

Abstract
α-Tocopheryl succinate (α-TOS) is a selective inducer of apoptosis in cancer cells, which involves the accumulation of reactive oxygen species (ROS). The molecular target of α-TOS has not been identified. Here we show that α-TOS inhibits succinate dehydrogenase (SDH) activity of complex II (CII) by interacting with the proximal and distal ubiquinone (UbQ) binding site (QP and QD, respectively). This is based on biochemical analyses and molecular modelling, revealing similar or stronger interaction energy of α-TOS compared to that of UbQ for the QP and QD sites, respectively. CybL-mutant cells with dysfunctional CII failed to accumulate ROS and undergo apoptosis in the presence of α-TOS. Similar resistance was observed when CybL was knocked down with siRNA. Reconstitution of functional CII rendered CybL-mutant cells susceptible to α-TOS. We propose that α-TOS displaces UbQ in CII causing electrons generated by SDH to recombine with molecular oxygen to yield ROS. Our data highlight CII, a known tumour suppressor, as a novel target for cancer therapy.
Keywords: α-tocopheryl succinate, complex II, ubiquinone-binding sites, reactive oxygen species, apoptosis, anti-cancer drug target
Introduction
Cytotoxic drugs that act by selectively affecting mitochondria in cancer cells, ‘mitocans’ (Ralph et al., 2007; Neuzil et al., 2006; Neuzil et al., 2007), are proving to be attractive for the treatment of cancer by virtue of their selective anti-cancer activity (Neuzil et al., 2007; Ko et al., 2004; Bonnet et al., 2007; Dong et al., in press). Prime examples of such drugs are α-tocopheryl succinate (α-TOS) (Neuzil et al., 2001a, 2001b), 3-bromopyruvate (3BP) (Ko et al., 2004; Geschwind et al., 2002) and dichloroacetate (DCA) (Bonnet et al., 2007). 3BP inhibits the glycolytic pathway enzyme hexokinase, and succinate dehydrogenase (SDH), suppressing cellular ATP production and mitochondrial respiration (Ko et al., 2001; Xu et al., 2005). DCA appears to target cancer cells by inhibiting pyruvate dehydrogenase kinase (Bonnet et al., 2007). Finally, β-phenylethyl isothiocyanate selectively kills cancer cells by eliciting the generation of reactive oxygen species (ROS) from mitochondria (Trachootham et al., 2006). Thus, mitocans are an emerging group of compounds with significant therapeutic potential as anti-cancer drugs.
α-TOS, an esterified, redox-silent analogue of vitamin E (VE), causes rapid production of ROS in different cancer cell lines, thereby triggering apoptosis (Weber et al., 2003; Stapelberg et al., 2005; Swettenham et al., 2005; Wang et al., 2005). α-TOS also binds and inhibits the anti-apoptotic function of Bcl-2 and Bcl-xL by blocking their Bcl-2 homology 3 (BH3)-binding domains (Shiau et al., 2006). This may explain, in part, how α-TOS sensitizes cancer cells to other anti-cancer drugs. However, it does not explain how the VE analogue causes accumulation of ROS, obligatory mediators of α-TOS-induced apoptosis. Indeed, antioxidants like superoxide dismutase (SOD) or the mitochondrially targeted coenzyme Q (MitoQ) (James et al., 2005, 2007) negate the apoptotic action of α-TOS on cancer cells (Weber et al., 2003; Stapelberg et al., 2005; Alleva et al., 2001). In addition, cancer cells that accumulate only low levels of ROS in response to α-TOS are less susceptible to apoptosis (Stapelberg et al., 2005; Swettenham et al., 2005; Kogure et al., 2002; Kang et al., 2004). Hence, α-TOS kills cancer cells through a mechanism that requires the production of ROS.
Additional evidence indicates that the effects of α-TOS on rapidly proliferating cancerous cells, combined with a reduced toxicity toward normal cells, results from a greater antioxidant defence for the latter (Church et al., 1993; Safford et al., 1994; Huang et al., 2006; Allen and Balin, 2003) and/or increased levels of esterases that inactivate the pro-oxidant α-TOS by hydrolysis of the succinate moiety, thereby converting the pro-vitamin to the redox-active, non-apoptogenic α-TOH (Fariss et al., 2001; Neuzil and Massa, 2005). This feature makes agents like α-TOS excellent candidates for cancer therapy.
