

Abstract
A number of image processing parameters in the 3D reconstruction of a ribosome complex from a cryo-EM data set were varied to test their effects on the final resolution. The parameters examined were pixel size, window size, and mode of Fourier amplitude enhancement at high spatial frequencies. In addition, the strategy of switching from large to small pixel size during angular refinement was explored. The relationship between resolution (in Fourier space) and the number of particles was observed to follow a lin-log dependence, a relationship that appears to hold for other data, as well. By optimizing the above parameters, and using a lin-log extrapolation to the full dataset in the estimation of resolution from half-sets, we obtained a 3D map from 131,599 ribosome particles at 6.7 Å resolution (FSC=0.5).
1. Introduction
Slowly but steadily, the resolution of single-particle reconstructions (see Frank, 2006) has improved, with particles of high symmetry (icosahedral viruses, GroEL) taking the lead. For instance, GroEL, having 14-fold symmetry, is now around 4 Å (Ludtke et al., 2008), and two reoviruses have been solved to close-to-atomic resolution (Yu et al., 2008; Zhang et al., 2008). While the electron-optical resolution of modern electron microscopes is well sufficient to achieve atomic resolution, the struggle to reach this goal faces extraordinary difficulties, especially in the absence of symmetries. If we assume a well-tuned instrument with high coherence, then the limiting factors are specimen stage instability, electrostatic charging effects, conformational heterogeneity, and strong background noise due to the low dose and the large amount of inelastic scattering in ice. There is undoubtedly room for improvement in the preparation of the specimen on the EM grid. For instance, thickness of ice has been identified as one of the factors determining the resolution of the reconstruction (Stagg et al., 2006). However, once a suitable protocol has been found, and a dataset has been collected, there are a large variety of ways to proceed toward a reconstruction. It is this latter problem that we have addressed with a large data set of ribosome images.
The problem can be phrased in terms of the optimization problem posed by the single-particle reconstruction from multiple projections with unknown angles (Yang et al., 2005). This optimization problem is usually addressed by an iterative processing method, called angular refinement (Penczek et al., 1994), in which alternately two different problems are solved, one being the orientation of the projections with reference to an existing density map, the other being the reconstruction of a density map from the projections set whose angles have been determined in the previous round (see the general formulation of this approach given in Yang et al., 2005). It has been pointed out (Penczek et al., 1994) that this method will not necessarily result in the global solution. For this reason, there is room and promise for a heuristic exploration.
It is difficult to explore the various factors experimentally/computationally since angular refinement, the most time-consuming step, can take days or even weeks even on a computer cluster. The approach we have taken is to explore the various strategies with a smaller subset of the data first, then apply the strategy yielding the best solution we found to the full data set available. Obviously, despite efforts to perform the exploration systematically, it is in the nature of the problem that our study must fall far short of an exhaustive treatment.
Nevertheless, the study has resulted in a much superior density map of a ribosomal complex, 70S•Phe-tRNAPhe•EF-Tu•GDP•kir than reported before (Valle et al., 2002; 2003). The quality of the map can be characterized by comparison of various components (ribosomal proteins or RNA) with X-ray structures, and is reflected by the experimental resolution in the range of 6.7 Å.
2. Investigation of Variables Limiting Resolution
2.1 Principle and Practice of Resolution Estimation
In order to investigate what processing method might lead to better results, we need to employ an appropriate measure of reconstruction quality. Conventionally, the Fourier shell correlation (FSC) curve (Saxton and Baumeister, 1982; Harauz and van Heel, 1986) is used, which is obtained by a comparison of reconstructions from randomly picked halfsets of the dataset. However, we were frustrated in our initial analysis since the FSC curve as a whole (not just the figure derived from it by setting a threshold) responded only marginally to changes in processing that produced a strongly different visual appearance in the reconstruction. In other words, we lacked a quantitative confirmation for improvements in quality quite obvious to the eye.
We then discovered that the improvements were not reflected by the FSC since they affected only that part of the density map that was occupied by the reconstructed molecule. In contrast, the FSC as applied to the whole volume measured the reproducibility of the density map as a whole, including the surroundings of the molecule.
Briefly, in a density map obtained by 3D reconstruction, the reconstructed molecule is usually surrounded by speckles of density termed “clutter.” The clutter becomes strongly visible at lowered density threshold. It is generated by superposition of projection density exterior to the particle itself, and carries a unique signature tied to the unique set of angles of the projections entering the reconstruction. Since the set of angles is different for the two halfsets, the clutter signature is irreproducible, leading to a steep falloff of the FSC for the periphery. Since the clutter occupies a relatively large portion of the total volume, it has a large effect on the resolution estimation. When resolution is measured by comparison of two halfset reconstructions, inconsistencies in the clutter will therefore lead to under-reported resolution. Using a similar rationale – to remove peripheral, disordered portions of the molecule structure -- Stewart and coworkers (2000) introduced soft masking to obtain a more realistic resolution estimate for the resolution of the icosahedrally ordered viral capsid of Ad2 virus (for another example, see Andersen et al., 2006). A smooth mask must be used, evidently, because application of a binary-valued mask to both reconstructions would lead to a false indication of reproducibility, resulting in a grossly over-reported resolution. The mask function which minimizes such artifacts for a global particle is a rotationally symmetric Gaussian mask, placed at the center of the particle, with a standard deviation corresponding to the particle radius.
The effect is demonstrated (Fig. 1) with one of the reconstructions (Fig. 2G) to be presented below. When using a reduced density threshold, the clutter outside the molecule becomes apparent. Without use of the Gaussian mask, the resolution is 12.4 Å, while with this mask in place, the resolution improves to 10.1 Å. Investigation of the part of the volume that was rejected by the mask (i.e., accepted by the mask complement) with the FSC gives a resolution of 19.7 Å, confirming that the reproducibility is much worse at the peripheral part of the volume. (The resolution of ~20 Å for the peripheral part of the volume may seem surprisingly high, but it is due to the fact that the soft mask complement inevitably catches some of the peripheral, well-reproducible ribosome mass).
Figure 1.
