia can lead to autoimmune responses to brain; furthermore, animals that develop a Th1 response to brain (MBP) after MCAO have worse outcomes than those that do not have such a response.16 Using more detailed behavioral testing in this study, we again show a relationship between neurological outcome and the immune response to MBP after MCAO. This relationship is most apparent in animals evidencing a Th1 response to MBP in brain (as opposed to spleen). Furthermore, there was a relationship between the robustness of the response to MBP and performance on the rotarod, suggesting that the autoimmune response to brain is more than an epiphenomenon. In addition, we show that animals have a humoral immune response to MBP after stroke and the titers of anti-MBP antibodies correlate with the degree of the systemic cellular immune response to this antigen. Clinical studies demonstrate the development of antibodies to brain in patients after stroke, but the pathological consequences of this humoral response are not known.37-39
Fractalkine (CX3CL1) is a membrane-bound chemokine that is constitutively expressed on the surface of neurons; its expression can also be induced on astrocytes and endothelium.40,41 Because of its unique structure, fractalkine can be cleaved from the cell surface and released into the circulation;42 excitotoxins are just one of the many different stimuli that lead to cleavage of fractalkine from the neuronal surface.43 Cells expressing receptors for fractalkine (CX3CR1) include monocytes, lymphocytes, and microglia, and fractalkine is a potent chemoattractant for these cells. Based on these properties of fractalkine, a role for the chemokine has been suggested in a number of neuroinflammatory disorders. Because fractalkine is upregulated in neurons and endothelial cells within the ischemic penumbra after experimental stroke, it could potentially contribute to the postischemic inflammatory response.40 In fact, animals deficient in fractalkine experience better outcomes after cerebral ischemia/reperfusion, although the mechanisms by which the absence of fractalkine provides benefit is unknown.44
Our data show that circulating levels of fractalkine are higher in animals with a Th1+ response to MBP in brain, which would be consistent with increased CNS expression (and cleavage) of this chemokine in brain. The fact that Th1 cells preferentially express the fractalkine receptor, and thereby preferentially respond to fractalkine, serves to polarize and augment the immune response.45 In patients with autoimmune disorders, serum levels of fractalkine correlate with disease activity.22-24 Given that the cellular immune response to MBP in spleen and circulating fractalkine levels were correlated in our study, it suggests that this chemokine could also be used as a marker of ongoing inflammation in the brain. Further studies to compare the histological expression of fractalkine and the cellular immune response in brain (ie, influx of lymphocytes) with circulating fractalkine levels and the Th1 response to brain are warranted.
Because infection is common after stroke and associated with worse outcome, our model of poststroke inflammation (MCAO+LPS) is clinically relevant. LPS, a component of the Gram-negative bacterial cell wall, is a potent inflammatory stimulus that initiates the innate immune response through stimulation of Toll-like receptor-4.46 We used LPS to initiate the innate immune response because most poststroke infections are caused by Gram-negative organisms.15 The ability of other inflammatory stimuli (which initiate inflammation through activation of other Toll-like receptors) to precipitate an autoimmune response after stroke needs to be investigated.
In these experiments we focused on the immune response to MBP, both as potentially damaging (Th1) and beneficial (Treg). It is possible that similar immunologic responses occur to other CNS antigens. Despite the fact that regulatory T cells are activated in an antigen-specific fashion, they secrete cytokines that influence the immune response in an antigen nonspecific fashion.47 This fact leads to the phenomenon known as “bystander suppression” and allows for induction of a therapeutic immunomodulatory response even when the pathogenic antigen leading to a harmful autoimmune response is unknown.48,49 Our findings may explain why infection is associated with worse outcome from stroke and suggest a possible therapeutic approach to limit the immunologic consequences of the infection.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by grants from the National Institutes of Neurological Disorders and Stroke (NINDS) (1RO1NS056457) and the American Heart Association Pacific Mountain Affiliate (0455505Z).
Footnotes
Disclosures
None.
