

Abstract
Inhalation is a common form of exposure to acrolein, a toxic reactive volatile aldehyde that is a ubiquitous environmental pollutant. Bronchial epithelial cells would be directly exposed to inhaled acrolein. The thioredoxin (Trx) system is essential for the maintenance of cellular thiol redox balance, and is critical for cell survival. Normally, thioredoxin reductase (TrxR) maintains the cytosolic (Trx1) and mitochondrial (Trx2) thioredoxins in the reduced state, and the thioredoxins keep the peroxiredoxins (Prx) reduced, thereby supporting their peroxidase function. The effects of acrolein on TrxR, Trx and Prx in human bronchial epithelial (BEAS-2B) cells were determined. A 30-min exposure to 5 μM acrolein oxidized both Trx1 and Trx2, although significant effects were noted for Trx1 at even lower acrolein concentrations. The effects on Trx1 and Trx2 could not be reversed by treatment with disulfide reductants. TrxR activity was inhibited 60% and >85% by 2.5 and 5 μM acrolein, respectively. The endogenous electron donor for TrxR, NADPH, could not restore its activity, and activity did not recover in cells during a 4-hr acrolein-free period in complete medium. The effects of acrolein on TrxR and Trx therefore extend beyond the duration of exposure. While there was a strong correlation between TrxR inhibition and Trx1 oxidation, the irreversible effects on Trx1 suggest direct effects of acrolein rather than loss of reducing equivalents from TrxR. Trx2 did not become oxidized until ≥90% of TrxR was inhibited, but irreversible effects on Trx2 also suggest direct effects of acrolein. Prx1 (cytosolic) and Prx3 (mitochondrial) shifted to a largely oxidized state only when >90 and 100% of their respective Trxs were oxidized. Prx oxidation was readily reversed with a disulfide reductant, suggesting that Prx oxidation resulted from lack of reducing equivalents from Trx and not direct reaction with acrolein. The effects of acrolein on the thioredoxin system and peroxiredoxins could have important implications for cell survival, redox-sensitive cell signaling, and tolerance to other oxidant insults.
Keywords: Acrolein, Peroxiredoxin-1, Peroxiredoxin-3, Thioredoxin, Thioredoxin Reductase, BEAS-2B cells
1. Introduction
Acrolein is a ubiquitous environmental pollutant, and inhalation is a prominent form of exposure. Major environmental sources include exhaust from internal combustion engines (Beauchamp et al., 1985) and wood combustion. Smoke from burning buildings and vegetative fires can have acrolein levels that are considered acutely dangerous (Beauchamp et al., 1985; Ghilarducci and Tjeerdema, 1995). Photochemical oxidation of airborne hydrocarbons also generates acrolein (Ghilarducci and Tjeerdema, 1995). Large amounts of acrolein are used as an aquatic pesticide and herbicide, and occupations with significant potential exposure include the manufacture of acrylate polymers, foundry operations, welding coated metals, coffee roasting, printing, linoleum production, tin plating, and rubber vulcanization (Beauchamp et al., 1985; Ghilarducci and Tjeerdema, 1995). Cigarette smoke is a major contributor to various lung diseases, and acrolein is a prominent component of the gas phase of cigarette smoke (Beauchamp et al., 1985), with 85–228 μg acrolein per cigarette (Esterbauer et al., 1991; Ghilarducci and Tjeerdema, 1995; Carnevali et al., 1998). Some of the inhaled acrolein can also be absorbed and distributed, causing systemic effects (Uchida et al., 1998a; Uchida et al., 1998b; Uchida, 2000; Barnoya and Glantz, 2005). Bronchial epithelial cells are directly exposed to inhaled acrolein and would therefore receive the highest doses. It is important to understand the effects of acrolein in these cells.
Acrolein is highly toxic to many cells including human bronchial epithelial cells and fibroblasts (Esterbauer et al., 1991), neurons (Lovell et al., 2001), vascular smooth muscle cells (Conklin et al., 1998), and endothelial cells (Patel and Block, 1993; Kachel and Martin, 1994; Szadkowski and Myers, 2007). Depending on the cell type, acrolein is 10–200 times more toxic than formaldehyde (Esterbauer et al., 1991; Conklin et al., 1998). Acrolein is more toxic than hydroxynonenal (4-HNE) (Lovell et al., 2001), and 100–150 times more reactive than 4-HNE (Esterbauer et al., 1991). Because it is a strong electrophile, acrolein can react nonenzymatically with nucleophiles including sulfhydryl ( SH) groups (Uchida et al., 1998b). This could have significant consequences for sulfhydryl-containing proteins and could disrupt the intracellular thiol redox equilibrium.
The maintenance of cellular thiol redox balance is critical for normal function and cell survival. Glutathione and the thioredoxins are major players in this maintenance, although these systems are not in redox equilibrium with each other (Nkabyo et al., 2002; Hansen et al., 2006a). The redox status of the thioredoxin (Trx) system may be more critical to cell survival than is glutathione (Hansen et al., 2006b). All mammalian cells have cytosolic (Trx1) and mitochondrial (Trx2) thioredoxins (Powis and Montfort, 2001). Trx1 and Trx2 are encoded by distinct genes, but they share a conserved active site (Trp-Cys-Gly-Pro-Cys-Lys) that is cycled between the reduced (dithiol) and oxidized (disulfide) forms (Watson et al., 2003). Trx2 has two Cys residues, both in the active site. Trx1 has five Cys residues, two in the active site (C32/C35), and another two in a second dithiol motif (C62/C69) (Watson et al., 2003). Of the two Trx1 dithiols, the C32/C35 active site is more readily oxidized (Watson et al., 2003). Cells normally maintain Trx1 and Trx2 largely in the reduced state (Nordberg and Arner, 2001; Powis and Montfort, 2001; Watson et al., 2003).
A major role of thioredoxins is to maintain the cysteine residues of intracellular proteins in a reduced state (Arner and Holmgren, 2000). In this way, they support the function of a variety of proteins including ribonucleotide reductase, protein disulfide isomerase, peroxiredoxins, and many others (Nordberg and Arner, 2001; Powis and Montfort, 2001). Peroxiredoxins (Prxs) are ubiquitous peroxidases that contain Cys at their active site. Peroxide substrates such as H2O2 and organic hydroperoxides oxidize the Prx active site Cys to sulfenic acid ( SOH). The 2-Cys Prxs (e.g. cytosolic Prx1 and Prx2, and mitochondrial Prx3) are homodimers in which this sulfenic acid reacts with the resolving Cys on the other subunit to form a disulfide-linked dimer (Wood et al., 2003a; Wood et al., 2003b; Peskin et al., 2007). The Trxs reduce these disulfides to regenerate the active Prx (Kim et al., 2005). The Trxs are therefore critical to supporting the peroxidase function of Prxs (Nordberg and Arner, 2001; Powis and Montfort, 2001).
In cells, the Trxs are kept reduced (active) by Trx reductases (TrxRs) (Powis and Montfort, 2001). Trxs are therefore directly dependent on TrxRs. Through Trxs, Prxs are also dependent on TrxRs. TrxR1 (cytosolic) and TrxR2 (mitochondrial) are NADPH-dependent homodimers (Nordberg and Arner, 2001; Powis and Montfort, 2001). They contain selenium (Se) as SeCys within the C-terminal Gly-Cys-SeCys-Gly active site (Nordberg and Arner, 2001).
TrxRs, Trxs, and Prxs are all important for cellular growth and survival. Knockout mice lacking either Trx1 or Trx2 do not survive (Powis and Montfort, 2001; Nonn et al., 2003). Inhibition or genetic suppression of Trx results in increased oxidant stress and apoptosis (Hansen et al., 2006a) and increased sensitivity to oxidants (Chen et al., 2006). Conversely, Trx overexpression enhances resistance to oxidant-induced apoptosis (Chen et al., 2006; Hansen et al., 2006a). TrxR inhibition increases oxidant susceptibility and favors apoptosis (Nordberg and Arner, 2001). Similarly, depletion of Prx3 enhances susceptibility to apoptotic insults (Chang et al., 2004). Overall, factors which oxidize or inhibit one or more of these proteins could decrease cell survival.