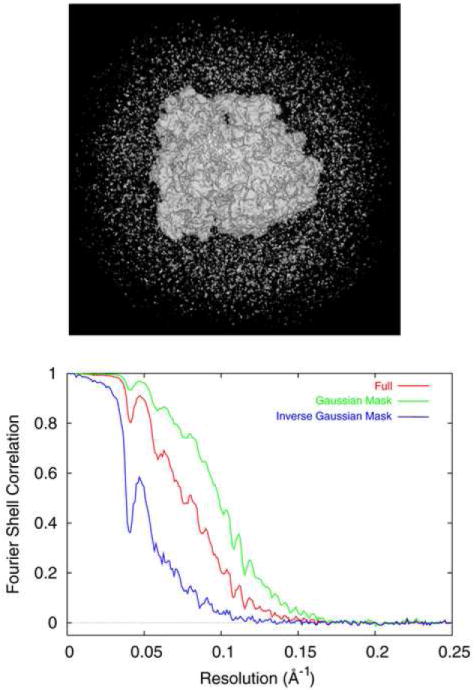
(A) Clutter surrounding the ribosome in reconstruction shown in Fig. 2G. (B) FSC curves computed for the full volume (red), after soft-masking (green), and for the clutter excluded by soft-masking (blue).
Figure 2.
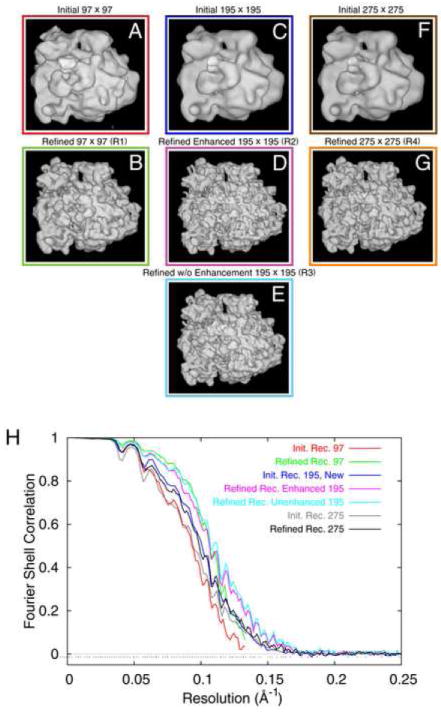
Reconstructions from a 52,000-particle test dataset, (A,B) initial and refined reconstruction, with arrays 2x decimated; (C,D) initial and refined reconstruction from the non-decimated data; (E) same as (D), except that amplitude enhancement was used in each step of the refinement; (F,G) same as (C,D) but with window size enlarged from 195 × 195 to 275 × 275. (H) FSC curves for (A-G), color-coded to match color surrounding each reconstruction.
A few tests with varying mask widths (+/-20% from the approximate radius of the ribosome, 130 Å) show that the reported resolution is to some extent, but not critically, dependent on the choice. By using the rule of Gaussian halfwidth = particle radius, we make sure that all nominal resolution figures for the ribosome are comparable. In the following, we therefore have used a half-width r = 130 Å throughout. Thus, for the purpose of this study, we give two resolution figures for each density map, the first with, and the second, in parenthesis, without application of the Gaussian mask to both half-set reconstructions being compared by the Fourier Shell Correlation. In the course of the presentation, it will became clear that only the former figure is sensitive to the various choices of parameters and strategies, and that it may best reflect the reproducible overall resolution of the particle region of the EM density map.
2.2 Creation of the Test Data Set
To determine which processing parameters affect resolution, a small test data set of 51,622 particles was created and processed using standard methods (Frank et al., 2000; Shaikh et al., 2008) to produce a baseline reconstruction. A sample of the complex described in Valle et al. (2003) and known to be quite stable was prepared (Grassucci et al., 2007a) and imaged on film in the standard manner (Grassucci et al., 2007b).
To briefly summarize, the sample consisted of E. coli ribosome stalled in the pre-accommodation state after GTP hydrolysis with the antibiotic kirromycin. Another preparation of this sample was previously (Valle et al., 2003) found to have tRNA in both the E and P sites, with aa-tRNA-EF-Tu-GDP also bound in greater than 70% of the ribosome population. Micrographs were recorded at low-dose conditions on an FEI Tecnai F30 Polara electron microscope operated with an accelerating voltage of 300kV, a magnification of ~38,000X and with the specimen cooled to liquid-nitrogen temperature (80° K). The micrographs were scanned with a Z/I Photoscan 2000 (Z/I Imaging Corp., Huntsville, Alabama) with a step size of 7 μm, resulting in a pixel size of 1.86 Å on the object scale.
Image processing was performed as described (Shaikh et al., 2008), with modifications described below. Prior to particle selection, all digitized micrographs were decimated by a factor of two using the SPIDER “DC S” command, resulting in a pixel size of 3.76 Å on the object scale. Digitized micrographs were grouped according to their defocus values into twelve defocus groups, with average defocus values ranging from 2.41 to 3.89 μm. The average range within a defocus group was 57 nm, and the maximum range 97 nm. This rather coarse grouping was chosen initially for expediency as it reduces the total number of refinement cycles in the test calculations. Defocus groups with narrower ranges were taken later on with the larger, undecimated data set (see below). Particle selection started in the standard fashion (Shaikh et al., 2008) including automated particle windowing from digitized micrographs (Rath and Frank, 2004; Roseman, 2004). After a 3D projection alignment of windowed particles to 83 projection views of a known reference (Penczek et al., 1994; Gabashvili et al., 2000) created using an angular spacing of 15°, aligned particles were chosen by using classification (Roseman, 2004; T.R. Shaikh and J. Frank, in prep.). Briefly, particles that best match a given reference projection are low-pass filtered and are subjected to correspondence analysis. Factorial coordinates from correspondence analysis are then used to classify the filtered particles via the K-means algorithm for each reference view. Particles are then selected either individually, or as an entire class. The total number of particles used was 51,622, with a window size of 97 × 97 pixels, and this data set will be referred to as Data Set 1. Image processing continued in the standard manner (Shaikh et al., 2008), resulting in an initial 3D reconstruction with a resolution of 10.7 Å (11.9 Å) using a cutoff of 0.5 in the Fourier shell correlation (FSC) curve (Fig. 2A).