References
- 1.Prass K, Meisel C, Hoflich C, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U, Volk HD, Meisel A. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke t helper cell type 1-like immunostimulation. J Exp Med. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neumann-Haefelin T, Kastrup A, de Crespigny A, Yenari MA, Ringer T, Sun GH, Moseley ME. Serial MRI after transient focal cerebral ischemia in rats: Dynamics of tissue injury, blood-brain barrier damage, and edema formation. Stroke. 2000;31:1965–1972. doi: 10.1161/01.str.31.8.1965. [DOI] [PubMed] [Google Scholar]
- 3.Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol. 1994;55:195–203. doi: 10.1016/0165-5728(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 4.Jander S, Kraemer M, Schroeter M, Witte OW, Stoll G. Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J Cereb Blood Flow Metab. 1995;15:42–51. doi: 10.1038/jcbfm.1995.5. [DOI] [PubMed] [Google Scholar]
- 5.Herrmann M, Vos P, Wunderlich MT, de Bruijn CH, Lamers KJ. Release of glial tissue-specific proteins after acute stroke: A comparative analysis of serum concentrations of protein s-100b and glial fibrillary acidic protein. Stroke. 2000;31:2670–2677. doi: 10.1161/01.str.31.11.2670. [DOI] [PubMed] [Google Scholar]
- 6.Wunderlich MT, Ebert AD, Kratz T, Goertler M, Jost S, Herrmann M. Early neurobehavioral outcome after stroke is related to release of neurobiochemical markers of brain damage. Stroke. 1999;30:1190–1195. doi: 10.1161/01.str.30.6.1190. [DOI] [PubMed] [Google Scholar]
- 7.Fassbender K, Schmidt R, Schreiner A, Fatar M, Muhlhauser F, Daffertshofer M, Hennerici M. Leakage of brain-originated proteins in peripheral blood: Temporal profile and diagnostic value in early ischemic stroke. J Neurol Sci. 1997;148:101–105. doi: 10.1016/s0022-510x(96)05351-8. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Wang X, Yang Z. Neuron-specific enolase in patients with acute ischemic stroke and related dementia. Chin Med J (Engl) 1995;108:221–223. [PubMed] [Google Scholar]
- 9.Abraha HD, Butterworth RJ, Bath PM, Wassif WS, Garthwaite J, Sherwood RA. Serum s-100 protein, relationship to clinical outcome in acute stroke. Ann Clin Biochem. 1997;34:366–370. doi: 10.1177/000456329703400405. [DOI] [PubMed] [Google Scholar]
- 10.Buttner T, Weyers S, Postert T, Sprengelmeyer R, Kuhn W. S-100 protein: Serum marker of focal brain damage after ischemic territorial mca infarction. Stroke. 1997;28:1961–1965. doi: 10.1161/01.str.28.10.1961. [DOI] [PubMed] [Google Scholar]
- 11.Pleines UE, Morganti-Kossmann MC, Rancan M, Joller H, Trentz O, Kossmann T. S-100 beta reflects the extent of injury and outcome, whereas neuronal specific enolase is a better indicator of neuroinflammation in patients with severe traumatic brain injury. J Neurotrauma. 2001;18:491–498. doi: 10.1089/089771501300227297. [DOI] [PubMed] [Google Scholar]
- 12.Prass K, Braun JS, Dirnagl U, Meisel C, Meisel A. Stroke propagates bacterial aspiration to pneumonia in a model of cerebral ischemia. Stroke. 2006;37:2607–2612. doi: 10.1161/01.STR.0000240409.68739.2b. [DOI] [PubMed] [Google Scholar]
- 13.Aslanyan S, Weir CJ, Diener HC, Kaste M, Lees KR. Pneumonia and urinary tract infection after acute ischaemic stroke: A tertiary analysis of the gain international trial. Eur J Neurol. 2004;11:49–53. doi: 10.1046/j.1468-1331.2003.00749.x. [DOI] [PubMed] [Google Scholar]
- 14.Langhorne P, Stott DJ, Robertson L, MacDonald J, Jones L, McAlpine C, Dick F, Taylor GS, Murray G. Medical complications after stroke: A multicenter study. Stroke. 2000;31:1223–1229. doi: 10.1161/01.str.31.6.1223. [DOI] [PubMed] [Google Scholar]
- 15.Hilker R, Poetter C, Findeisen N, Sobesky J, Jacobs A, Neveling M, Heiss WD. Nosocomial pneumonia after acute stroke: Implications for neurological intensive care medicine. Stroke. 2003;34:975–981. doi: 10.1161/01.STR.0000063373.70993.CD. [DOI] [PubMed] [Google Scholar]
