

Abstract
Increased levels of homocysteine (Hcy), recognized as hyperhomocysteinemia (HHcy), were associated with cardiovascular diseases. There was controversy regarding the detrimental versus cardio protective role of inducible nitric oxide synthase (iNOS) in ischemic heart disease. The aim of this study was to test the hypothesis that the Hcy generated nitrotyrosine by inducing the endothelial nitric oxide synthase, causing endothelial-myocyte (E–M) coupling. To differentiate the role of iNOS versus constitutive nitric oxide synthase (eNOS and nNOS) in Hcy-mediated nitrotyrosine generation and matrix remodeling in cardiac dysfunction, left ventricular (LV) tissue was analyzed from cystathionine beta synthase (CBS) heterozygote knockout, iNOS homozygote knockout, CBS−/+/iNOS−/− double knockout, and wild-type (WT) mice. The levels of nitrotyrosine, MMP-2 and -9 (zymographic analysis), and fibrosis (by trichrome stain) were measured. The endothelial-myocyte function was determined in cardiac rings. In CBS−/+ mice, homocysteine was elevated and in iNOS−/− mice, nitric oxide was significantly reduced. The nitrotyrosine and matrix metalloproteinase-9 (MMP-9) levels were elevated in double knockout and CBS−/+ as compared to WT mice. Although MMP-2 levels were similar in CBS−/+, iNOS−/−, and CBS−/+/iNOS−/−, the levels were three- to fourfold higher than WT. The levels of collagen were similar in CBS−/+ and iNOS−/−, but they were threefold higher than WT. Interesting, the levels of collagen increased sixfold in double knockouts, compared to WT, suggesting synergism between high Hcy and lack of iNOS. Left ventricular hypertrophy was exaggerated in the iNOS−/− and double knockout, and mildly increased in the CBS−/+, compared to WT mice. The endothelial-dependent relaxation was attenuated to the same extent in the CBS−/+ and iNOS−/−, compared to WT, but it was robustly blunted in double knockouts. The results concluded that homocysteine generated nitrotyrosine in the vicinity of endothelium, caused MMP activation and endothelium-myocyte uncoupling. The generation of nitrotyrosine was independent of iNOS.
Keywords: FIBROSIS, EXTRACELLULAR MATRIX, MMP-2, MMP-9, LVH, COLLAGEN, ACETYLCHOLINE, BRADYKININ, NITROPRUSSIDE, HOMO-CYSTEINE, HYPERHOMOCYSTEINEMIA
Every capillary and/or vascular endothelium resides in the basement membrane (BM) which is rich in extracellular matrix (ECM). In general, the endothelial cell derived matrix metalloproteinase (MMP), tissue inhibitor of metalloproteinase (TIMP) and nitric oxide (NO) are in ternary complex [Tyagi et al., 2005]. The ECM separates the endothelium from the under lying muscle [Harker et al., 1974; Hashimoto et al., 2003]. The thickness of BM dictates the endothelial-derived factors to interact with the muscle. NO, particularly generated by constitutive eNOS or nNOS, plays a critical role not only in the physiology of cardiac, vascular endothelial function and blood flow but also plays a protective role in hypertensive heart disease [Shesely et al., 1996; Tyagi and Hayden, 2003]. The acute and/or chronic deficiency in NO by inhibitor, L-NAME, in vitro and in vivo causes hypertension and cardiovascular, renal and neuronal remodeling [Massion and Balligand, 2007].
The location of NO generation probably is the most important aspect to determine the protective versus detrimental effects of NO. The NO generated by inducible nitric oxide synthase (iNOS) in the vicinity of super oxides generates peroxynitrite, nitration of the tyrosine residues in protein such as the TIMP in ternary MMP/NO/TIMP complex. This event depletes the NO in the vicinity of endothelium, causing decrease in NO bio-availability and activating the latent MMP [Tyagi et al., 1993]. Although iNOS is induced in many pathological conditions, the studies in ischemia reperfusion demonstrated cardioprotective role of iNOS-generated NO [Jones et al., 2005]. Therefore, a controversy exists regarding the detrimental versus cardio protective role of iNOS in ischemic heart disease [Lefer, 2006]. Although increased levels of homocysteine (Hcy) known as hyperhomocysteinemia (HHcy) induces iNOS, facilitates nitrotyrosine formation, the differential role of iNOS versus constitutive nitric oxide synthase (eNOS and nNOS) in Hcy-mediated nitrotyrosine generation and matrix remodeling is unclear [Jin et al., 2007]. In addition, it is unclear whether nNOS derived from muscle contributes relaxation and blood flow regulation [Tyagi et al., 2005]. We hypothesized that Hcy generated nitrotyrosine by inducing nitric oxide in the vicinity of endothelium and thereby attenuated endothelial-myocyte (E-M) coupling (i.e., endothelial-dependent relaxation), by sequestering available nitric oxide.