A cycle of angular refinement is defined in the following way: defocus group reconstructions from the previous cycle are merged and CTF-corrected, to yield an intermediate reconstruction. This reconstruction is used as a 3D reference to refine data from each defocus group. For this purpose, to maximize the correlation signal, the 3D reference is first convoluted (via Fourier relationship) with the CTF of the current defocus group. In this manner, a new set of alignment parameters and Eulerian angles is obtained for each defocus group, and a new reconstruction is computed for each.
Several cycles of angular refinement were performed, gradually restricting the angular search range from completely unrestricted to 5°. Midway through the refinement the step size of the angular search was reduced from 2° to 1.5° (Shaikh et al., 2008). One of the parameters tested in the refinement was the mode of amplitude enhancement. Such enhancement is used to bring the average experimental amplitudes in each shell of the 3D Fourier transform to the level of low-angle solution scattering data (Gabashvili et al., 2000). This enhancement is normally used at the very end of the refinement (Gabashvili et al., 2000) but it has also lately been used after every iteration of the refinement (e.g., Connell et al., 2007). In this mode, the intermediate reconstruction from the previous step is first amplitude-enhanced before the individual CTFs are applied to it.
The idea of applying the enhancement after every step is that the accuracy of alignment may be enhanced due to the higher weight of high spatial frequencies in the correlation expressions. However, a concern and possible disadvantage is that the concurrent amplification of noise may result in erratic alignment errors. For Data Set 1, we used the repeated amplitude enhancement option, each time filtering the resulting density map to the nominal resolution calculated with the FSC=0.5 cutoff. After eleven iterations of refinement, a final reconstruction from Data Set 1 was obtained with a resolution of 9.4 Å (10.2 Å) at 0.5 FSC (Fig.2B). In the following, this reconstruction will be referred to as Reconstruction 1.
2.3 Examination of the Effect of Pixel Size
According to sampling theory, a continuous signal can be represented faithfully by a set of discrete samples if the signal is sampled at a rate of at least twice the signal’s highest spatial frequency. In digital image processing, numerical errors make it necessary to use at least a factor of three instead (see Frank, 2006). With a sampling rate of 3.76 Å per pixel, the highest resolution possible from the decimated micrographs used in Data Set 1 is therefore somewhere in the range between 7.5 and 11.3 Å. Since the resolution of Reconstruction 1 was toward the end of this range (9.4 Å (10.2Å)), the micrograph decimation was thought to be likely a limiting factor. The initial reconstruction of the test data set was therefore redone using the non-decimated, raw micrograph data.
The micrographs had originally been scanned at 7 μm, resulting in arrays with 1.86 Å per pixel. The positions of the final ~52,000 particles within their respective source micrographs were already known from the creation of Data Set 1 using the decimated micrographs. Using this information, and multiplying each coordinate by two to compensate for the initial decimation, it was possible to window each particle from the non-decimated micrographs with high precision. The new non-decimated particles were contained in arrays of 195 × 195 pixels. This data set will be referred to as Data Set 2. A new initial 3D reconstruction was created using Data Set 2 and the final refined alignment angles, rotations, and translations from the processing that led to Reconstruction 1. The resulting initial reconstruction (i.e., using the initial coarse angular grid of 15°) had a resolution of 9.9 Å (10.9 Å) (Fig. 2C).
Because this initial reconstruction was created using refined alignment information, the use of a large angular search range during the subsequent refinement was considered superfluous. The first two angular search ranges, unrestricted and 15°, were therefore not employed in this instance. After nine iterations of refinement, using amplitude enhancement after each iteration as described previously, the resolution of the refined density map went to 9.4 Å (10.3Å) at FSC=0.5 (Fig. 2D). This density map will be referred to as Reconstruction 2. Thus, after comparable efforts in image processing and refinement, doubling the micrograph’s sampling resolution by switching to the non-decimated data set produced a nominal resolution improvement of merely 0.05 Å (0.01 Å), which is insignificant. This leads to the conclusion that with the limited (N~ 52,000) dataset used in the test, the 2-fold coarser sampling, at 3.72 Å per pixel, is not a resolution-limiting factor.
2.4 Examination of the Effect of the Mode of Amplitude Enhancement
To investigate the effect of the iterative use of amplitude enhancement, the processing of Data Set 2 was repeated without employing it. The density map used as the initial reference was the 9.9-Å reconstruction from Data Set 2. Refinement processing for Data Set 2 was repeated as before, without the procedure of amplitude enhancement and low-pass filtering between each iteration that was used to obtain Reconstruction 2. The density map resulting from this processing method will be referred to as Reconstruction 3. According to the FSC=0.5 cutoff, the refined map of Reconstruction 3 had a nominal resolution of 8.9 Å (10.1Å) (Fig. 2E). Thus, with otherwise identical steps of image processing, the addition of iterative amplitude enhancement decreased the resolution achieved with this data set by ~0.5 Å (0.2Å). We conclude from this result that the application of amplitude enhancement after each iteration of angular refinement produced no benefit; on the contrary, it reduced the final resolution significantly.
All final reconstructions were routinely amplitude-enhanced, following the protocol spelled out by Gabashvili et al. (2000): by boosting their Fourier amplitudes so that they conformed to the amplitude profile measured by low-angle X-ray solution scattering of a 70S ribosome sample. As application of a final mere multiplicative factor, this correction has no effect on resolution.
2.5 Examination of the Effect of the Window Size
The choice of window size affects the CTF correction in a complicated way, which requires a brief explanation. There exist two different strategies for CTF correction. Following one strategy, the digitized micrographs are CTF-corrected before any additional processing is done, the other where uncorrected data are processed and corrected at the very end (e.g., Zhu et al.,1997; Gabashvili et al., 2000). We routinely use the second strategy, for various reasons which mostly have to do with the numerical behavior of filters for low SNR (see Frank, 2006). This strategy requires that the window containing the particle include sufficient surrounding background, equivalent in size to the range of the point-spread function placed on the boundary of the particle. For high defocus values, the radius of the point-spread function can be 10 Å and even more. Calculations on phantom data (unpublished results) were performed to determine the minimum window size needed to capture all information required in the CTF correction.