- 16.Becker KJ, Kindrick DL, Lester MP, Shea C, Ye ZC. Sensitization to brain antigens after stroke is augmented by lipopolysaccharide. J Cereb Blood Flow Metab. 2005;25:1634–1644. doi: 10.1038/sj.jcbfm.9600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Ruetzler C, Pandipati S, Spatz M, McCarron RM, Becker K, Hallenbeck JM. Mucosal tolerance to e-selectin provides cell-mediated protection against ischemic brain injury. Proc Natl Acad Sci U S A. 2003;100:15107–15112. doi: 10.1073/pnas.2436538100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeda H, Spatz M, Ruetzler C, McCarron R, Becker K, Hallenbeck J. Induction of mucosal tolerance to e-selectin prevents ischemic and hemorrhagic stroke in spontaneously hypertensive genetically stroke-prone rats. Stroke. 2002;33:2156–2163. doi: 10.1161/01.str.0000029821.82531.8b. [DOI] [PubMed] [Google Scholar]
- 19.Frenkel D, Huang Z, Maron R, Koldzic DN, Moskowitz MA, Weiner HL. Neuroprotection by il-10-producing MOG CD4+ t cells following ischemic stroke. J Neurol Sci. 2005;233:125–132. doi: 10.1016/j.jns.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 20.Becker K, Kindrick D, McCarron R, Hallenbeck J, Winn R. Adoptive transfer of myelin basic protein-tolerized splenocytes to naive animals reduces infarct size: A role for lymphocytes in ischemic brain injury? Stroke. 2003;34:1809–1815. doi: 10.1161/01.STR.0000078308.77727.EA. [DOI] [PubMed] [Google Scholar]
- 21.Becker KJ, McCarron RM, Ruetzler C, Laban O, Sternberg E, Flanders KC, Hallenbeck JM. Immunologic tolerance to myelin basic protein decreases stroke size after transient focal cerebral ischemia. Proc Natl Acad Sci U S A. 1997;94:10873–10878. doi: 10.1073/pnas.94.20.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsunawa M, Isozaki T, Odai T, Yajima N, Takeuchi HT, Negishi M, Ide H, Adachi M, Kasama T. Increased serum levels of soluble fractalkine (cx3cl1) correlate with disease activity in rheumatoid vasculitis. Arthritis Rheum. 2006;54:3408–3416. doi: 10.1002/art.22208. [DOI] [PubMed] [Google Scholar]
- 23.Yajima N, Kasama T, Isozaki T, Odai T, Matsunawa M, Negishi M, Ide H, Kameoka Y, Hirohata S, Adachi M. Elevated levels of soluble fractalkine in active systemic lupus erythematosus: Potential involvement in neuropsychiatric manifestations. Arthritis Rheum. 2005;52:1670–1675. doi: 10.1002/art.21042. [DOI] [PubMed] [Google Scholar]
- 24.Kastenbauer S, Koedel U, Wick M, Kieseier BC, Hartung HP, Pfister HW. Csf and serum levels of soluble fractalkine (cx3cl1) in inflammatory diseases of the nervous system. J Neuroimmunol. 2003;137:210–217. doi: 10.1016/s0165-5728(03)00085-7. [DOI] [PubMed] [Google Scholar]
- 25.Miller A, Lider O, al-Sabbagh A, Weiner HL. Suppression of experimental autoimmune encephalomyelitis by oral administration of myelin basic protein. V. Hierarchy of suppression by myelin basic protein from different species. J Neuroimmunol. 1992;39:243–250. doi: 10.1016/0165-5728(92)90258-m. [DOI] [PubMed] [Google Scholar]
- 26.al-Sabbagh AM, Goad EP, Weiner HL, Nelson PA. Decreased CNS inflammation and absence of clinical exacerbation of disease after six months oral administration of bovine myelin in diseased SJL/J mice with chronic relapsing experimental autoimmune encephalomyelitis. J Neurosci Res. 1996;45:424–429. doi: 10.1002/(SICI)1097-4547(19960815)45:4<424::AID-JNR11>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 27.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 28.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: Evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- 29.Schallert T, Whishaw IQ. Bilateral cutaneous stimulation of the somato-sensory system in hemidecorticate rats. Behav Neurosci. 1984;98:518–540. doi: 10.1037//0735-7044.98.3.518. [DOI] [PubMed] [Google Scholar]
- 30.Boggs JM. Myelin basic protein: A multifunctional protein. Cell Mol Life Sci. 2006;63:1945–1961. doi: 10.1007/s00018-006-6094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hofstetter HH, Targoni OS, Karulin AY, Forsthuber TG, Tary-Lehmann M, Lehmann PV. Does the frequency and avidity spectrum of the neuroantigen-specific t cells in the blood mirror the autoimmune process in the central nervous system of mice undergoing experimental allergic encephalomyelitis? J Immunol. 2005;174:4598–4605. doi: 10.4049/jimmunol.174.8.4598. [DOI] [PubMed] [Google Scholar]
- 32.Taylor AW, Streilein JW. Inhibition of antigen-stimulated effector t cells by human cerebrospinal fluid. Neuroimmunomodulation. 1996;3:112–118. doi: 10.1159/000097235. [DOI] [PubMed] [Google Scholar]