MATERIALS AND METHODS
ANIMAL MODELS
The CBS−/+ (heterozygote knockout) mice were used as an experimental model of mild hyperhomocysteinemia (HHcy). The CBS−/+ and iNOS−/− mice were obtained from Jackson Laboratories. Because CBS homozygote (−/−) dies at the age of 3–4 weeks [Watanabe et al., 1995], the heterozygous knockouts are appropriate to make cross bred line with iNOS KO mice. Therefore, we created mild HHcy in iNOS−/− by crossing CBS−/+ mice with iNOS−/−. This created double knockout (CBS−/+/iNOS−/−) line after six times back crossing at the breeding facility of the University Of Louisville School Of Medicine. The breeding protocol provided four groups of mice: (1) CBS−/+; (2) iNOS−/−; (3) CBS−/+/iNOS−/− (double knockout); and (4) CBS+/+/iNOS+/+ (wild type, WT).
GENOTYPE AND PHENOTYPE DETERMINATION
Tail biopsies were obtained from mice at 4 weeks of age. Total genomic DNA was isolated by DNAZolTM solution according to the manufacturer’s recommendation. The concentration of isolated DNA was measured using an optical density of one at 260 nm of 50 μg/ml of DNA. DNA was amplified by PCR for sequences in intron 3 and Neo insert in CBS−/+ containing mouse lines. The PCR primers were as follows: 5′-GCCTCTGTCTGCTAACCTA-3′; 5′-GAG-GTCGACGGTATCGATA-3′ [Watanabe et al., 1995]. The product was identified by two bands (CBS−/+) with differing electrophoretic mobility. The phenotype was determined by measuring the Hcy levels in the blood obtained from the tail vein, by HPLC separation and spectrophometric titration with dithio-bis-nitrobenzoate with absorption measured at 412 nm, using ε412 nm of 13,600 M−1 cm−1 [Tyagi et al., 1998]. The iNOS genotyping was carried out using three primer sets: (1) 5′-ACATGCAGAATGAGTACCGG-3′ (oIMR1216); (2) 5′-TCAACATCTCCTGGTGGAAC-3′ (oIMR1217); and (3) 5′-AATAT-GCGAAGTGGACCTCG-3′ (oIMR1218), as described by Jackson laboratory.
ANIMAL HOUSING
Animal room temperature was maintained between 22 and 24°C. A 12-h light-dark cycle was maintained by artificial illumination. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Louisville School of Medicine, according to the National Institute of Health Guidelines for animal research. The animals were fed standard rodent chow and water ad libitum. Levels of water and food intake, and changes in body weight were measured every other day.
NO2/NO3 MEASUREMENTS
Nitric oxide (NO) generation was determined by measuring total NO2/NO3 contents by Griess method, using microplate reader.
NITROTYROSINE DETERMINATION
The LV tissue homogenates were prepared. The levels of total 3-nitrotyrosine was measured by Western blot analysis using antinitrotyrosine antibody (Upstate Technologies). The intensities of color generated by the nitrotyrosine and secondary antibody (alkaline phosphatase conjugate) were scanned by a densitometer. The amounts of total nitrotyrosine in the LV tissue homogenates were estimated by comparing with standards, using 3-nitrotyrosine as standard dot-blot on membranes.
MMP ACTIVITY
Gelatinolytic activity of MMP-2 and 9 was measured by 1%-gelatin gel zymography as described earlier [Tyagi et al., 1993]. The lytic activities in the bands were scanned. The amounts related to standard purified MMP-2 and -9 were calculated in the samples [Tyagi et al., 1993].
HISTOLOGY
The heart was excised and weighed for comparisons between control and experimental groups. Portions of the LV were fixed in 10% zinc formalin for histology, or frozen in liquid nitrogen for tissue analysis of MMP, nitrotyrosine, and collagen. Serial tissue sections were compared with routine H&E stain for tissue orientation and cellularity. Masson’s trichrome stains were employed for characterizing collagen and elastic fibers (fibrosis) [Tyagi et al., 1995].
COLLAGEN
The pro-fibrotic collagen contents of the LV tissues were measured biochemically [Hodgkin et al., 1992]. LV rings used for functional studies were used to isolate and estimate collagen contents. Tissue was autoclaved twice in double-distilled water at 228°F for 3 h. The supernatant was collected to remove soluble collagenous material. The rest of the tissue was defatted by a chloroform/methanol solution (2:1) and dried to a constant weight. The collagen-free and fat-free specimen were then dissolved in 0.1 N NaOH at 100°C for 30-min. This procedure removed nonelastic proteins and gives maximum recovery of major elastin specific crosslink desmosines. After rehydration in Tris-Cl (pH 7.5), samples were digested with 1 mg tissue/20 μg thermolysin. The digested solution was analyzed for desmosine absorption at 320 nm (ε320 nm 7,850 M−1 cm−1). There are 7.5 desmosines per tropoelastin molecule. From this absorption, the concentration of elastic proteins was measured. Non-elastic tissue (cartilage) was used as reference. Another set of tissue was analyzed biochemically for collagen content. Samples were defatted as described above. Each sample was hydrolyzed in 0.5 ml of 6 N HCl under vacuum at 115°C for 24 h. After reconstitution in 4 ml of double-distilled water, the hydroxyproline content was determined spectrophotometrically [Hodgkin et al., 1992]. The collagen content was calculated by multiplying hydroxyproline levels by a factor of 7.46, assuming that hydroxyproline constitutes 13.4% of the collagen molecule.