Based on these findings another data set, Data Set 3, was created. The particles for this data set were windowed with a size of 275 × 275 pixels (corresponding to 511 × 511 Å, in contrast to 363 × 363 Å for the smaller non-decimated window size) from the non-decimated micrographs. By applying the final alignment data from the refinement that created Reconstruction 1, the initial reconstruction of Data Set 3 was created. The initial reconstruction had a nominal resolution of 10.6Å (14.8 Å) (Fig. 2F). Using identical steps of image processing, the use of a larger particle window size led to a deterioration of the resolution by ~0.7 Å (~3.9Å), when compared to the initial reconstruction of Reconstruction 3 (see Discussion). Refinement of this data set proceeded in a manner similar to the refinement of Reconstruction 3; one refinement of 9 iterations was used without repeated amplitude enhancement. The nominal resolution (FSC=0.5) of this refined density map, Reconstruction 4, was 10.1 Å (12.4 Å) (Fig. 2G).
3.0 Application of Reconstruction Strategy to the Large Data Set
To reiterate, image processing on the non-decimated Data Set 2 without iterative amplitude enhancement produced a refined reconstruction, Reconstruction 3, with the best nominal resolution (8.9 Å (10.1 Å). To capitalize on the findings, another, larger data set of ~132,000 particles was then recorded for the same specimen with higher magnification, and image processing was used similarly as in the creation of Reconstruction 1. The larger data set was processed using two rounds of refinement without amplitude enhancement and successively smaller pixel sizes, similar to the way Reconstruction 3 had been obtained. The use of different pixel sizes within the same refinement protocol, proceeding from coarse (or decimated) to fine (or “raw”), was adopted in order to accelerate the processing. For this reason, we quote CPU times separately for both steps of the reconstruction procedure.
3.1 Initial Reconstruction and First Refinement
The biological sample used was identical to the one used in the creation of the Data Set 1 (Valle et al., 2003). Micrographs were recorded in a similar manner as for Data Set 1, except with the increased magnification of ~58,000x. In this experiment, a provision was made to reduce electron backscatter and thereby reduce the fog level, by backing the film used to record the images by an extra film. This provision requires a brief explanation: film cassettes by the manufacturer of the electron microscope were originally developed for 100 kV. These cassettes have steel backing. For this voltage, and even up to 200 kV, backscattering is minimal, and no problems due to the increase in fog level arise. Use of such cassettes becomes problematic, however, when the acceleration voltage reaches 300 kV, the voltage used in the experiment. To minimize this effect, before cassettes without metal backing were available, we placed an extra film behind the recording film.
The micrographs were scanned using a sampling of 7 microns on a Z/I Photoscan 2000 (Z/I Imaging Corp, Huntsville, Alabama) resulting in a pixel size of 1.20 Å on the object scale. Digitized micrographs were organized into 92 defocus groups with average defocus values ranging from 1.20 to 4.52 μm. The average defocus range within a defocus group was 164 nm, with a maximum range of 348 nm. The envelope function corresponding to this maximum defocus range falls off to ~50% at the spatial frequency corresponding to 6.7Å; thus, although the choice of defocus grouping does contribute to the final resolution limitation, it is not the decisive factor.
The first steps of image processing were chosen similar to the steps used in creating Data Set 1, except for differences outlined below. A decimation factor of 3x resulted in a pixel size of 3.6 Å, which was used for the initial reconstruction and the first round of refinement. Particle picking and alignment proceeded as before for creation of the Data Set 1, resulting in 131,599 particles with a window size of 103 × 103 pixels, corresponding to 371 × 371 Å on the object scale. The initial reconstruction was obtained in the standard manner, with a resolution of 10.7 Å (13.8 Å) at FSC=0.5 (Fig. 3A). Refinement of this initial reconstruction was continued for eleven iterations without the use of repeated amplitude enhancement. The final density map after refinement had a nominal resolution of 8.7 Å (9.3 Å) (Fig. 3B). This refinement was performed on a Beowulf cluster using 35 nodes (1.6Ghz dual-core, dual Opteron; 4 cores per node) in less than 125 hours.
Figure 3.
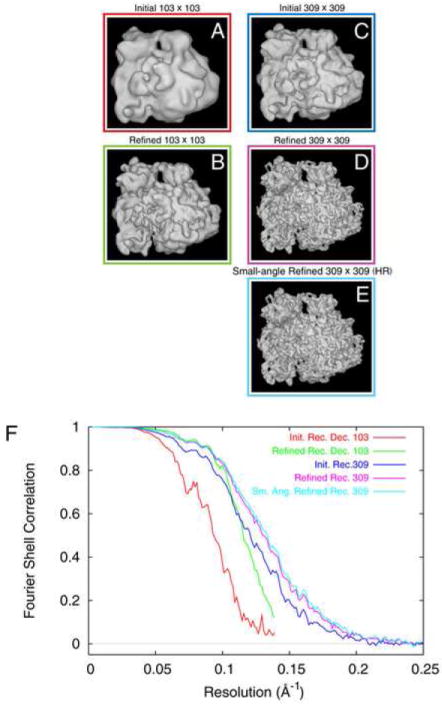
Reconstructions from the full ~132,000-particle dataset. (A,B) initial and refined reconstruction, with arrays 3x decimated; (C,D,E) initial, refined, and fine-angle refined reconstruction from the non-decimated data. (F) FSC curves for (A-G), color-coded to match color surrounding each reconstruction.
3.2 Non-decimated Dataset and Second Refinement
In a manner similar to that used to create Reconstruction 3, a new non-decimated data set was created. The ~132,000 particles were windowed from non-decimated micrographs while compensating for the previous 3-fold decimation. The window size used was 309 × 309 pixels.
The initial reconstruction for this refinement was based on the non-decimated data set, and the alignment information from the final iteration of the 3-fold decimated data set refinement. The initial reconstruction immediately had a resolution of 8.3 Å (9.4 Å) at FSC=0.5 (Fig. 3C). Refinement continued through ten iterations in total, producing a density map with a resolution of 7.7 Å (8.7 Å) at FSC=0.5 (Fig. 3D). The refinement required less than 724 hours to conclude, on a Beowulf cluster of 27 nodes.
Small-angle refinement was initiated using this 8.7-Å density map as a reference. Small-angle refinement operates functionally identical to ‘standard’ refinement, except that the angular search range is restricted to 2° and the angular step size used is 0.5°. The small-angle refinement was allowed to proceed for 7 iterations, requiring approximately 920 hours on a 48-node cluster, and resulting in a final map with a resolution of 7.5 Å (8.4 Å) (Fig. 3E). This final map will be referred to as the “High-resolution Reconstruction.”