- 33.Irani DN. Brain-derived gangliosides induce cell cycle arrest in a murine T cell line. J Neuroimmunol. 1998;87:11–16. doi: 10.1016/s0165-5728(98)00038-1. [DOI] [PubMed] [Google Scholar]
- 34.Irani DN, Lin KI, Griffin DE. Brain-derived gangliosides regulate the cytokine production and proliferation of activated t cells. J Immunol. 1996;157:4333–4340. [PubMed] [Google Scholar]
- 35.Trajkovic V, Vuckovic O, Stosic-Grujicic S, Miljkovic D, Popadic D, Markovic M, Bumbasirevic V, Backovic A, Cvetkovic I, Harhaji L, Ramic Z, Mostarica Stojkovic M. Astrocyte-induced regulatory T cells mitigate CNS autoimmunity. Glia. 2004;47:168–179. doi: 10.1002/glia.20046. [DOI] [PubMed] [Google Scholar]
- 36.Gimsa U, A OR, Pandiyan P, Teichmann D, Bechmann I, Nitsch R, Brunner-Weinzierl MC. Astrocytes protect the CNS: Antigen-specific t helper cell responses are inhibited by astrocyte-induced upregulation of ctla-4 (cd152) J Mol Med. 2004;82:364–372. doi: 10.1007/s00109-004-0531-6. [DOI] [PubMed] [Google Scholar]
- 37.Bornstein NM, Aronovich B, Korczyn AD, Shavit S, Michaelson DM, Chapman J. Antibodies to brain antigens following stroke. Neurology. 2001;56:529–530. doi: 10.1212/wnl.56.4.529. [DOI] [PubMed] [Google Scholar]
- 38.Gromadzka G, Zielinska J, Ryglewicz D, Fiszer U, Czlonkowska A. Elevated levels of anti-heat shock protein antibodies in patients with cerebral ischemia. Cerebrovasc Dis. 2001;12:235–239. doi: 10.1159/000047709. [DOI] [PubMed] [Google Scholar]
- 39.Dambinova SA, Khounteev GA, Izykenova GA, Zavolokov IG, Ilyukhina AY, Skoromets AA. Blood test detecting autoantibodies to n-methyl-d-aspartate neuroreceptors for evaluation of patients with transient ischemic attack and stroke. Clin Chem. 2003;49:1752–1762. doi: 10.1373/49.10.1752. [DOI] [PubMed] [Google Scholar]
- 40.Tarozzo G, Campanella M, Ghiani M, Bulfone A, Beltramo M. Expression of fractalkine and its receptor, cx3cr1, in response to ischaemia-reperfusion brain injury in the rat. Eur J Neurosci. 2002;15:1663–1668. doi: 10.1046/j.1460-9568.2002.02007.x. [DOI] [PubMed] [Google Scholar]
- 41.Tarozzo G, Bortolazzi S, Crochemore C, Chen SC, Lira AS, Abrams JS, Beltramo M. Fractalkine protein localization and gene expression in mouse brain. J Neurosci Res. 2003;73:81–88. doi: 10.1002/jnr.10645. [DOI] [PubMed] [Google Scholar]
- 42.Hundhausen C, Schulte A, Schulz B, Andrzejewski MG, Schwarz N, von Hundelshausen P, Winter U, Paliga K, Reiss K, Saftig P, Weber C, Ludwig A. Regulated shedding of transmembrane chemokines by the disintegrin and metalloproteinase 10 facilitates detachment of adherent leukocytes. J Immunol. 2007;178:8064–8072. doi: 10.4049/jimmunol.178.12.8064. [DOI] [PubMed] [Google Scholar]
- 43.Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci. 2000;20:RC87. doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soriano SG, Amaravadi LS, Wang YF, Zhou H, Yu GX, Tonra JR, Fairchild-Huntress V, Fang Q, Dunmore JH, Huszar D, Pan Y. Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J Neuroimmunol. 2002;125:59–65. doi: 10.1016/s0165-5728(02)00033-4. [DOI] [PubMed] [Google Scholar]
- 45.Fraticelli P, Sironi M, Bianchi G, D’Ambrosio D, Albanesi C, Stop-pacciaro A, Chieppa M, Allavena P, Ruco L, Girolomoni G, Sinigaglia F, Vecchi A, Mantovani A. Fractalkine (cx3cl1) as an amplification circuit of polarized th1 responses. J Clin Invest. 2001;107:1173–1181. doi: 10.1172/JCI11517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 47.Weiner HL. Induction and mechanism of action of transforming growth factor-beta-secreting th3 regulatory cells. Immunol Rev. 2001;182:207–214. doi: 10.1034/j.1600-065x.2001.1820117.x. [DOI] [PubMed] [Google Scholar]
- 48.Teng YT, Gorczynski RM, Hozumi N. The function of TGF-beta-mediated innocent bystander suppression associated with physiological self-tolerance in vivo. Cell Immunol. 1998;190:51–60. doi: 10.1006/cimm.1998.1389. [DOI] [PubMed] [Google Scholar]
- 49.Lundin BS, Dahlgren UI, Hanson LA, Telemo E. Oral tolerization leads to active suppression and bystander tolerance in adult rats while anergy dominates in young rats. Scand J Immunol. 1996;43:56–63. doi: 10.1046/j.1365-3083.1996.d01-15.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.