Initial studies noted that acrolein inhibited the activity of Trx1 in vitro (Yang et al., 2004), and caused a dose-dependent decline in Trx activity in A549 cancer cells, with maximal inhibition after 30 min with ≥25 μM (Yang et al., 2004). However, neither the specific Trx(s) affected or the redox status of the Trxs were determined (Yang et al., 2004). Subsequent studies with human endothelial cells showed a much greater sensitivity to acrolein with 5 μM sufficient to oxidize both Trx1 and Trx2 (Szadkowski and Myers, 2007). Trx1 was sensitive to even lower concentrations (Szadkowski and Myers, 2007). Go et al. (2007) noted that adduction of acrolein to Cys73 of Trx1 can inhibit Trx1 activity and change the redox state of Trx1 in bovine endothelial cells.
Relatively high doses of acrolein have been shown to inhibit TrxR activity in cells, e.g. 50–75 μM for 30 min caused ~65% inhibition in A549 cells (Yang et al., 2004), whereas 25 μM for 30 min inhibited 88% of TrxR activity in human umbilical vein endothelial cells (HUVECs) (Park et al., 2005). No studies to date have examined the effects of acrolein on Prxs.
The effects of acrolein on one or more of these proteins in cells may be direct, whereas effects on others may be indirect, e.g. inhibition of TrxR could result in an inability to keep the Trxs and Prxs reduced. While previous papers have looked at TrxR and Trxs in different systems, none have examined the coordinated effects on TrxR, Trxs, and Prxs within the same system. Furthermore, none have examined the effects of acrolein on these proteins in normal bronchial epithelium, which would receive the highest dose of inhaled acrolein in vivo.
This paper examines the effects of low micromolar concentrations of acrolein on the activity of TrxR and on the redox status of Trxs and Prxs in human bronchial epithelial cells. All three protein types are oxidized/inhibited by ≥5 μM acrolein, with significant effects on TrxR and Trx1 at even lower concentrations. The effects on TrxR and Trx were consistent with acrolein-protein adducts: they were not reversible in vitro, and TrxR activity did not recover in cells after acrolein removal. In contrast, the oxidation of Prxs did not occur until nearly all of the respective Trx was oxidized, and Prx oxidation was reversible in vitro by disulfide reduction. The effects of acrolein on TrxR and Trx may therefore result from direct effects of acrolein, whereas Prx oxidation likely results from loss of function of the upstream Trxs.
2. Materials and methods
2.1 Reagents
The following were purchased from Invitrogen Corp. (Carlsbad, CA): Hank’s balanced salt solution (HBSS), 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonate (AMS), and pre-cast gels (12% Bis-Tris, 16% Tris-Glycine) and matching electrophoresis and loading buffers. Phenylmethylsulfonyl fluoride (PMSF) and Tris were from Research Organics (Cleveland, OH). EDTA, guanidine-HCl, and trichloroacetic acid (TCA) were obtained from Fisher Scientific (Hampton, NH). Primary antibodies specific for Trx1, Trx2, TrxR1, Prx1, Prx3, and Prx-SO3 were obtained from Abcam (Cambridge, MA). The Prx-SO3 antibody recognizes the over oxidized (sulfinic and sulfonic acid) forms of Prx-1, Prx-2, Prx-3, and Prx-4. HRP-conjugated secondary antibodies were purchased from Bio-Rad (Hercules, CA) or Promega (Madison, WI). The Supersignal West Pico Chemiluminescent substrate was obtained from Pierce (Rockford, IL). Sodium chromate (Na2CrO4; 99+%) was the highest purity available from Aldrich Chemical (Milwaukee, WI). Chromates are known carcinogens and should be handled accordingly. Purified rat liver thioredoxin reductase (TrxR1) was from Sigma Chemical (St. Louis, MO). Acrolein and all other chemicals and reagents were purchased from Sigma Chemical, or from the sources indicated. Acrolein is volatile, potentially toxic, and has a low flash point. It was stored under nitrogen gas in a flammable-materials storage refrigerator, and was handled in a ventilated fume hood to prevent inhalational exposure.
2.2 Cell culture
BEAS-2B cells were obtained from the American Type Culture Collection (ATCC no. CRL-9609). They were grown at 37°C in humidified air containing 5% CO2 in Dulbecco's Modified Eagle's Medium (DMEM) with 25 mM HEPES and 4.5 g/L glucose (BioWhittaker 12-709F, Lonza, Inc.), 10% fetal bovine serum (Valley Biomedical, Winchester, VA), penicillin (100 U/ml), and streptomycin (100 μg/ml). The cells were fed every 48 h, and were subcultured prior to reaching confluence using the Reagent Pak system (Clonetics, CC-5034). Normal plating density was 3000 to 5000 cells/cm2.
2.3 Redox blots for thioredoxins and peroxiredoxins
Cells were grown to 70–90% of confluence (typically in T-75 or T-25 flasks), washed once in pre-warmed HBSS, and treated with acrolein in HBSS at 37°C for the indicated times. Untreated cells were exposed to acrolein without HBSS. Following treatment, the cells were washed once in HBSS and immediately processed for redox blots. The redox status of Trx1 was determined as adapted from (Halvey et al., 2005). Adherent cells (60–70% confluent) were washed once in pre-warmed HBSS, and treated with acrolein in HBSS at 37°C for the indicated times. Following treatment, they were washed once in HBSS and scraped into chilled 6 M guanidine-HCl, 100 mM Tris-HCl pH 8.3, 3 mM EDTA, 0.5 % Triton X-100, 50 mM iodoacetic acid. The samples were briefly sonicated on ice to shear the DNA, and guanidine and unreacted iodoacetate were removed using Microspin G25 columns (GE Healthcare, Piscataway NJ). The column eluates were mixed with an equal volume of loading buffer, separated by native PAGE (16% Tris-Glycine), transferred to nitrocellulose, blocked with 5% bovine serum albumin (BSA) and probed with anti-Trx1 and HRP-conjugated goat anti-rabbit IgG (Bio-Rad, Hercules, CA).
Trx2 redox status was determined according to (Chen et al., 2006). Cells were washed and treated with Cr(VI) as for Trx1 studies (above), but were then scraped into 10% TCA. Following incubation on ice for ≥30 min, they were briefly sonicated on ice, and centrifuged for 10 min at 4°C (12000 x g). The pellets were resuspended in ice-cold 100% acetone, iced for 30 min, and centrifuged for 15 min (12000 x g). The pellets were dried and then resuspended in 80 mM Tris-HCl pH 7, 2% SDS, 1 mM PMSF, 25 mM AMS. After incubation (15 min on ice, 10 min at 37°C) (Chen et al., 2006), the samples were centrifuged for 5 min (12000 x g), mixed with LDS (lithium dodecyl sulfate) loading buffer and separated by SDS-PAGE (12% Bis-Tris) under non-reducing conditions, and transferred to nitrocellulose. The blots were blocked with 5% milk (Carnation nonfat) and probed with anti-Trx2 and HRP-conjugated goat anti-rabbit IgG. The isolation of mitochondria and their use as standards for oxidized and reduced Trx2 has been previously described (Szadkowski and Myers, 2007).
Redox western blots for Prx1 and Prx3 were done according to the method of Cox et al. (Cox et al., 2008). After acrolein treatment, the cells were washed in HBSS and scraped into the following buffer: 40 mM HEPES pH 7.4, 50 mM NaCl, 100 mM N-ethylmaleimide (NEM), 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, and 10 μg/ml catalase. Following a 15-min incubation at room temperature, the cells were lysed by adding CHAPS (3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate) to 1%. The supernatants were run on non-reducing SDS-PAGE, and the blots were probed with anti-Prx1 or anti-Prx3.
2.4 TrxR activity
TrxR activity was measured as the NADPH-dependent reduction of DTNB (5,5′-dithiobis(2-nitrobenzoic) acid) (Holmgren, 1977; Arner et al., 1995), except that the assay was done at 37°C rather than room temperature. The sample to be tested was pre-incubated (10 min 37°C) in buffer (0.1 M sodium phosphate pH 7.4, 5 mM EDTA) for 10 min. Ethanol, the vehicle for the TrxR inhibitor auranofin, was included at a volume equal to that for the auranofin used below. DTNB was added to 3 mM and incubation was continued for 5 min to allow non-specific thiols to react with DTNB. NADPH was added to 0.2 mM to the sample cuvet and the rate of increase in absorbance at 412 nm was measured. The assay was repeated in the presence of 4 μM auranofin, a strong inhibitor of TrxR (Gromer et al., 1998). The amount of NADPH-dependent activity that was inhibited by auranofin is attributed to TrxR. Auranofin inhibited essentially all the activity in the samples we examined.