3.3 Estimation of Resolution Using Extrapolation
The estimation of resolution via Fourier shell correlation is based on a comparison of two reconstructions from randomly picked halfsets of the total particle set. Because of this fact, the resolution is usually underestimated since the total set has better statistics than either half-set (see Frank, 2006). It is however possible to estimate the “true” resolution for the full data set by extrapolation from multiple resolution tests with increasing numbers of particles, up to the total number in the data set. We have indications, to be detailed in the following section, that this estimation by extrapolation is justified.
To estimate the “true” resolution through extrapolation, five resolution tests were performed, each with a different number of particles. Halfset reconstructions were either left unmasked or were masked using a Gaussian mask with r = 130 Å.
The resulting FSC=0.5 values follow Rosenthal and Henderson’s (2003) observed linear behavior when plotted as ln(Nd) versus 1/d2, where d denotes the resolution distance (i.e., the distance corresponding to the spatial frequency where FSC=0.5), and N the number of particles. This behavior was previously confirmed in other studies (e.g., Liu et al., 2007). Tentatively, the data appear to follow two regimes, with B=560 A2 and B=300 Å 2, respectively. Extrapolation to the total number present in the data set (131,599 particles) along the latter line would yield 6.7 Å (7.6 Å) (Fig. 4). However, due to measurement errors in the reported FSC resolution, fitting of the data by a single straight line may be more realistic, and in that case the extrapolation would yield a slightly different value, 7 Å.
Figure 4.
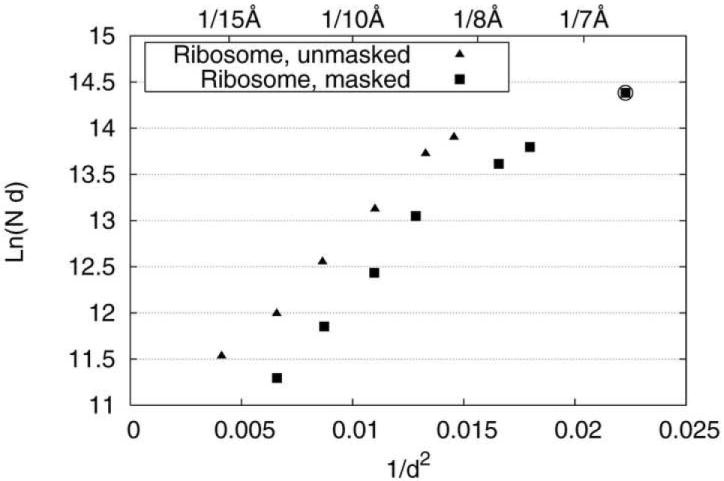
Plot of ln(Nd) as a function of 1/d2, with d [Å] estimated from FSC=0.5. The curve shows two linear regimes, one below, the other above N~40,000. The data point marked by a circle, corresponding to d = 6.7 Å, was derived from extrapolation in the latter regime.
Based on the results of this estimation, we filtered the high-resolution reconstruction to 6.7 Å. We then applied amplitude correction using an empirical curve obtained by low-angle X-ray scattering of 70S E. coli ribosomes in solution which extends to 8 Å (Gabashvili et al., 2000). To deal with the extra span between 8 and 6.7 Å, we used a simple polynomial extrapolation. This empirically-guided correction should be more accurate than a correction via negative B-factor (Frank, 2006).
3.4 The Ribosome at 6.7 Å Resolution: a Preliminary Assessment
Compared to the previously published cryo-EM map of the E. coli ribosome from an identical specimen (Valle et al., 2003), at a resolution of 10.3 Å resolution (FSC=0.5), the new map (Fig. 5A) represents a significant advance. A comparison with X-ray structures of the E. coli ribosome (Schuwirth et al., 2005) – albeit in a different state -- enables us to assess how individual components such as peripheral ribosomal proteins, A-form RNA helices, and tRNA are rendered (Fig. 5B). Of high interest is finally the additional detail observed in the interactions among EF-Tu, GDP, aminoacyl-tRNA, and the ribosome, which advances our understanding of the tRNA selection step during decoding (see Valle et al., 2002; 2003; Frank et al., 2005; Sengupta et al., in preparation). While a more detailed evaluation of the new map using molecular-dynamics fitting (Trabuco et al., 2008) is pending (Sengupta et al., in preparation), some preliminary observations are provided in the following.
Figure 5.
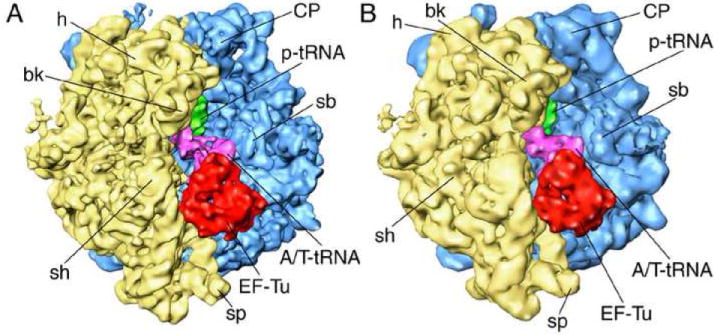
70S ribosome in complex with EF-Tu•aa-tRNA ternary complex, GDP, and antibiotic kirromycin, imaged at different resolutions. (A) 6.7Å map obtained in the current study; (B) previous 10-Å map of the identical sample (Valle et al 2003). Landmarks for 30S subunit (yellow): h, head; bk, beak; sh, shoulder; for 50S subunit (blue): CP, central protuberance; sb, stalk base.
The best indicator of the quality and effective resolution of the map is provided by observation of known features. In addition to the more prominent appearance of major and minor grooves in double- helical rRNA, other secondary structural features are identifiable in the 6.7-Å map. To demonstrate some of these features, close-up views of 16S rRNA helices 16 and 17 situated at the shoulder region of the small subunit, and 5S rRNA of the large subunit are provided (Fig. 6). In addition to regions of the map filtered to the nominal resolution (Butterworth filter with start and stop at +/- 0.05 around the spatial frequency (in Nyquist units) corresponding to 6.7 Å), we show the same regions filtered with a Butterworth filter starting at 6.7 Å and extending to ~4 Å, with the halfwidth of the filter lying in the vicinity of 5 Å.