LVH
The myocyte volume was estimated by a digital microscope with 100× objectives, in fixed grid, for both the control and the experimental groups [Tamura et al., 2000]. The increase in myocyte volume was considered as LVH [Tamura et al., 2000].
ENDOTHELIAL-MYOCYTE FUNCTION
The hearts were arrested in diastole by injecting 0.2 ml/100 g body weight of 20% KCl solution (I.V.). The LV and RV were separated. LV wall thickness and diameter (mm) were measured by a digital micrometer. The “deli” (i.e., doughnut) shape LV rings were mounted in a tissue myobath [Cox et al., 2002]. One of two mounting wires was connected to a force transducer. The ring was stretched and brought to resting tension at which 10 mM endothelin-1 (ET-1) was added to a bath. At the maximum ET-1 contraction, acetylcholine and/or BK (endothelial-dependent) or nitroprusside (endothelial-independent) were added.
Dose-response curves were generated. The % relaxation was calculated based on 100% contraction to ET-1. The data was fitted to a non-linear least squares equation: % Relaxation = (A/(1+expB(Dose–C))) + D, where A, C, and D are constants. The validity of measurements regarding endocardial endothelial function using “deli” shape LV rings was established by measuring responses to various cardiotonic agents [Mujumdar et al., 2001].
STATISTICAL ANALYSIS
Values are given as average ± SD from n = 6 in each group. Differences between groups were evaluated by using a two-way ANOVA, followed by the Bonferroni post-hoc test [Tarone, 1990], focusing on the respective effects of CBS−/+ on iNOS−/−. The results of the two-way comparisons are reported as: *, compared to CBS−/+ or iNOS. **or ***, compared to wild type. A P < 0.05 was considered significant.
RESULTS
GENOTYPE AND PHENOTYPE OF DOUBLE TRANSGENICS
The PCR analysis revealed knockout for iNOS (homozygote) and CBS (heterozygote) genes, as identified by one and two PCR products, respectively (Fig. 1). Although we did not measured activity levels of eNOS and/or nNOS, the levels of NO2/NO3 were higher, and Hcy were lower in WT mice. Interestingly, the levels of NO2/NO3 were lower and Hcy were higher in double KO mice (Table I and Fig. 2). These results suggested the match of genotype with phenotype.
Fig. 1.

Genotype of iNOS (A), and CBS (B): Tail tissue DNA was analyzed for iNOS and CBS using respective primers by PCR. In iNOS, wild-type (+/+) copy at kb and disrupted DNA copy (−/−) at 6.3 kb was observed. In CBS, 1.5 kb DNA was observed in wild-type, however, in heterozygote (−/+) two DNA bands were observed.
TABLE I.
Plasma Hcy, μmol/L, NO2/NO3 nmol/L, and Gravimetric Parameters: Body Weight (BW), Heart Weight (HW), and Kidney Weight (KW) in Grams
| Double KO | CBS−/+ | iNOS−/− | WT | |
|---|---|---|---|---|
| Hcy | 12.3 ± 2.9* | 11.5 ± 3.1 | 3.4 ± 1.2 | 3.2 ± 1.0 |
| NO2/NO3 | 2.5 ± 1.3* | 45.0 ± 5.6 | 3.1 ± 1.1 | 42.0 ± 4.8 |
| BW | 23 ± 1 | 22 ± 1 | 23 ± 1 | 25 ± 1 |
| HW | 0.22 ± 0.02* | 0.19 ± 0.01 | 0.22 ± 0.01* | 0.18 ± 0.02 |
| KW | 0.23 ± 0.01* | 0.24 ± 0.02* | 0.21 ± 0.01 | 0.21 ± 0.02 |
P <0.05 compared with wild-type.
Fig. 2.