2.5 Miscellaneous
Protein was determined by a modified Lowry method, with bovine serum albumin as the standard (Myers and Myers, 1997).
Differences between multiple groups were assessed using one-way ANOVA and the Tukey-Kramer post test (Prism software, Graphpad). Significance was assumed at p < 0.05.
3. Results
3.1 Acrolein oxidizes Trx1 and Trx2
The effects of acrolein on the redox status of Trx1 (cytosolic) were analyzed by redox western blot. Iodoacetate covalently labels the –SH groups of reduced Trx1; the resulting negative charge increases migration in native PAGE relative to oxidized Trx1 which does not react with iodoacetate. Reduced Trx1 refers to the form in which both dithiols are reduced and represents the active form. The active site dithiol is the more easily oxidized, so partially oxidized Trx1 likely represents oxidation of the active site C32/C35, whereas both dithiols (C32/C35, C62/C69) are oxidized in the fully oxidized form (Watson et al., 2003). In untreated cells, 98% of Trx1 was fully reduced (Fig. 1). Acrolein oxidized Trx1 in a dose-dependent manner. At 1 and 2.5 μM acrolein, 19 and 50%, respectively, of Trx1 was partially oxidized with the remainder reduced. At 5 μM acrolein only 7% was reduced, with 67 and 26% in the partially and fully oxidized states, respectively. At 12.5 and 25 μM acrolein, 76 and 94% of the Trx1 was in the fully oxidized form, respectively (Fig. 1).
Fig. 1.

(A) Redox western blot of Trx1 in HMEC-1 cells treated with the indicated concentrations of acrolein for 30 min, washed once in HBSS and immediately processed for Trx1 redox status. (B) The relative abundance of the three redox states of Trx1 as determined by densitometry of redox blots. Points and error bars represent the mean ± S.D. (n = 3 independent experiments). For some, the error bars are smaller than the points as shown. *(p < 0.05), **(p < 0.01), or ***(p < 0.001) relative to the corresponding results for 0 μM acrolein.
The redox status of the mitochondrial form (Trx2) was also examined by redox western blot. In contrast to Trx1, Trx2 only has one dithiol. Reduced Trx2 migrates slower because AMS bonds to the two –SH groups of the active site thereby increasing the mass of Trx2 (Hansen et al., 2006a). In untreated cells and those treated with 1 μM acrolein, all of the Trx2 was reduced (–SH,–SH), i.e. it migrated coincident with Trx2 in DTT-treated mitochondria (Fig. 2). In cells treated with 2.5 μM acrolein, the bulk of Trx2 ran as reduced although careful examination of enlarged photos suggested some increased migration which might suggest an onset of oxidation. In cells treated with 5 μM acrolein, Trx2 migrated faster than reduced Trx2, but lagged slightly behind the fully oxidized Trx2 band in the cells treated with 12.5 μM acrolein (Fig. 2). The 5 μM samples therefore suggest some Trx2 oxidation. These results are similar to those observed in HMEC-1 cells (Szadkowski and Myers, 2007). It is possible that one of the two –SH groups of Trx2 in the 5 μM samples remains reduced, i.e. it binds one AMS rather than the two AMS bound by fully reduced Trx2, which could account for the migration between the reduced and fully oxidized forms. With ≥12.5 μM acrolein, all of the Trx2 was oxidized, i.e. it migrated coincident with Trx2 in air-oxidized mitochondria (Fig. 2).
Fig. 2.

Redox western blot of Trx2. BEAS-2B cells were either untreated or treated with the indicated concentrations of acrolein for 30 min, washed once in HBSS and immediately processed for Trx2 redox status. Standards for the migration of oxidized (oxid.) and reduced (red.) Trx2 are included, and consist of isolated mitochondria that had either been stored frozen (“mito, oxid.”) (Trx2 oxidizes in storage), or had been pre-treated with DTT which reduces Trx2 (“mito, DTT”). The AMS bonds to the –SH groups of the active site of reduced Trx2, increasing its mass. Reduced (AMS-adducted) Trx2 therefore migrates slower (Hansen et al., 2006a). Oxidized Trx2 migrated at 12 kDa consistent with its known mass. Relative to the cell lysates, only small amounts of the purified mitochondria were loaded so that the Trx2 band intensity better matched that of cell lysates. The blot shown is representative of three independent experiments.
Overall, the results suggest significant oxidation of Trx1 and Trx2 in cells exposed to ≥5 μM acrolein, with some oxidation of Trx1 evident after even 1 and 2.5 μM acrolein.
3.2 Acrolein results in Prx1 and Prx3 oxidation
For Prxs to function as peroxidases, Trxs must keep the Prxs reduced. The oxidation of Trx1 and Trx2 in acrolein-treated cells might therefore prevent the cells from keeping their Prxs reduced. We therefore examined the redox status of Prx3 (mitochondrial) and Prx1 (cytosolic) in acrolein-treated cells. An approximately equal mix of reduced and oxidized Prx3 was seen with 0–2.5 μM acrolein (Fig. 3A,B), probably because Trx2 remained largely reduced (Fig. 2). A significant increase in oxidized Prx3 was seen after 30 min with 5 μM acrolein, (Fig. 3A,B); this increase coincides with the shift in Trx2 redox state (Fig. 2). Longer (1 hr) incubation of cells with 5 μM acrolein resulted in complete oxidation of Prx3 (not shown). With ≥12.5 μM acrolein, >90% of Prx3 was oxidized (Fig. 3A,B).
Fig. 3.

Redox western blots of Prx3 (A) and Prx1 (C) in cells treated for 30 min with acrolein as indicated. In this redox blot method, N-ethylmaleimide (NEM) reacts with the –SH groups in reduced Prx to prevent their oxidation during cell harvest/lysis. Reduced Prx therefore runs as monomers. If one Cys-SH of Prx is oxidized to Cys-SOH, it will react with the nearby Cys-SH of the other subunit to create an intermolecular Prx disulfide (Prx-S-S-Prx). Oxidized Prx therefore runs as dimers. The relative abundance of the reduced and oxidized states of Prx3 (B) and Prx1 (D) were determined by densitometry of redox blots. Points and error bars represent the mean ± S.D. (n = 3 independent experiments). *(p < 0.05) or ***(p < 0.001) relative to the corresponding results for 0 μM acrolein.
Approximately 75% of Prx1 was in the reduced state in cells treated with ≤2.5 μM acrolein (Fig. 3C,D). In these cells, ≥50% of Trx1 remained reduced (Fig. 1). However, at ≥5 μM acrolein, the point at which 93% of Trx1 was oxidized (Fig. 1), there was a significant shift in Prx1 redox state such that ≥70% was oxidized (Fig. 3C,D). Therefore, the shift in Prx1 redox status only occurs once there is little to no reduced Trx1. The oxidation of Prx1 and Prx3 therefore likely results from the inability of Trx1 and Trx2 to keep their respective Prxs reduced.
Over-oxidized Prx (e.g. Prx-SO2H or Prx-SO3H) cannot react with the NEM, and does not form Prx dimers. Over-oxidized Prx would therefore run as a monomer, similar to reduced Prx. However, blots probed with anti-Prx-SO3 were negative (not shown). Since this antibody recognizes the over oxidized forms of Prx-1 through Prx-4, over-oxidized peroxiredoxins were not a significant component in these cells regardless of the acrolein concentration.
3.3 Acrolein inhibits TrxR
For Trxs to keep Prxs and other proteins reduced, TrxR must maintain Trx1 and Trx2 in their reduced/active state. The effect of acrolein on TrxR activity in cells was therefore examined. A small but significant inhibition was seen with 1 μM acrolein, whereas 2.5 and 5 μM acrolein inhibited 60 and >85% of TrxR activity, respectively (Fig. 4A). Further increases in acrolein inhibited 94–100% of TrxR. The TrxR activity assay includes NADPH, the endogenous electron donor for TrxR. Simple oxidation of TrxR, such as occurs when it reduces Trx, is reversed by electrons donated by NADPH. The loss of activity in acrolein-treated cells therefore represents activity that is not restored by NADPH. Also, the cell lysates were dialyzed overnight prior to the activity assay, so free acrolein had been removed. This suggests prolonged inhibition that is not dependent on the continued presence of acrolein. The amount of TrxR1 protein in cells did not change with acrolein treatment (Fig. 4B) so the activity declines represent inhibition and not loss of protein.