Figure 6.
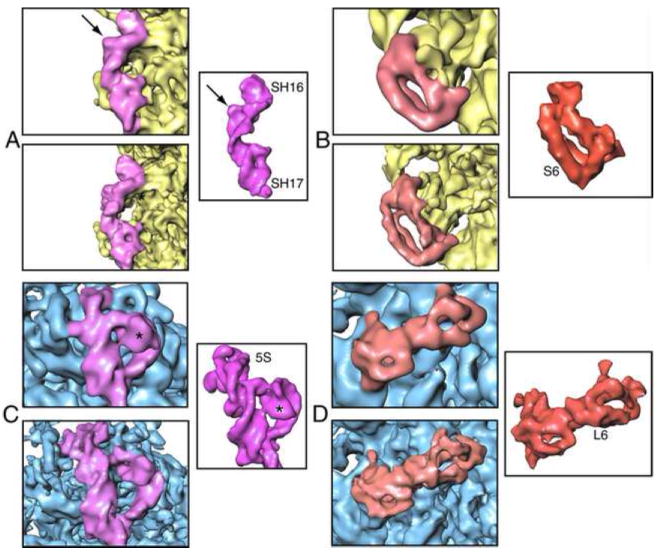
Close-up views of secondary structural features of selected ribosomal proteins and rRNA regions in the 6.7-Å map. (A) Helices 16 and 17 (purple) of 16S rRNA; (B) protein S6 (red) of the 30S subunit (yellow); (C) 5S rRNA; (D) protein L6 of the 50S subunit (blue). For each of these components, an inset shows same structure from the E. coli X-ray coordinates (Schuwirth et al., 2005) for comparison. Atomic coordinates were converted to density maps at 6.7Å resolution (PDB codes 30S: 2AVY, 50S: 2AW4).
These density regions of the cryo-EM map can be compared with density maps generated by converting the coordinates of these regions taken from the crystal structure of the E. coli 70S ribosome (PDB code 2AVY or 2AW7 for 30S; 2AW4 or 2AWB for 50S; Schuwirth et al 2005), and filtering them to the same resolution. All molecular surfaces are solid, without interruption by spikes or other noisy features. The fact that the molecular features are discernable with comparable definition as in the X-ray map filtered to the same resolution gives us confidence that the method of resolution estimation, by extrapolation from the half-set to the full data set, is well justified.
In helix 16 of 16S rRNA, an ‘interior loop’ region (marked with an arrow in Fig. 7A) is clearly visible. Another example for molecular features visible with high definition is a very unique fold in the 5S rRNA structure, marked with an asterisk in Fig. 7C. Similarly, enlarged views of protein S6 at the platform of small subunit, and protein L6 near the factor-binding region of the large subunit manifest distinct features of alpha-helical and beta-sheeted regions previously not visible (Fig.6 B,D). In addition, a comparison with the crystal structures reveals local conformational changes in the rRNA helices and intra-domain rearrangements in individual proteins -- changes that must be attributed to differences in the functional state between the ribosome in the crystal (as shown by X-ray crystallography) and in solution (by cryo-EM).
Figure 7.
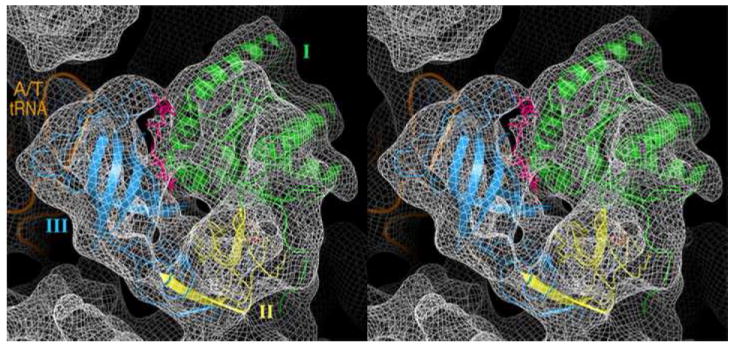
Stereo view of elongation factor Tu (EF-Tu) fitted into the corresponding density in the 6.7-Å map of the ribosome complex (represented as mesh). The features of alpha-helical (see domain I, green) and beta-sheeted (see domain III, golden) regions of the protein are clearly seen. In addition, partial density corresponding to the antibiotic kirromycin (pink; molecular formula C43H60N2O12, formula weight ~800 (794.96) dalton,) is distinctly visible.
Of particular interest in this complex is the binding interaction between the ribosome and the aa-tRNA•EF-Tu•GDP•kir ternary complex. An inspection of the ligand density in the map (viewed with the crystal structure [PDB code 1OB2] previously fitted into the lower-resolution map of the identical complex (Valle et al 2003)) (Fig. 7) indicates a much better definition of the alpha helices (see domain I) and beta-sheeted (see domain III) regions of EF-Tu. Furthermore, this map demonstrates densities with distinct features corresponding to the nucleotide (bound to domain I, close to the sarcin-ricin loop; Fig. 7A), and the antibiotic kirromycin (at the interface of domain I and III; Fig. 7B), in contrast to the lower resolution map where densities corresponding to these molecules were inseparable from the rest of the ligand mass. It is a further reflection of the high quality of the map that phosphorus atoms show up as “bumps” at the expected places along the phosphate backbones of RNA when the low-pass filter is further opened to 5 Å (Fig. 6).
4 Discussion
Resolution determination
One caveat that was reaffirmed in this study is that the FSC computed for the whole volume of the reconstruction consistently underestimates the resolution of the molecule reconstructed, and that it should be applied, instead, to a soft-masked version of the density map. Without this measure, it is virtually impossible to gauge the effects of image processing parameters on the reconstruction quality. While soft masking was introduced by Stewart et al. (2000) as a means to suppress irreproducible parts of the molecule in the resolution estimation, we would like to point out that in addition, reconstruction artifacts unrelated to the molecule structure are intrinsically irreproducible in a single-particle reconstruction, due to the fact that the sets of angles do not exactly match in two randomly drawn halfsets.
Standardized data set
The creation of a standardized data set is an important step in the current and future development and optimization of image processing algorithms. The biological sample was selected because it is known to be quite homogeneous, from the fact that it could be previously reconstructed to high resolution without resort to classification (Valle et al., 2003).