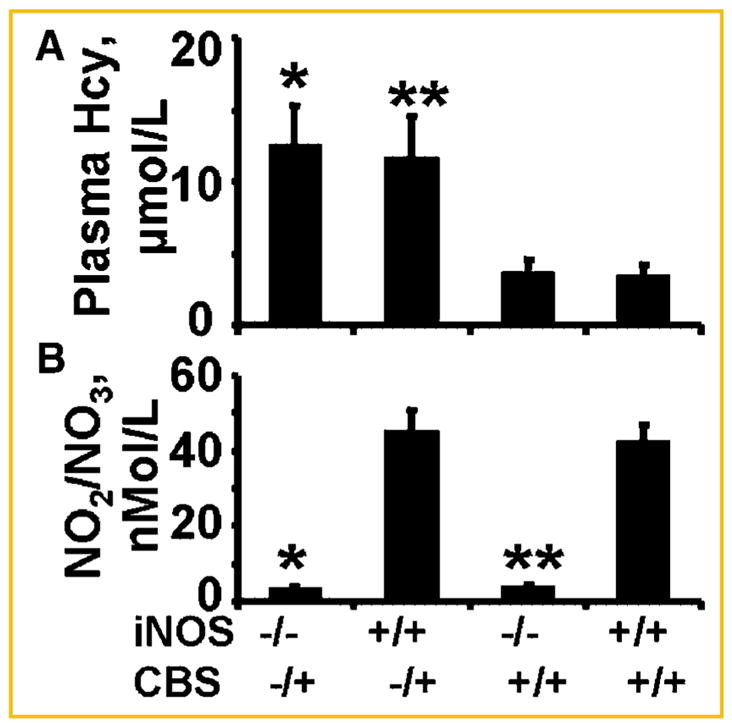
Phenotype of iNOS−/− by the measurements of NO2/NO3 (B) and CBS−/+ by the measurements of Hcy (A) in tail vein blood. The plasma was analyzed by Griess method for NO and Hcy by HPLC and spectrophotometer. N = 6 in each group; *P <0.05 compared with iNOS−/− for NO2/NO3 levels, and compared with CBS−/+ for Hcy levels. **P <0.05 compared with wild-type.
NITROTYROSINE
The levels of nitrotyrosine in CBS−/+ mice was significantly higher than WT as well as in any other group, suggesting that CBS−/+ (HHcy) is sufficient in generating nitrotyrosine with or without the iNOS (Fig. 3).
Fig. 3.

The LV tissue homogenates was analyzed for total 3-nitrotyrosine levels. A: Representative the band at 66 kDa in SDS–PAGE followed by Western blot analysis using 3-nitrotyrosine antibody was shown as marker of nitrityrosinylation. Identical amount of total protein was loaded onto each lane. Membranes were re-probed for beta actin. B: Scanned bands unit data of total 3-nitrityrosine/actin ratio in double KO (iNOS−/−/CBS−/+), CBS−/+, iNOS−/−, and wild-type (iNOS+/+/CBS+/+) mice. N = 6 in each group; *P <0.05 compared with iNOS−/− or with CBS−/+ group. **P <0.05 compared with wild-type.
MMP-2 AND -9
There was significant increase in MMP-2 activity in iNOS−/−, CBS−/+, and double KO as compared to WT mice hearts, suggesting constitutive role of MMP-2 in remodeling. However, in double KO and CBS−/+, there was higher induction of MMP-9 activity (Fig. 4). The results suggested that CBS−/+ induced MMP-9 activity with or without the iNOS.
Fig. 4.
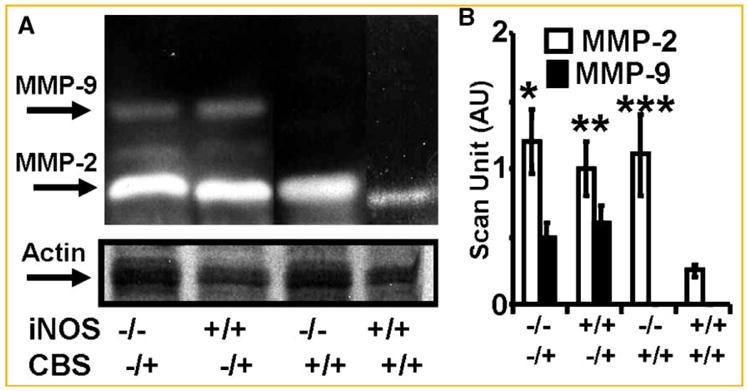
The LV tissue homogenates was analyzed for MMP activity by gelatin gel zymography. A: The bands at 92 and 72 kDa for MMP-9 and -2, in SDS–PAGE-zymography, were analyzed for MMP activity. Identical amount of total protein was loaded onto each lane. Identical gels were blotted for beta actin. B: Scan unit data of MMP-2 and -9 activity normalized with actin in double KO (iNOS−/−/CBS−/+), CBS−/+, iNOS−/−, and wild type (iNOS+/+/CBS+/+) mice. N = 6 in each group; *P <0.05 compared with iNOS−/− or with CBS−/+ group. **P <0.05 compared with wild type. ***P <0.05 compared with WT.
CARDIAC FIBROSIS, COLLAGEN, AND LVH
Although there was fibrosis in iNOS−/− as compared to WT, but the fibrosis was higher in perivascular and interstitial areas in CBS−/+ and CBS−/+/iNOS−/− double KO as compared to iNOS−/+ or WT mice (Fig. 5). The collagen measurements suggested the similar trend in collagen contents (Fig. 6A). In contrast, there was LVH in iNOS−/−, and there was very little effect on LVH due to CBS−/+ (Fig. 6B). These results suggested that cardiac fibrosis and collagen deposition are influenced by Hcy, however, the LVH and myocyte hypertrophy were primarily influenced by iNOS.