Fig. 4.

(A) Acrolein inhibits the activity of TrxR in cells. Cells were treated with the indicated concentrations of acrolein for 30 min, washed and scraped into HBSS, pelleted by centrifugation (800 x g, 5 min) and frozen. The pellets were thawed, suspended in 0.1 M sodium phosphate pH 7.4/5 mM EDTA and sonicated twice (15 sec each) on ice. Following centrifugation (30 min at 12000 x g), the supernatants were dialyzed overnight against 0.1 M sodium phosphate pH 7.4. The TrxR activity in each dialysate was determined by the DTNB assay as described in the methods. (B) Representative western blot for TrxR1 in the cell lysate dialysates used in A. Each lane was loaded with 6.75 μg total protein. (C) Acrolein inhibits the activity of purified TrxR1. Purified TrxR1 (2.66 pmol) was incubated 20 min at 37°C in a total volume of 1.0 ml of 0.1 M sodium phosphate pH 7.4 with 0.2 mM NADPH and acrolein as indicated. DTNB was added and its rate of reduction was followed by increased absorbance at 412 nm. A and C show the mean ± S.D. (n = 3 independent experiments). *(p < 0.05) or ***(p < 0.001) relative to the corresponding results for 0 μM acrolein.
Incubation of acrolein with purified TrxR1 in vitro for 20 min similarly caused pronounced concentration-dependent inhibition of TrxR activity (Fig. 4C). The inhibition was observed at even lower concentrations of acrolein, e.g. 1 μM acrolein in cells inhibited only 12% of activity (Fig. 4A) whereas 1 μM in vitro inhibited 80% (Fig. 4C). The greater sensitivity of the purified enzyme likely reflects that, unlike the cellular environment, there were no competing proteins for reaction with acrolein in vitro.
3.4 Reversibility of the effects of acrolein
If the oxidation/inhibition of Trxs and Prxs by acrolein represents conversion of the active site sulfhydryls to disulfides, then disulfide reductants should reverse these effects. Alternatively, if acrolein reacts directly with the sulfhydryls, or causes other changes that do not result in the disulfide forms of these proteins, then disulfide reductants would not be expected to reverse these effects.
For Trx1, cells treated with 12.5 μM acrolein for 30 min exhibited a mix of partially and fully oxidized forms of Trx1 (Fig. 5). The disulfide reductant Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (Getz et al., 1999) could not reverse this oxidation of Trx1 (Fig. 5). However, TCEP could reverse the Trx1 oxidation caused by treatment of cells with 50 μM Cr(VI) as Na2CrO4 (Fig. 5). Thus, the oxidation of Trx1 following Na2CrO4 exposure is consistent with a disulfide form, whereas acrolein exposure causes changes to Trx1 that are not disulfide in nature. For cells treated with 12.5 μM acrolein for 30 min, DTT or TCEP could not reverse the oxidation of Trx2 (not shown). This was true regardless of when the TCEP was added, which included: (a) treated cells were washed and scraped in HBSS with 1 mM TCEP (plus or minus 1% Triton X-100) and incubated for 15 min and then TCA precipitated, or (b) if TCEP was added after the TCA precipitate was washed and dried, but before the AMS labeling step. As a control, TCEP was able to reduce Trx2 in air-oxidized mitochondria (not shown). Thus, the effects of acrolein on Trx1 and Trx2 are not reversible by disulfide reductants.
Fig. 5.
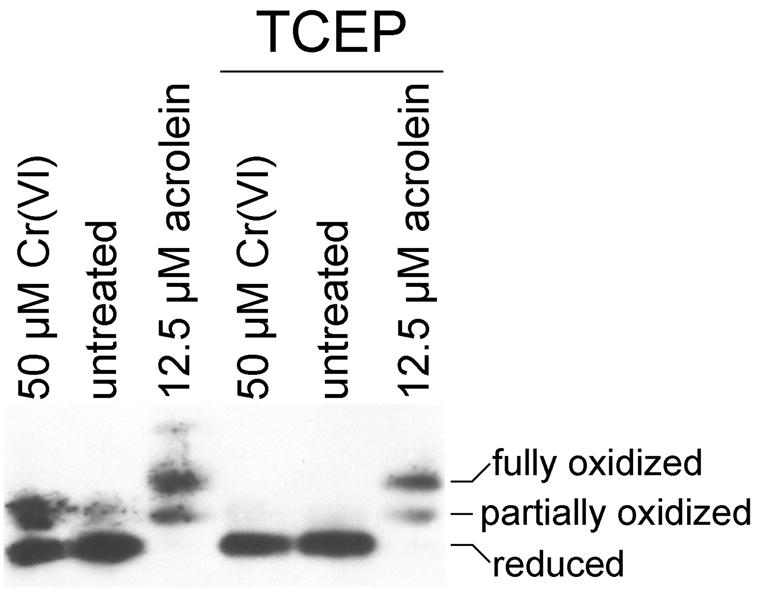
The oxidation of Trx1 by acrolein is not reversible by the disulfide reducing agent TCEP. In the three lanes at left, the redox state of Trx1 was assessed in cells immediately following treatment with 50 μM Cr(VI) as Na2CrO4 for 3 hr, HBSS for 30 min (untreated), or 12.5 μM acrolein for 30 min. For the three lanes at right, cells were given these same treatments, but were then washed in HBSS, incubated in HBSS with 1 mM TCEP for 15 min at room temp. Following a final wash, the Trx1 redox status was determined.
In contrast to Trx, the oxidation of Prx3 in acrolein-treated cells could be reversed by a disulfide reductant (Fig. 6). This implies that the oxidation of Prx in these cells represents the formation of disulfide links between the two subunits to form the Prx dimers observed in the gels.
Fig. 6.

The oxidation of Prx3 by acrolein is reversible by disulfide reduction. In untreated cells or cells treated for 30 min with 12.5 μM acrolein, the redox state of Prx3 was assessed using the standard protocol with non-reducing gel conditions (top). Alternatively, aliquots of the same samples were first incubated for 10 min at 70°C with NuPAGE reducing agent (Invitrogen) prior to loading on the gel (bottom).
Since TrxR is directly responsible for keeping the Trxs reduced in cells, and is therefore indirectly responsible for keeping the Prxs reduced, additional experiments were conducted to determine if the TrxR activity could recover in cells after acrolein was removed. As in Fig. 4, immediately following a 30-min acrolein exposure, TrxR activity was markedly inhibited in a dose-dependent manner, with >90% inhibited by ≥5 μM acrolein (Fig. 7A). When acrolein was removed and the cells were exposed to acrolein-free medium for 4 hr, there was no recovery of TrxR activity, either with or without the protein synthesis inhibitor geneticin (Fig. 7A). The effects of acrolein on TrxR in cells therefore persist well after acrolein removal. The levels of TrxR protein in these cells as determined by western blot did not change with acrolein exposure or recovery (Fig. 7B). Acrolein therefore affects the activity but not the amount of TrxR protein.
Fig. 7.

(A) TrxR activity in acrolein-treated cells and following a 4-hr acrolein-free recovery. For “no recovery”, the cells were treated with the indicated concentrations of acrolein for 30 min, washed in HBSS and the pellets were frozen and later processed for TrxR activity as described for Fig. 4. For the “4 hr recovery”, the cells were treated with the indicated concentrations of acrolein for 30 min, washed in HBSS and then incubated for 4 hr in DMEM medium with 10% FBS. The “4 hr + geneticin” was an identical to the 4 hr recovery except that the protein synthesis inhibitor geneticin (800 μg per ml) was included during the recovery. All values represent the mean ± S.D. (n = 3 independent experiments, except for the no recovery for 2.5 μM acrolein for which n = 2). There was no significant difference between the “no recovery”, “4 hr recovery”, and “4 hr geneticin” samples for the 2.5, 5, and 12.5 μM acrolein treatments. (B) Representative western blots for TrxR1 in the cell lysate dialysates used in A. Each lane was loaded with 4.2 μg total protein.