Effect of pixel size
The pixel size was changed during the course of refinement, from “decimated” to “raw,” in order to accelerate the computation. As expected, the non-decimated data leads to improved resolution. However, we found that the transfer of alignment parameters from a coarsely- to a finely-sampled dataset can be done without a setback in resolution. Thus, the strategy of doing the refinement in two parts, first with a decimated version of the dataset, then with the finely sampled dataset and jump-starting with the alignment parameters of the first part, was successful.
Effect of Amplitude Enhancement
Our results indicate that that refinement with amplitude enhancement in each step brings no benefit, but rather, the resolution deteriorates slightly. We used a filter that boosts the Fourier amplitudes to the values reported by low-angle X-ray scattering (Gabashvili et al., 2000) in each step. The resulting final map had resolution worse than the map in which no amplitude enhancement was used. The stated purpose of increasing Fourier amplitudes after each iteration is to increase the contribution of high spatial frequencies in the 3D projection alignment, which should translate into higher resolution of the density map. This result suggests that the concomitant amplification of noise in the high spatial frequency range counteracts the putative improvement.
Effect of Window Size
The final variable examined was particle window size. This was to determine if larger windows, and therefore information within the total reach of the point spread function, would improve resolution. The standard particle window size used for Reconstructions 2 & 3 was 195 × 195 pixels, corresponding to 363 × 363 Å on the object scale. Our larger window size was 275 × 275 pixels, corresponding to 511 × 511 Å on the object scale. This window size was chosen because it would be adequate to capture high spatial frequency information contained in the side ripples of the point spread function, according to our simulations (unpublished). In comparable refinements where the only variable modified was particle window size, Reconstructions 3 and 4, the nominal resolution deteriorated by ~2.3 Å when the larger window size was used. Even with the Gaussian mask applied to the larger window size, the resolution was significantly worse than for the smaller window. Our results show that, judged by the resolution criterion, the effect of the side ripples of the point spread function beyond the 363 × 363 Å box is negligible. We can explain the deterioration of nominal resolution both with and without soft masking by the extra amount of clutter admitted by the larger window.
Resolution by Extrapolation to the Full Data Set
The curve of ln(Nd) versus 1/d2 is seen to follow two regimes, with an improved B-factor toward higher particle numbers. Since the subsets were drawn randomly, with equal representations of the defocus groups, the result cannot be explained as an effect of statistical bias. However, as demonstrated by comparison with the X-ray data, the resolution estimated on the basis of the extrapolation from half to full data set is borne out by the molecular features visualized. In some portions of the ribosome, these features appear even crisper and more informative than in the X-ray map. Surprisingly, phosphate atoms are visible in a 5-Å version of the map, not just as undulations along the backbone (as expected when solely the information on the 6- to 6.5-Å distance along the backbone were transmitted) but by distinct bumps, showing that there exists valid structural information up to 5 Å.
Conclusions
Our exploration of parameters and processing methods that might lead to improved results has produced a density map of the ribosome with greatly improved resolution and definition, compared with previously published maps. As has been pointed out, this study falls far short of a systematic exploration of a wide range of options, which might produce even better results but would require much larger computational expenditures. The new biologically relevant information contained in the ribosome map will be expounded elsewhere. The appearance of phosphorus atoms along the phosphate backbone on the map when limited to 5Å resolution indicates the presence of information well beyond the FSC=0.5 cutoff, although we can say for sure that the signal-to-noise ratio at this spatial frequency falls short of the SNR=1 cutoff (see Malhotra et al., 1998; Rosenthal and Henderson, 2003). In the course of our inquiry, we confirmed the validity of a relationship initially postulated by Rosenthal and Henderson (2003), which may allow extrapolation to the full data set as well as the prediction of the minimum data collection requirements toward a more ambitious target resolution.
Materials and Methods
SPIDER (Frank et al., 1996) was used for all image processing. The visualization of some figures was done using IRIS Explorer (Numerical Algorithm Group, Downers, IL). For molecular modeling, Pymol (DeLano Scientific, San Carlos, CA) was used.
Table 1.
For resolution figures, the second number in parentheses refers to the resolution obtained before application of the soft Gaussian mask to both half-set reconstructions compared by FSC.
| Reconstruction | Pixel Size (Å) | Window Size (Pixels;Å) | Initial Resolution (Å) | Iterative Amplitude Enhancement | No. of Refinement Iterations | Reconstruction Resolution (Å) |
|---|---|---|---|---|---|---|
| 1 | 3.76 | 97 ; 365 | 10.7 (11.9) | Yes | 11 | 9.4 (10.2) |
| 2 | 1.86 | 195 ; 363 | 9.9 (10.9) | Yes | 9 | 9.4 (10.3) |
| 3 | 1.86 | 195 ; 363 | 9.9 (10.9) | No | 9 | 8.9 (10.1) |
| 4 | 1.86 | 275 ; 512 | 10.6 (14.8) | No | 9 | 10.1 (12.4) |
| High Resolution | ||||||
| 1st Refinement | 3.6 | 103 ; 371 | 10.7 (13.8) | No | 11 | 8.7 (9.3) |
| 2nd Refinement | 1.2 | 309 ; 371 | 8.3 (9.4) | No | 10 | 7.7 (8.7) |
| Small Angle | - | - | - | No | 7 | 7.5 (8.4) |
Acknowledgments
We would like to thank Måns Ehrenberg, Uppsala University, Sweden for the specimen of the pre-accommodated E. coli 70S•Phe-tRNAPhe•EF-Tu•GDP•kir complex. We would like to thank Hstau Liao and Jie Fu for discussions of the extrapolation of resolution. We acknowledge the assistance of Richard Hall, UC Berkeley, for his contribution to the selection of high-quality particles, based on his analysis of micrograph quality (unpublished). We thank Michael Watters for assistance with the preparation of the illustrations. This work was supported by HHMI and NIH grants P41 RR 01219, R01 GM29169 and R01 GM55440 (to J.F.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen CBF, Becker T, Blau M, Anand M, Halic M, Balar B, Mielke T, Boesen T, Pedersen JS, Spahn CMT, Kinzy TG, Andersen GR, Beckmann R. Structure of eEF3 and the mechanism of transfer RNA release from the E-site. Nature. 2006;443:663–668. doi: 10.1038/nature05126. [DOI] [PubMed] [Google Scholar]