Fig. 5.

Histological analysis of the hearts from (1) double KO (iNOS−/−/CBS−/+), (2) CBS−/+, (3) iNOS−/−, and (4) wild-type (iNOS+/+/CBS+/+) mice. The 10 μm paraffin imbedded tissue sections were stained by Trichrome-blue for collagen.
Fig. 6.
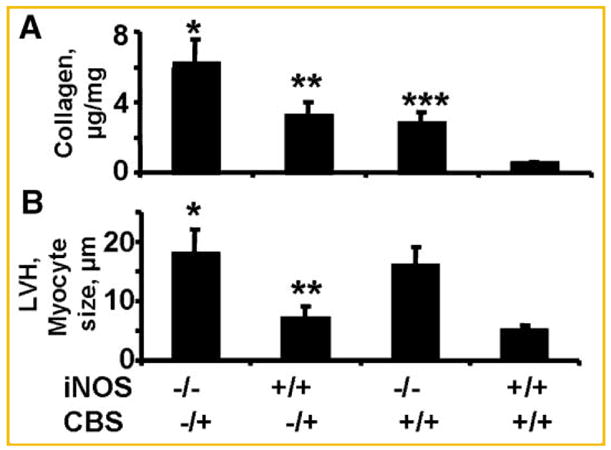
A: Biochemical analysis to total hydroxyl praline collagen, and (B) LVH by myocyte size measured by a micro meter in: (1) double KO (iNOS−/−/CBS−/+), (2) CBS−/+, (3) iNOS−/−, and (4) wild-type (iNOS+/+/CBS+/+) mice. N = 6 in each group; *P <0.05 compared with iNOS−/− or with CBS−/+ group. **P <0.05 compared with wild-type. ***P <0.05 compared with WT.
ENDOTHELIAL-MYOCYTE FUNCTION
Because acetylcholine (ACH) is endothelial-dependent relaxing factor, bradykinin (BK) is endothelial-dependent hyperpolarizing factor, and nitroprusside (NPR) is endothelial-independent relaxing factor, it is possible to determine the distinct contribution of endothelial and myocyte cell function through the use of the exogenous relaxing factors (ACH, BK, and NPR). We observed normal relaxation responses, in ET-1 precontracted cardiac rings, from WT hearts, however, the responses in double KO rings were paradoxic contraction (Fig. 7). Although the responses to ACH were attenuated in CBS−/+, iNOS−/−, and double KO as compared to WT, the responses to NPR were only attenuated in double KO (Fig. 8). These results suggested endothelial dysfunction occurred in CBS−/+, iNOS−/−, and double KO mice, whereas myocytes were dysfunctional only in CBS−/+/iNOS−/− double KO.
Fig. 7.
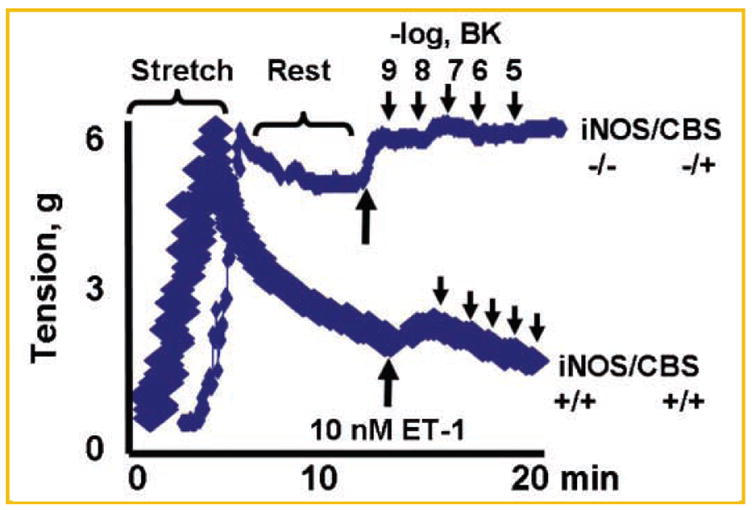
A typical experiment of E-M function in cardiac rings: The LV rings were stretched, rested, and activated by calcium. The bradykinin (BK) was used to determine the endothelial-dependent (EDHF) cardiotonic agent to relax the cardiac rings from double KO (iNOS−/−/CBS−/+) and WT (iNOS+/+/CBS+/+) mice. Note: the ring from double KO has no response (tone) to BK, where as ring from WT relaxed normally with BK.
Fig. 8.

The dose response curves: (A) for bradykinin (BK); (B) for acytylcholine (ACH); and (C) for sodium nitroprusside (Npr) in cardiac rings from (1) double KO (iNOS−/−/CBS−), (2) CBS −, (3) iNOS−/−, and (4) wild-type (iNOS+/+/CBS+/+) mice. N = 6 in each group; *P <0.05 compared with iNOS−/− or with CBS−/+ group.