4. Discussion
The acrolein exposures that oxidized/inhibited these proteins in BEAS-2B cells are consistent with those that cause cytotoxicity in human endothelial cells (Szadkowski and Myers, 2007), porcine pulmonary cells (Patel and Block, 1993), and murine lymphocytes (Kern and Kehrer, 2002). The effects on the redox state of Trx1 and Trx2 in BEAS-2B cells are similar both quantitatively and qualitatively to those reported for human endothelial cells (Szadkowski and Myers, 2007). These effects may therefore apply to other human cell types as well. Acrolein also causes redox changes to Trx1 in bovine aortic endothelial cells (BAECs) (Go et al., 2007) and to purified Trx1 in vitro (Go et al., 2007). In BAECs, Trx1-acrolein adducts were noted with just 1 μM acrolein (Go et al., 2007), but acrolein exposure was twice as long as in our studies. Further redox shifts in Trx1, similar to those observed here, were seen in BAECs following 5 and 10 μM acrolein (Go et al., 2007).
Trx1 and Trx2 are not in redox equilibrium with each other, so they have been used to differentially assess the impacts of other oxidants on the thiol redox status of the cytosolic and mitochondrial compartments (Halvey et al., 2005; Hansen et al., 2006a). For the results reported here, Trx1 was more sensitive to oxidation by acrolein than is Trx2. At 1 and 2.5 μM acrolein, 19 and 50% of the Trx1 was oxidized, whereas Trx2 maintained a reduced state until 5 μM acrolein. Similar differential effects were reported in human microvascular endothelial cells treated with acrolein (Szadkowski and Myers, 2007). The relative susceptibility is in contrast to that for t-butyl hydroperoxide or diamide which preferentially oxidize Trx2 relative to Trx1 (Chen et al., 2006; Hansen et al., 2006a).
When compared to another human lung cell line (A549 lung adenocarcinoma cells), the effects on Trxs in BEAS-2B cells occurred at much lower concentrations. Whereas essentially all of the Trx1 and Trx2 was oxidized in BEAS-2B exposed to 5 μM acrolein, ≥25 μM acrolein was required to inhibit >90% of total Trx activity in A549 cells (Yang et al., 2004). Only ca. 50% inhibition was observed with 10 μM acrolein (Yang et al., 2004)
TrxR activity in BEAS-2B cells was also about 10 times more sensitive to acrolein than that in A549 cells. In A549, 50–75 μM acrolein for 30 min caused ca. 65% inhibition (Yang et al., 2004), whereas in BEAS-2B cells just 2.5 and 5 μM caused 60% and >85% inhibition, respectively (Fig. 4A). TrxR in BEAS-2B cells was also more sensitive to acrolein than that in human umbilical vein endothelial cells (HUVECs), which required 25 μM acrolein for 30 min to inhibit 88% of TrxR (Park et al., 2005). While TrxR protein levels were not examined in the HUVECs or A549 cells, we noted that the amount of TrxR protein remained constant (Fig. 4B).
The declines in activity do not therefore result from declines in TrxR protein. We also noted that TrxR activity remained suppressed in BEAS-2B cells even 4 hr after the 2.5 to 12.5 μM acrolein treatment is removed (Fig. 7). The effects on TrxR in BEAS-2B cells are therefore prolonged and TrxR activity is not easily restored in these cells. Even intermittent exposure to acrolein may therefore have prolonged effects in normal bronchial epithelial cells. This is in contrast to A549 cells, in which TrxR activity recovered to 70% and 100% of normal 2 hrs and 4 hrs after removal of the 50–75 μM acrolein treatment (Yang et al., 2004). It was not determined if this recovery was due to new protein synthesis or reactivation of existing TrxR (Yang et al., 2004). The differences between the A549 and BEAS-2B cells could reflect their different origins and are consistent with their relative resistance to oxidant stress. A549 cells have proven considerably more resistant to other oxidants than other cell types (Watjen and Beyersmann, 2004). The A549s consist of four different cell types which can be distinguished both morphologically and biochemically (Croce et al., 1999), and one or more of these subpopulations could account for the enhanced oxidant tolerance. It is important to note that differences between BEAS-2B cells and other cell types may not necessarily reflect inherent differences in acrolein susceptibility. Differences in growth medium, stage of growth, and other specifics of culture conditions could contribute to differential susceptibility to acrolein. The findings with BEAS-2B cells do, however, indicate that acrolein can cause significant effects at low micromolar concentrations.
Since the BEAS-2B cells originate from normal human bronchial epithelium, they should better reflect normal cells. BEAS-2B cells are a recognized model of normal human bronchial epithelium, and they have been used as a model for cytotoxicity associated with other airborne toxins (Frampton et al., 1999; Ghio et al., 1999; Van Vleet et al., 2002; Gurr et al., 2005). Whereas A549s are tumorigenic, BEAS-2B cells are not (Reddel et al., 1988). Overall, the Trxs and TrxR in BEAS-2B cells were considerably more sensitive to the effects of acrolein than would have been predicted by the A549 cells. TrxR also proved more sensitive to acrolein than another unsaturated aldehyde 4-hydroxynonenal (4-HNE). After 6 hr, 10 μM 4-HNE caused no inhibition of TrxR in HeLa cells, whereas 50 μM caused ca. 60% inhibition (Fang and Holmgren, 2006). These 4-HNE treatments are higher and longer than the 5 μM acrolein which causes >85% inhibition after just 30 min in BEAS-2B cells. These differences between acrolein and 4-HNE directly reflect their differential reactivity with other species, i.e. acrolein is 100–150 times more reactive than 4-HNE (Esterbauer et al., 1991).
The detailed mechanism by which acrolein mediates inhibition/oxidation of TrxR and Trxs remains to be determined in these cells. However, given that acrolein can react with some thiols, it is plausible that acrolein is reacting with one or more of the sulfhydryls in the Trxs. Human Trx1 has 5 thiols (C32/C35, C62/C69, and C73), and the detection of at least 3 redox states (Fig. 1) implies effects on more than one of the thiols. One possibility is that acrolein reacts directly with one or more of the thiols to form Trx-S-acrolein adducts. While different from conventional oxidation in which disulfides (–S–S–) are formed, Trx-acrolein adducts in essence represent Trx oxidation because they block its sulfhydryl groups, which is reflected in altered mobility in the redox blots. Since disulfide reductants could not reverse the redox changes to Trx1 or Trx2, Trx oxidation following acrolein exposure is not disulfide in nature. Whatever the nature of the changes to Trx, they are not readily reversed. Trx-S-acrolein adducts are one possibility that could explain the results. Adducts of acrolein to Cys73 of Trx1 have been reported following in vitro incubation of equal amounts of Trx1 and acrolein, while adducts to dithiols were not observed (Go et al., 2007). Adducts to the disulfides may occur in cells, however, and would explain the inability of disulfide reductants to shift either of the oxidized states to a more reduced form (Fig. 5). Similarly, in BAECs exposed to acrolein, dithiothreitol could not reverse the putative acrolein-Trx adducts (Go et al., 2007). Since thiol-acrolein adducts are generally quite stable (Esterbauer et al., 1991), Trx-S-acrolein adducts could be difficult to reverse. The Trx1 redox changes may also be due to other effects of acrolein, however. It has been suggested that some of the changes in BAECs may result from oxidation by ROS or potential inhibition of TrxR (Go et al., 2007). Other as yet undescribed effects of acrolein may be responsible for the Trx oxidation. Regardless of the mechanism, the inability to readily reverse Trx oxidation suggests long-term effects that could impact the redox status and functions of proteins that are dependent on Trx for keeping their thiols reduced, and could alter Trx-mediated signaling.
Acrolein did inhibit TrxR activity in these cells (Fig. 4). Redox-active sites within TrxR include the flavin (FAD), the active site Cys-SeCys (C-terminal) and the N-terminal domain dithiol. NADPH donates electrons to the FAD which then reduces the N-terminal dithiol which then reduces the active site Cys-SeCys. Disruption or inhibition of one or more of these sites should inhibit TrxR activity. This disruption was not reversible. Unreacted acrolein was removed by prolonged dialysis of the cell lysates, and activity was not reversed by NADPH, the native electron donor for TrxR. TrxR inhibition is therefore not likely due to a simple oxidation of the redox-active thiols or selenol as would occur when TrxR reduces endogenous substrates such as Trx. The lack of recovery in cells after a 4-hr acrolein-free period also shows that the changes to TrxR are not easily reversed, and implies that new protein synthesis does not significantly restore TrxR activity during this 4-hr recovery. One plausible mechanism for prolonged TrxR inhibition would be covalent adduction of acrolein to active site Cys-SeCys or to the N-terminal dithiol. Of these, the active site is likely more reactive. It is exposed on the surface of the enzyme (Sandalova et al., 2001) and the selenol of the SeCys residue (pKa ca. 5.2) (Jacob et al., 2003) should be ionized to selenolate at physiological pH making it more reactive. 2,4-dinitrochlorobenzene covalently binds the Cys-SeCys, irreversibly inhibiting the enzyme (Arner et al., 1995), but it does not modify the N-terminal dithiol (Nordberg et al., 1998). Both curcumin and 4-HNE covalently bind the active site Cys and SeCys residues of TrxR, causing irreversible inactivation (Fang et al., 2005; Cassidy et al., 2006; Fang and Holmgren, 2006). However, the mechanism by which acrolein inhibits TrxR remains to be determined, and non-adduct mechanisms of inhibition must also be considered at this point.