- Connell SR, Takemoto C, Wilson DN, Wang H, Murayama K, Terada T, Shirouzu M, Rost M, Schüler M, Giesebrecht J, Dabrowski M, Mielke T, Fucini P, Yokoyama S, Spahn CMT. Structural basis for interaction of the ribosome with the switch regions of GTP-bound elongation factors. Mol Cell. 2007;25:751–764. doi: 10.1016/j.molcel.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Frank J. Three-dimensional Electron Microscopy of Macromolecular Assemblies. Oxford University Press; 2006. [Google Scholar]
- Frank J, Radermacher R, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Frank J, Penczek P, Agraweal RK, Grassucci RA, Heagle AB. Three-dimensional Cryoelectron Microscopy of Ribosomes. Meth Enzymol. 2000;317:276–291. doi: 10.1016/s0076-6879(00)17020-x. [DOI] [PubMed] [Google Scholar]
- Frank J, Sengupta J, Gao H, Li W, Valle M, Zavialov A, Ehrenberg M. The role of tRNA as a molecular spring in decoding, accommodation, and peptidyl transfer. FEBS Lett. 2005;579:939–963. doi: 10.1016/j.febslet.2004.10.105. [DOI] [PubMed] [Google Scholar]
- Gabashvili IS, Agrawal RK, Spahn CMT, Grassucci RA, Svergun DI, Frank J, Penczek P. Solution structure of the E. coli 70S ribosome at 11.5 Å resolution. Cell. 2000;100:537–549. doi: 10.1016/s0092-8674(00)80690-x. [DOI] [PubMed] [Google Scholar]
- Grassucci RA, Taylor TD, Frank J. Preparation of macromolecular complexes for cryo-electron microscopy. Nature Prot. 2007a;2:3238–3246. doi: 10.1038/nprot.2007.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassucci RA, Taylor TD, Frank J. Visualization of macromolecular complexes using cryo-electron microscopy with FEI Tecnai TEMs. Nature Prot. 2007b;3:330–339. doi: 10.1038/nprot.2007.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauz G, van Heel M. Exact filters for general geometry three-dimensional reconstruction. Optik. 1986;73:146–156. [Google Scholar]
- Liu X, Jiang W, Jakana J, Chiu W. Averaging tens to hundreds of icosahedral particle images to resolve protein secondary structure elements using multi-path simulated annealing optimization algorithm. J Struct Biol. 2007;160:11–27. doi: 10.1016/j.jsb.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baker M, Chen D, Song J-L, Chuang D, Chiu W. De Novo backbone trace of GroEL from single particle electron cryomicroscopy. Structure. 2008;16:441–448. doi: 10.1016/j.str.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Penczek P, Grassucci RA, Frank J. The ribosome at improved resolution: New techniques for merging and orientation refinement in 3D cryoelectron microscopy of biological particles. Ultramicroscopy. 1994;53:251–270. doi: 10.1016/0304-3991(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Penczek PA, Zhu J, Schroeder R, Frank J. Three-dimensional reconstruction with contrast transfer compensation from defocus series. Scanning Microsc. 1997;11:147–154. [Google Scholar]
- Rath BK, Frank J. Fast automatic particle picking from cryo-electron micrographs using a locally normalized cross-correlation function: a case study. J Struct Biol. 2004;145:84–90. doi: 10.1016/j.jsb.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Roseman AM. FindEM—a fast, efficient program for automatic selection of particles from electron micrographs. J Struct Biol. 2004;145:91–99. doi: 10.1016/j.jsb.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Saxton WO, Baumeister W. The correlation averaging of a regularly arranged bacterial cell envelope protein. J Microsc. 1982;127:127–138. doi: 10.1111/j.1365-2818.1982.tb00405.x. [DOI] [PubMed] [Google Scholar]
- Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JD. Structures of the bacterial ribosome at 3.5Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- Shaikh TR, Gao H, Baxter W, Asturias F, Boisset N, Leith A, Frank J. SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. 2008 doi: 10.1038/nprot.2008.156. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg SM, Lander GC, Pulokas J, Fellmann D, Cheng A, Quispe JD, Mallick SP, Avila RM, Carragher B, Potter CS. Automated cryoEM data acquisition and analysis of 284 742 particles of GroEL. J Struct Biol. 2006;155:470–481. doi: 10.1016/j.jsb.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Stewart PL, Cary RB, Peterson SR, Chiu CY. Digitally collected cryo-electron micrographs for single particle reconstruction. Microscopy Research and Technique. 2000;49:224–232. doi: 10.1002/(SICI)1097-0029(20000501)49:3<224::AID-JEMT2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle M, Sengupta J, Swami NK, Grassucci RA, Burkhardt N, Nierhaus KH, Agrawal RK, Frank J. Cryo-EM reveals an active role for aminoacyl-tRNA in the accommodated state. EMBO J. 2002;21:3557–3567. doi: 10.1093/emboj/cdf326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle M, Zavialov A, Li W, Stagg SM, Sengupta J, Nielsen RC, Nissen P, Harvey SC, Ehrenberg M, Frank J. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat Struct Biol. 2003;10:899–906. doi: 10.1038/nsb1003. [DOI] [PubMed] [Google Scholar]
- Yang C, Ng EG, Penczek PA. Unified 3-D structure and projection orientation refinement using quasi-Newton algorithm. J Struct Biol. 2005;149:53–64. doi: 10.1016/j.jsb.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Yu X, Jin L, Zhou ZH. 3.88 Å structure of cytoplasmic polyhedrosis virus by cryo-electron microscopy. Nature advance online publication. 2008 doi: 10.1038/nature06893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Settembre E, Xu C, Dormitzer PR, Bellamy R, Harrison SC, Grigorieff N. Near-atomic resolution using electron cryomicroscopy and single-particle reconstruction. PNAS. 2008;105:1867–1872. doi: 10.1073/pnas.0711623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Penczek P, Schroeder R, Frank J. Three-dimensional reconstruction with contrast transfer-function correction from energy-filtered cryo-electron micrographs – procedures and applications to the 70S Escherichia coli ribosome. J Struct Biol. 1997;118:197–219. doi: 10.1006/jsbi.1997.3845. [DOI] [PubMed] [Google Scholar]