DISCUSSION
It was novel that we demonstrated Hcy generated the nitrotyrosine with or without the iNOS (Fig. 3). This may indicate a prominent role of constitutive, eNOS or nNOS, nitric oxide in generating nitrotyrosine by Hcy. This suggested that during HHcy, Hcy in the vicinity of endothelium utilized the NO from endothelium in causing the structural and functional abnormalities in the endomyocardium.
There was fibrosis in CBS−/+ as compared to WT mice, the fibrosis was higher in double knockouts. There was marginal LVH in CBS−/+ mice, the LVH was higher in iNOS−/− mice. Although the levels of MMP-2 were increased in CBS−/+ as well as in iNOS−/− as compared to WT mice, there was higher induction of MMP-9 expression in CBS−/+ with or without iNOS as compared to WT. Our data suggested a coordination between LVH, fibrosis and possibly angiogenesis as the hallmark of compensatory response to heart failure; however, a disruption in coordination between LVH and angiogenesis contributed to the transition to cardiac fibrosis and failure [Shiojima et al., 2005]. However, the mechanism of this coordination was unclear. We suggested that, in the absence of blood supply to the myocardium at risk, the lack of angiogenesis by increase angiostatic factors leads to fibrosis and LVH. Although, it was a paradox that activation of MMP was needed to disruption of the matrix during angiogenesis and to create new blood vessels, or opening the collaterals, however, the peptides generated by MMP activation were angiostatic. Studies by O’Reilly [1997] and O’Reilly et al. [1997], suggested endostatin and angiostatin generated by MMP action on collagen-18 and plasminogen, respectively, inhibit tumor angiogenesis. In addition, the vasostatin was generated by degradation of calreticulin [Pike et al., 1998]. Tumstatin [Maeshima et al., 2002], arresten [Colorado et al., 2000], and canstatin [Kamphaus et al., 2000] generated by degradation of collagen-4. Restin [Ramchandran et al., 1999] was generated by degradation of collagen-15. The increase in MMP (Fig. 4) was consistent with the notion that MMPs cause discoordinated matrix degradation leading to decompensatory LVH and heart failure by generating angiostatic factors, explaining how MMP, TIMP, and LVH co-exist.
Sixteen percent of the myocardial mass is capillaries, including the lumen and endothelium [Hoppeler and Kayar, 1988]. The capillary endothelium is embedded in the muscle, and plays a very important role in myocardial diastolic relaxation [Mebazaa et al., 1995]. Nitric oxide (NO) generation from the endocardial endothelium contributes to myocyte contraction, relaxation, and heart rate [Pinsky et al., 1997]. A gradient of NO concentration (i.e., high in endocardium and low in midmyocardium) has been depicted [Pinsky et al., 1997] that is consistent with the notion that there is more capillary endothelium in the endocardium than in epi- or mid-myocardium [Scarabelli et al., 2001]. The importance of endocardial endothelium in cardiac contraction/relaxation is illustrated by attenuation of the responses to CaCl2 and acetylcholine in the endothelium-denuded myocardium [Gattuso et al., 1999]. Furthermore, the Hcy-mediated contractile dysfunction was amplified by angiotensin II and endothelin-1 [Tyagi et al., 1999]. Here we suggested that endothelial dependent bradykinin (BK) and acetylcholine (ACH) responses were severely attenuated in double knockouts as compared to WT mice. The responses to endothelial independent nitroprusside (NPR) were only attenuated in double knockouts and not in CBS−/+ or iNOS−/− mice.
We studied two forms of endothelium-myocyte (E-M) coupling. The structure coupling implies the thickness of the peri-capillary matrix between the E and M. Accumulation of interstitial collagen between E and M increases distance from E to M. The functional coupling implies the efficiency of transport of endothelial-derived cardio-active agents to the cardiac muscle. The increase in distance from E to M impairs local endothelial-derived NO diffusion to the cardiac muscle and interferes with cardiac diastolic relaxation.
LIMITATIONS
Although the source of superoxide in the hyperhomocysteinemic mice was not determined in this study, previously we demonstrated that Hcy induced NADPH oxidase that promoted ONOO- and derivatized 3-nitotyrosine. Although the study may reflect a simple series of observations without clear mechanistic links; such as what proteins undergo 3-nitrotyrosination, and what effect does this modification have on protein function? Previously we demonstrated that TIMP-4 was nitrated in oxidative stress [Tyagi et al., 2005]. Here we measured total levels of 3-nitrotyrosination that correlated with fibrosis, myocardial dysfunction, endothelial dysfunction, and LVH in transgenic animals.
CONCLUSION
The results of this study suggested that Hcy generated nitrotyrosine in the vicinity of endothelium, causing nitration of the matrix proteins and fibrosis. This led to impair myocyte’s ability to relax, and caused myocyte hypertrophy and disconnect between endothelium and myocyte. The schematics in Figure 9 showed the hypothesis and molecular connection in endothelial myocyte matrix in cardiac remodeling during hyperhomocysteinemia.
Fig. 9.