In these studies, the examination of TrxR, Trxs, and Prxs within the same cells allowed for direct examination of the relationship of the effects of acrolein on these proteins. Given the dependence of Prxs on Trxs, the oxidation of Trxs could influence the redox status and function of the Prxs. Similarly, inhibition of TrxR could influence the redox status and function of the Trxs. There was a strong relationship between the inhibition of TrxR and the oxidation of Trx1 (Fig. 8A). This might imply that Trx1 oxidation results from the inability of TrxR to maintain Trx1 in a reduced state. However, this type of Trx1 oxidation should be reversible by disulfide reductants, which proved not to be true (Fig. 5). As discussed above, the data are more consistent with some other type of modification that is not reversed by disulfide reductants, of which Trx1-S-acrolein adducts are one possibility.
Fig. 8.
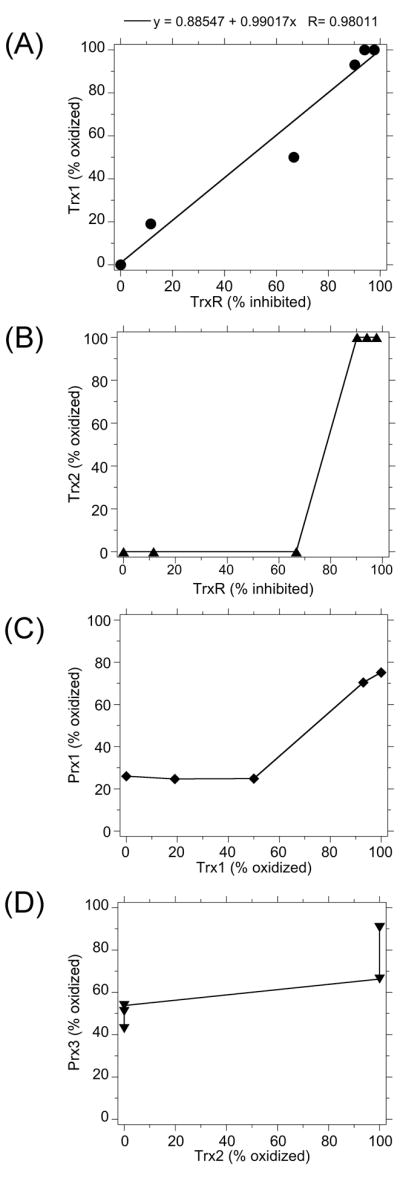
Relationship between the inhibition/oxidation of various proteins in acrolein-exposed cells: TrxR and Trx1 (A), TrxR and Trx2 (B), Trx1 and Prx1 (C), and Trx2 and Prx3 (D).
Unlike Trx1 which exhibited progressive oxidation with increasing acrolein concentrations, Trx2 remained reduced until 5 μM acrolein at which point there was an abrupt shift to the oxidized form. This abrupt shift is apparent in the relationship between TrxR inhibition and Trx2 oxidation (Fig. 8B). At the point of this shift, 90% of total TrxR was inhibited. There is not therefore a linear relationship between TrxR inhibition and Trx2 oxidation. However, the assay measured total TrxR activity; it did not distinguish between TrxR1 and TrxR2. It is possible that Trx2 oxidation might result from TrxR2 inhibition. However, the inability to reverse Trx2 oxidation by disulfide reductants argues that Trx2 oxidation results from other effects of acrolein and not from loss of reducing equivalents from TrxR2.
The cytosolic Prxs such as Prx1 should be dependent on Trx1 to maintain their redox state. At ≤2.5 μM acrolein, Prx1 redox state resembled that of untreated cells, but at 5 μM acrolein it abruptly shifted to approximately 75% oxidized (Fig. 3). This is the point at which >90% of Trx1 is oxidized (Fig. 8C). In contrast, the normal Prx1 redox state is maintained when 50% of Trx1 is oxidized (Fig. 8C). Thus, these cells can maintain Prx1 redox status until almost all of the Trx1 is oxidized.
Mitochondrial Prxs such as Prx3 should be dependent on Trx2 to maintain their redox state. At 5 μM acrolein, Trx2 shifted to the oxidized form and at this point a significant increase in Prx3 oxidation was noted (Fig. 3). Prx3 oxidation increased to >90% at the next acrolein concentration (12.5 μM) (Fig. 3). Fig. 8D further illustrates the graphical relationship between Trx2 and Prx3 redox state. Thus, for both Prx1 and Prx3, increased Prx oxidation is not seen until the respective Trxs are almost completely oxidized. The data suggest that Prx oxidation occurs because the Trxs can no longer maintain the redox status of their Prxs. For Prx1 and Prx3, this type of Prx oxidation should yield intermolecular disulfide homodimers in non-reducing gels, which is exactly what was observed (Fig. 3). Such intermolecular disulfides should be reduced by disulfide reductants, which proved to be the case (Fig. 6). An alternative mechanism of Prx oxidation would be the direct reaction of acrolein with Prx to form Prx-S-acrolein adducts. Such adducts should prevent dimer formation and should not be readily reversed by disulfide reductants. The formation of dimers and their reversal in reducing gels (Figs. 3, 6) are therefore inconsistent with Prx-S-acrolein adducts. While it is possible that a small percentage of Prx sulfhydryls react with acrolein, the data are consistent with the formation of intermolecular disulfides as the prominent form of oxidized Prx. The conformation of the Prxs may protect their –SH groups from direct reaction with acrolein. While Prx2 shows a high rate of reaction with peroxide substrates, its sulfhydryls are not readily alkylated with iodoacetamide or various amino acid chloroamines, despite the high reactivity of these agents with other cellular –SH groups (Peskin et al., 2007). A third possible form of Prx oxidation is overoxidation of the sulfhydryls to sulfinic (Prx-SO2) and sulfonic (Prx-SO3) forms. For example, for Prx oxidation mediated by lipid hydroperoxide metabolites of arachidonic acid, about 65% is reversible (disulfide) whereas the remainder represents overoxidation to Prx-SO2 and Prx-SO3 (Cordray et al., 2007). However, using an antibody that recognizes the Prx-SO2 and Prx-SO3 forms of Prx1, Prx2, Prx3, and Prx4, we found no evidence for overoxidation in acrolein-exposed cells. Furthermore, overoxidation should diminish Prx dimerization. Together, the data imply that Prx oxidation in acrolein-exposed cells largely generates intermolecular disulfides which are reversible by disulfide reduction. Since Prx oxidation only occurs once the Trxs are completely or almost completely oxidized, the underlying cause is likely the inability of the Trx system to maintain the redox state of the Prxs. Consistent with this, inhibition of TrxR in erythrocytes causes Prx oxidation (Low et al., 2008). In acrolein-exposed cells, therefore, the redox status of Prx3 and Prx1 may largely reflect the functional state of the respective Trx systems. The reversibility of the Prx redox changes also underscores the fundamental differences in the effects on Prxs (reversible) vs. Trxs and TrxR (irreversible).
Acrolein-mediated oxidation of Trxs and Prxs, and inhibition of TrxR, could conceivably be mediated by increased reactive oxygen species (ROS). Since Prxs function as peroxidases, increases in peroxides could result following Prx oxidation. Acrolein-mediated increases in ROS have usually been reported at much higher concentrations of acrolein, however. 30 to 100 μM acrolein has been reported to significantly increase superoxide (O2−·) and H2O2 in cells (Jaimes et al., 2004; Wu et al., 2006), but this is well above the 5 μM acrolein that caused prominent effects on the Trx system and Prxs reported here. However, a 52% increase in dichlorofluorescein fluorescence was seen in BAECs following 5 μM acrolein for 1 hr (Go et al., 2007), although the responsible oxidants were not determined. However, millimolar levels of H2O2 have not resulted in significant Trx oxidation in some other cells (Fernando et al., 1992; Szadkowski and Myers, 2007), and Trx2 was not significantly oxidized by epidermal growth factor-induced reactive oxygen species (ROS) (Halvey et al., 2005; Hansen et al., 2006a). Furthermore, oxidation mediated by H2O2 would be expected to be readily reversible, which was not the case for Trx and TrxR. Therefore, acrolein-mediated increases in ROS may not account for the bulk of the effects on TrxR and Trx, but the inhibition of these enzymes may result in Prx oxidation and contribute to increases in ROS. Since the Prx oxidation was reversible, it is plausible that endogenous peroxide substrates contributed to, or were largely responsible for, shifting the Prxs to a more oxidized state following acrolein exposure. The ability of various aldehydes to increase peroxide production in cells (Uchida, 2000) may in part be due to the oxidation of the Prxs following Trx oxidation.
The acrolein-induced changes to the redox state/activity could have important implications for cell growth and survival. Inhibition of TrxR increases the susceptibility to oxidants and favors apoptosis (Nordberg and Arner, 2001). Genetic suppression or inhibition of Trx results in increased oxidant stress and apoptosis (Hansen et al., 2006a) and increased sensitivity to oxidants (Chen et al., 2006). Conversely, overexpression of Trx enhances resistance to oxidant-induced apoptosis (Chen et al., 2006; Hansen et al., 2006a). The overoxidation of Prx1 following 6-hydroxydopamine exposure enhances apoptosis, whereas overexpression of Prx1 protects cells from 6-hydroxydopamine (Lee et al., 2008). Prx3 may form a primary defense against peroxides in mitochondria and depletion of Prx3 renders HeLa cells more susceptible to apoptosis caused by tumor necrosis factor-α and staurosporine (Chang et al., 2004). Loss of protection from oxidant stress is just one of the potential consequences of the effects of acrolein on the Trx/Prx system. Other more direct effects are also possible. For example, deletion of, or adduction to, the SeCys residue of TrxR can have negative effects that extend beyond those of just blocking TrxR activity (Anestål et al., 2008). Further studies on the consequences of the effects of acrolein on the TrxR/Trx/Prx system are warranted.
In summary, this report describes the effects of acrolein on the thioredoxins, peroxiredoxins, and TrxR in human bronchial epithelial cells. Low micromolar concentrations of acrolein caused oxidation of cytosolic and mitochondrial Trxs and Prxs and inhibited TrxR. The effects on Trxs and TrxR were not reversible in vitro and TrxR activity did not recover following acrolein removal. Redox changes to Prxs were not observed until their respective Trxs were oxidized. Prx oxidation was readily reversed with a disulfide reductant, suggesting that Prx oxidation resulted from lack of reducing equivalents from Trx. These effects of acrolein on the Trx system and Prxs could have important implications for the normal function and survival of the bronchial epithelium, and the ability of these cells to tolerate other oxidant insults.
Acknowledgments
This research was supported by grant ES012707 from the National Institute of Environmental Health Sciences (NIEHS), and by the Dept. of Pharmacology & Toxicology of the Medical College of Wisconsin.
References
- Anestål K, Prast-Nielsen S, Cenas N, Arnér ES. Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells. PLoS ONE. 2008;3:e1846. doi: 10.1371/journal.pone.0001846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnér ES, Bjornstedt M, Holmgren A. 1-Chloro-2,4-dinitrobenzene is an irreversible inhibitor of human thioredoxin reductase. Loss of thioredoxin disulfide reductase activity is accompanied by a large increase in NADPH oxidase activity. J Biol Chem. 1995;270:3479–3482. doi: 10.1074/jbc.270.8.3479. [DOI] [PubMed] [Google Scholar]
- Arnér ESJ, Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- Barnoya J, Glantz SA. Cardiovascular effects of secondhand smoke: nearly as large as smoking. Circulation. 2005;111:2684–2698. doi: 10.1161/CIRCULATIONAHA.104.492215. [DOI] [PubMed] [Google Scholar]
- Beauchamp RO, Andjelkovich DA, Kligerman AD, Morgan KT, Heck HA. A critical review of the literature on acrolein toxicity. CRC Crit Rev Toxicol. 1985;14:309–380. doi: 10.3109/10408448509037461. [DOI] [PubMed] [Google Scholar]
- Carnevali S, Nakamura Y, Mio T, Liu X, Takigawa K, Romberger DJ, Spurzem JR, Rennard SI. Cigarette smoke extract inhibits fibroblast-mediated collagen gel contraction. Am J Physiol Lung Cell Mol Physiol. 1998;274:L591–L598. doi: 10.1152/ajplung.1998.274.4.L591. [DOI] [PubMed] [Google Scholar]
- Cassidy PB, Edes K, Nelson CC, Parsawar K, Fitzpatrick FA, Moos PJ. Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles. Carcinogenesis. 2006;27:2538–2549. doi: 10.1093/carcin/bgl111. [DOI] [PubMed] [Google Scholar]
- Chang TS, Cho CS, Park S, Yu S, Kang SW, Rhee SG. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem. 2004;279:41975–41984. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Cai J, Jones DP. Mitochondrial thioredoxin in regulation of oxidant-induced cell death. FEBS Lett. 2006;580:6596–6602. doi: 10.1016/j.febslet.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin DJ, Langford SD, Boor PJ. Contribution of serum and cellular semicarbazide-sensitive amine oxidase to amine metabolism and cardiovascular toxicity. Toxicol Sci. 1998;46:386–392. doi: 10.1006/toxs.1998.2528. [DOI] [PubMed] [Google Scholar]
- Cordray P, Doyle K, Edes K, Moos PJ, Fitzpatrick FA. Oxidation of 2-Cys-peroxiredoxins by arachidonic acid peroxide metabolites of lipoxygenases and cyclooxygenase-2. J Biol Chem. 2007;282:32623–32629. doi: 10.1074/jbc.M704369200. [DOI] [PubMed] [Google Scholar]
- Cox AG, Pullar JM, Hughes G, Ledgerwood EC, Hampton MB. Oxidation of mitochondrial peroxiredoxin 3 during the initiation of receptor-mediated apoptosis. Free Radic Biol Med. 2008;44:1001–1009. doi: 10.1016/j.freeradbiomed.2007.11.017. [DOI] [PubMed] [Google Scholar]
- Croce MV, Colussi AG, Price MR, Segal-Eiras A. Identification and characterization of different subpopulations in a human lung adenocarcinoma cell line (A549) Pathol Oncol Res. 1999;5:197–204. doi: 10.1053/paor.1999.0212. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Fang J, Holmgren A. Inhibition of thioredoxin and thioredoxin reductase by 4-hydroxy- 2-nonenal in vitro and in vivo. J Am Chem Soc. 2006;128:1879–1885. doi: 10.1021/ja057358l. [DOI] [PubMed] [Google Scholar]
- Fang J, Lu J, Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J Biol Chem. 2005;280:25284–25290. doi: 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- Fernando MR, Nanri H, Yoshitake S, Nagata-Kuno K, Minakami S. Thioredoxin regenerates proteins inactivated by oxidative stress in endothelial cells. Eur J Biochem. 1992;209:917–922. doi: 10.1111/j.1432-1033.1992.tb17363.x. [DOI] [PubMed] [Google Scholar]
- Frampton MW, Ghio AJ, Samet JM, Carson JL, Carter JD, Devlin RB. Effects of aqueous extracts of PM10 filters from the Utah Valley on human airway epithelial cells. Am J Physiol. 1999;277(21):L960–L967. doi: 10.1152/ajplung.1999.277.5.L960. Lung Cell. Mol. Physiol. [DOI] [PubMed] [Google Scholar]