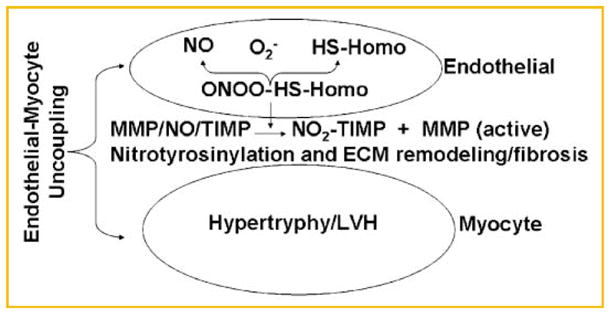
Hypothesis suggesting a plausible mechanism of generation of nitrotyrosine in the vicinity of endothelium and the formation of fibrosis in between E and M, causing E-M uncoupling.
Acknowledgments
NIH; Grant numbers: HL-71010, HL-74185, HL-88012, NS-51568.
A part of this study were supported by NIH grants HL-71010, HL-74185, HL-88012 and NS-51568.
References
- Colorado PC, Torre A, Kamphaus G, Maeshima Y, Hopfer H, Takahashi K, Volk R, Zamborsky ED, Herman S, Sarkar PK, Ericksen MB, Dhanabal M, Simons M, Post M, Kufe DW, Weichselbaum RR, Sukhatme VP, Kalluri R. Anti-angiogenic clues from vascular basement membrane collagen. Cancer Res. 2000;6:2520–2526. [PubMed] [Google Scholar]
- Cox M, Sood H, Hunt M, Chandler D, Henegar J, Aru G, Tyagi SC. Apoptosis in the left ventricle of chronic volume overload causes endocardial endothelial dysfunction in rats. Am J Physiol. 2002;282:H1197–H1206. doi: 10.1152/ajpheart.00483.2001. [DOI] [PubMed] [Google Scholar]
- Gattuso A, Mazza R, Pellegrino D, Tota B. Endocardial endothelium (EE) mediates luminal acetylcholine-nitric oxide signaling in isolated frog heart. Am J Physiol. 1999;276:H633–H641. doi: 10.1152/ajpheart.1999.276.2.H633. [DOI] [PubMed] [Google Scholar]
- Harker LA, Slichter SJ, Scott CR, Ross R. Homocysteinemia, vascular injury and arterial thrombosis. N Engl J Med. 1974;291:537–543. doi: 10.1056/NEJM197409122911101. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Wen G, Lawton MT, Boudreau NJ, Bollen AW, Yang GY, Barbaro NM, Higashida RT, Dowd CF, Halbach VV, Young WL. Abnormal expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in brain arteriovenous malformations. Stroke. 2003;34:925–931. doi: 10.1161/01.STR.0000061888.71524.DF. [DOI] [PubMed] [Google Scholar]
- Hodgkin DD, Gibert RD, Ros PJ, Sandberg LB, Boucek RJ. Dietary lipid modulation of connective tissue matrix in rat abdominal arota. Am J physiol. 1992;262:R389–R394. doi: 10.1152/ajpregu.1992.262.3.R389. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Kayar SR. Capillary and oxidative capacity of muscles. News Physiol Sci. 1988;3:113–116. [Google Scholar]
- Jin L, Caldwell RB, Li-Masters T, Caldwell RW. Homocysteine induces endothelial dysfunction via inhibition of arginine transport. J Physiol Pharmacol. 2007;58:191–206. [PubMed] [Google Scholar]
- Jones SP, Greer JJ, Ware PD, Yang J, Walsh K, Lefer DJ. Deficiency of iNOS does not attenuate severe congestive heart failure in mice. Am J Physiol Heart Circ Physiol. 2005;288:H365–H370. doi: 10.1152/ajpheart.00245.2004. [DOI] [PubMed] [Google Scholar]
- Kamphaus GD, Colorado PC, Panka DJ, Hopfer H, Ramchandran R, Torre A, Maeshima Y, Mier JW, Sukhatme VP, Kalluri R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J Biol Chem. 2000;275:1209–1215. doi: 10.1074/jbc.275.2.1209. [DOI] [PubMed] [Google Scholar]
- Lefer DJ. Induction of HIF-1alpha and iNOS with siRNA: A novel mechanism for myocardial protection. Circ Res. 2006;98:10–11. doi: 10.1161/01.RES.0000200398.52220.cc. [DOI] [PubMed] [Google Scholar]
- Maeshima Y, Sudhakar A, Lively JC, Ueki K, Kharbanda S, Kahn CR, Sonenberg N, Hynes RO, Kalluri R. Tumstatin, an endothelial cell specific inhibitor of protein synthesis. Science. 2002;295:140–143. doi: 10.1126/science.1065298. [DOI] [PubMed] [Google Scholar]
- Massion PB, Balligand JL. Relevance of nitric oxide for myocardial remodeling. Curr Heart Fail Rep. 2007;4:18–25. doi: 10.1007/s11897-007-0021-6. [DOI] [PubMed] [Google Scholar]
- Mebazaa A, Wetzel R, Cherian M, Abraham M. Comparison between endothelial and great vessel endothelial cells: Morphology, growth, and prostaglandin release. Am J Physiol. 1995;268:H250–H259. doi: 10.1152/ajpheart.1995.268.1.H250. [DOI] [PubMed] [Google Scholar]