- Getz EB, Xiao M, Chakrabarty T, Cooke R, Selvin PR. A comparison between the sulfhydryl reductants tris(2-carboxyethyl)phosphine and dithiothreitol for use in protein biochemistry. Anal Biochem. 1999;273:73–80. doi: 10.1006/abio.1999.4203. [DOI] [PubMed] [Google Scholar]
- Ghilarducci DP, Tjeerdema RS. Fate and effects of acrolein. Rev Environ Contam Toxicol. 1995;144:95–146. doi: 10.1007/978-1-4612-2550-8_2. [DOI] [PubMed] [Google Scholar]
- Ghio AJ, Carter JD, Dailey LA, Devlin RB, Samet JM. Respiratory epithelial cells demonstrate lactoferrin receptors that increase after metal exposure. Am J Physiol. 1999;276(20):L933–L940. doi: 10.1152/ajplung.1999.276.6.L933. Lung Cell. Mol. Physiol. [DOI] [PubMed] [Google Scholar]
- Go YM, Halvey PJ, Hansen JM, Reed M, Pohl J, Jones DP. Reactive aldehyde modification of thioredoxin-1 activates early steps of inflammation and cell adhesion. Am J Pathol. 2007;171:1670–1681. doi: 10.2353/ajpath.2007.070218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromer S, Arscott LD, Williams CH, Jr, Schirmer RH, Becker K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J Biol Chem. 1998;273:20096–20101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- Gurr JR, Wang AS, Chen CH, Jan KY. Ultrafine titanium dioxide particles in the absence of photoactivation can induce oxidative damage to human bronchial epithelial cells. Toxicology. 2005;213:66–73. doi: 10.1016/j.tox.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Halvey PJ, Watson WH, Hansen JM, Go YM, Samali A, Jones DP. Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem J. 2005;386:215–219. doi: 10.1042/BJ20041829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol. 2006a;46:215–234. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- Hansen JM, Zhang H, Jones DP. Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions. Free Radic Biol Med. 2006b;40:138–145. doi: 10.1016/j.freeradbiomed.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Holmgren A. Bovine thioredoxin system. Purification of thioredoxin reductase from calf liver and thymus and studies of its function in disulfide reduction. J Biol Chem. 1977;252:4600–4606. [PubMed] [Google Scholar]
- Jacob C, Giles GI, Giles NM, Sies H. Sulfur and selenium: the role of oxidation state in protein structure and function. Angew Chem Int Ed Engl. 2003;42:4742–4758. doi: 10.1002/anie.200300573. [DOI] [PubMed] [Google Scholar]
- Jaimes EA, DeMaster EG, Tian RX, Raij L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler Thromb Vasc Biol. 2004;24:1031–1036. doi: 10.1161/01.ATV.0000127083.88549.58. [DOI] [PubMed] [Google Scholar]
- Kachel DL, Martin WJ., II Cyclophosphamide-induced lung toxicity: mechanism of endothelial cell injury. J Pharmacol Exp Ther. 1994;268:42–46. [PubMed] [Google Scholar]
- Kern JC, Kehrer JP. Acrolein-induced cell death: a caspase-influenced decision between apoptosis and oncosis/necrosis. Chem Biol Interact. 2002;139:79–95. doi: 10.1016/s0009-2797(01)00295-2. [DOI] [PubMed] [Google Scholar]
- Kim JA, Park S, Kim K, Rhee SG, Kang SW. Activity assay of mammalian 2-cys peroxiredoxins using yeast thioredoxin reductase system. Anal Biochem. 2005;338:216–223. doi: 10.1016/j.ab.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Lee YM, Park SH, Shin DI, Hwang JY, Park B, Park YJ, Lee TH, Chae HZ, Jin BK, Oh TH, Oh YJ. Oxidative modification of peroxiredoxin is associated with drug-induced apoptotic signaling in experimental models of Parkinson disease. J Biol Chem. 2008;283:9986–9998. doi: 10.1074/jbc.M800426200. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer's disease brain and is toxic to primary hippocampal cultures. Neurobiol Aging. 2001;22:187–194. doi: 10.1016/s0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- Low FM, Hamptom MB, Peskin AV, Winterbourn CC. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood. 2008;109:2611–2617. doi: 10.1182/blood-2006-09-048728. [DOI] [PubMed] [Google Scholar]
- Myers CR, Myers JM. Cloning and sequence of cymA, a gene encoding a tetraheme cytochrome c required for reduction of iron(III), fumarate, and nitrate by Shewanella putrefaciens MR-1. J Bacteriol. 1997;179:1143–1152. doi: 10.1128/jb.179.4.1143-1152.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkabyo YS, Ziegler TR, Gu LH, Watson WH, Jones DP. Glutathione and thioredoxin redox during differentiation in human colon epithelial (Caco-2) cells. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1352–G1359. doi: 10.1152/ajpgi.00183.2002. [DOI] [PubMed] [Google Scholar]
- Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol. 2003;23:916–922. doi: 10.1128/MCB.23.3.916-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg J, Arnér ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- Nordberg J, Zhong L, Holmgren A, Arnér ES. Mammalian thioredoxin reductase is irreversibly inhibited by dinitrohalobenzenes by alkylation of both the redox active selenocysteine and its neighboring cysteine residue. J Biol Chem. 1998;273:10835–10842. doi: 10.1074/jbc.273.18.10835. [DOI] [PubMed] [Google Scholar]
- Park YS, Misonou Y, Fujiwara N, Takahashi M, Miyamoto Y, Koh YH, Suzuki K, Taniguchi N. Induction of thioredoxin reductase as an adaptive response to acrolein in human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2005;327:1058–1065. doi: 10.1016/j.bbrc.2004.12.104. [DOI] [PubMed] [Google Scholar]
- Patel JM, Block ER. Acrolein-induced injury to cultured pulmonary artery endothelial cells. Toxicol Appl Pharmacol. 1993;122:46–53. doi: 10.1006/taap.1993.1170. [DOI] [PubMed] [Google Scholar]
- Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annu Rev Biophys Biomol Struct. 2001;30:421–455. doi: 10.1146/annurev.biophys.30.1.421. [DOI] [PubMed] [Google Scholar]
- Reddel RR, Ke Y, Gerwin BI, McMenamin MG, Lechner JF, Su RT, Brash DE, Park JB, Rhim JS, Harris CC. Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Res. 1988;48:1904–1909. [PubMed] [Google Scholar]
- Sandalova T, Zhong L, Lindqvist Y, Holmgren A, Schneider G. Three-dimensional structure of a mammalian thioredoxin reductase: implications for mechanism and evolution of a selenocysteine-dependent enzyme. Proc Natl Acad Sci USA. 2001;98:9533–9538. doi: 10.1073/pnas.171178698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szadkowski A, Myers CR. Acrolein oxidizes the cytosolic and mitochondrial thioredoxins in human endothelial cells. Toxicology. 2007;243:164–176. doi: 10.1016/j.tox.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K. Role of reactive aldehyde in cardiovascular diseases. Free Radic Biol Med. 2000;28:1685–1696. doi: 10.1016/s0891-5849(00)00226-4. [DOI] [PubMed] [Google Scholar]
- Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J Biol Chem. 1998a;273:16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- Uchida K, Kanematsu M, Sakai K, Matsuda T, Hattori N, Mizuno Y, Suzuki D, Miyata T, Noguchi N, Niki E, Osawa T. Protein-bound acrolein: potential markers for oxidative stress. Proc Natl Acad Sci USA. 1998b;95:4882–4887. doi: 10.1073/pnas.95.9.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vleet T, Macé K, Coulombe RJ. Comparative aflatoxin B1 activation and cytotoxicity in human bronchial cells expressing cytochromes P450–1A2 and 3A4. Cancer Res. 2002;62:105–112. [PubMed] [Google Scholar]
- Watjen W, Beyersmann D. Cadmium-induced apoptosis in C6 glioma cells: influence of oxidative stress. BioMetals. 2004;17:65–78. doi: 10.1023/a:1024405119018. [DOI] [PubMed] [Google Scholar]
- Watson WH, Pohl J, Montfort WR, Stuchlik O, Reed MS, Powis G, Jones DP. Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J Biol Chem. 2003;278:33408–33415. doi: 10.1074/jbc.M211107200. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003a;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003b;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- Wu CC, Hsieh CW, Lai PH, Lin JB, Liu YC, Wung BS. Upregulation of endothelial heme oxygenase-1 expression through the activation of the JNK pathway by sublethal concentrations of acrolein. Toxicol Appl Pharmacol. 2006;214:244–252. doi: 10.1016/j.taap.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Yang X, Wu X, Choi YE, Kern JC, Kehrer JP. Effect of acrolein and glutathione depleting agents on thioredoxin. Toxicology. 2004;204:209–218. doi: 10.1016/j.tox.2004.06.056. [DOI] [PubMed] [Google Scholar]