- Mujumdar VS, Smiley LM, Tyagi SC. Activation of matrix metallo-proteinase dilates and decreases cardiac tensile strength. Intern J Cardiol. 2001;79:277–286. doi: 10.1016/s0167-5273(01)00449-1. [DOI] [PubMed] [Google Scholar]
- O’Reilly MS. Angiostatin: An endogenous inhibitor of angiogenesis and tumor growth. EXS. 1997;79:273–294. [PubMed] [Google Scholar]
- O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, Nakhasi H, Teruya-Feldstein J, Wirth P, Gupta G, Tosato G. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med. 1998;188:2349–2356. doi: 10.1084/jem.188.12.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinsky DJ, Patton S, Mesaros S, Brovkovych V, Kubaszewski E, Grunfeld S, Malinski T. Mechanical transduction of NO synthesis in the beating heart. Circ Res. 1997;81:372–379. doi: 10.1161/01.res.81.3.372. [DOI] [PubMed] [Google Scholar]
- Ramchandran R, Dhanabal M, Volk R, Waterman MJ, Segal M, Lu H, Knebelmann B, Sukhatme VP. Antiangiogenic activity of restin, NC10 domain of human collagen XV: Comparison to endostatin. Biochem Biophys Res Commun. 1999;255:735–739. doi: 10.1006/bbrc.1999.0248. [DOI] [PubMed] [Google Scholar]
- Scarabelli T, Stephanou A, Rayment N, Pasini E, Comini L, Curello S, Ferrari R, Knight R, Latchman D. Apoptosis of endothelial cells precedes myocytes cell apoptosis in ischemia/reperfusion injury. Circulation. 2001;104:353–256. doi: 10.1161/01.cir.104.3.253. [DOI] [PubMed] [Google Scholar]
- Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressure in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2059–2064. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Said S, Harris J, Lu W, Gerdes AM. Reverse remodeling of cardiac myocyte hypertrophy in hypertension and failure by targeting of the renin-angiotensin system. Circulation. 2000;102(2):253. doi: 10.1161/01.cir.102.2.253. [DOI] [PubMed] [Google Scholar]
- Tarone RE. A modified Bonferroni method for discrete data. Biometrics. 1990;46:515. [PubMed] [Google Scholar]
- Tyagi SC, Hayden MR. Role of nitric oxide in matrix remodeling in diabetes and heart failure. Heart Fail Rev. 2003;8:23–28. doi: 10.1023/a:1022138803293. [DOI] [PubMed] [Google Scholar]
- Tyagi SC, Ratajska A, Weber KT. Myocardial matrix metalloproteinase(s): Localization and activation. Mol Cell Biochem. 1993;126:49–59. doi: 10.1007/BF01772207. [DOI] [PubMed] [Google Scholar]
- Tyagi SC, Meyer L, Schmaltz RA, Reddy HK, Voelker DJ. Proteinases and restenosis in human coronary artery: Extracellular matrix production exceeds the expression of proteolytic activity. Atherosclerosis. 1995;116:43–57. doi: 10.1016/0021-9150(95)05520-7. [DOI] [PubMed] [Google Scholar]
- Tyagi SC, Smiley LM, Mujumdar VS, Clonts B, Parker JL. Reduction-oxidation (redox) and vascular tissue level of homocyst(e)ine in human coronary atherosclerotic lesions and role in vascular extracellular matrix remodeling and vascular tone. Mol Cell Biochem. 1998;181:107–116. doi: 10.1023/a:1006882014593. [DOI] [PubMed] [Google Scholar]
- Tyagi SC, Smiley LM, Mujumdar VS. Homocyst(e)ine impairs endocardial endothelial function. Canad J Physiol Pharmacol. 1999;77:950–957. [PubMed] [Google Scholar]
- Tyagi N, Moshal KS, Ovechkin AV, Rodriguez W, Steed M, Henderson B, Roberts AM, Joshua IG, Tyagi SC. Mitochondrial mechanism of oxidative stress and systemic hypertension in hyperhomocysteinemia. J Cell Biochem. 2005a;96:665–671. doi: 10.1002/jcb.20578. [DOI] [PubMed] [Google Scholar]
- Tyagi SC, Rodriguez W, Patel AM, Roberts AM, Falcone JC, Passmore JC, Fleming JT, Joshua IG. Hyperhomocysteinemic diabetic cardiomyopathy: Oxidative stress, remodeling, and endothelial-myocyte uncoupling. J Cardiovasc Pharmacol Ther. 2005b;10(1):1–10. doi: 10.1177/107424840501000101. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR, Maeda N. Mice deficient in cystathionine beta-synthase: Animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci. 1995;92:1585–1589. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]