The target of α-TOS in cancer cells that results in ROS production has not previously been defined. One possibility whereby the VE analogue could give rise to ROS and ensuing apoptosis is by interfering with the ROS-generating centres along the mitochondrial electron redox chain. Of particular interest is complex II, since previous data showed that toxicity of α-TOS on cancer cells can be suppressed by the mitochondrially targeted coenzyme Q (MitoQ) (Stapelber et al., 2005; Swettenham et al, 2005), that is known to interact with the ubiquinone (UbQ)-binding sites of complex II (James et al., 2005, 2007).
Here we examined the effect of α-TOS on the respiratory chain, identifying the UbQ-binding pockets in complex II (CII) as the key molecular target for the agent, and propose that interaction of the VE analogue with the UbQ-binding sites results in ROS generation. The UbQ-binding sites of CII represent a novel, thus far unrecognized target for anti-cancer drugs.
Results
Cancer cells accumulate ROS and undergo apoptosis when exposed to α-TOS
We first tested whether exposure of breast cancer cells to α-TOS caused generation of ROS, which translates to induction of apoptosis. The results obtained by flow cytometry (Figure 1a,b,e) show that both erbB2-low MCF7 and erbB2-high MDA-MB-453 cells accumulated ROS and underwent apoptosis when challenged with α-TOS. EPR spectroscopy confirmed ROS generation occurring in cells exposed to α-TOS (Figure 1c,d). Pre-treatment of the cells with MitoQ or their co-treatment with SOD significantly suppressed ROS accumulation and decreased the extent of apoptosis (Figure 1). MCF7 ρ0 cells with dysfunctional electron redox chain, when exposed to α-TOS, showed relatively low levels of ROS accumulation and, consequently, the induction of apoptosis was reduced (Figure 1f,g).
Figure 1.

ROS generation and induction of apoptosis in cancer cells by α-TOS. ErbB2-low MCF7 and -high MDA-MB-453 cells were exposed to α-TOS at 50 μM and for times shown, and assessed for DHE- (a) and annexin V-positive cells (%) (b). Jurkat cells were treated with 50 μM α-TOS for 2 h or as shown and assessed for ROS accumulation using EPR spectroscopy (c – evaluation of the DMPO-OH adduct level in nmol/mg protein; d – representative EPR spectra of cells exposed to α-TOS in the absence (1) or presence of MitoQ (2) or to MitoQ only (3)). Panel e shows the kinetics of apoptosis induction in Jurkat cells exposed to 50 μM α-TOS in the absence or presence of MitoQ. Control (C) MCF7 parental and ρ0 cells exposed to 50 μM α-TOS for 2 h (T) were assessed for ROS accumulation (f) and apoptosis (g). Where indicated, the cells were pre-treated for 1 h with 2 μM MitoQ (MQ) or co-treated with SOD (PEG-SOD, 750 units/mg). The data shown represent mean values ± S.D. (n=3). The symbol ‘*’ denotes significant difference (p<0.05) in the level of apoptosis and ROS of treated cells compared with the untreated control cell population.
α-TOS targets the UbQ-binding pockets on complex II
Studies with α-tocopherol (α-TOH) indicated that the anti-oxidant was capable of binding to the UbQ site in CII where it acts as a competitive inhibitor of succinate dehydrogenase (SDH) (Yu and Yu, 1982). Therefore, we investigated whether α-TOS, with its related structure but opposing biological activity as a pro-oxidant and an apoptogen, also interferes with UbQ binding to CII. Using high levels of succinate (20 mM) that is capable of entering cells (Spencer, 1976; Maehara et al., 1988) to promote both SDH activity (Maehara et al., 1998; Berridge and Tan, 1993) and mitochondrial respiration proceeding via CII, NeuTL cells were assayed for their ability to reduce MTT in the presence or absence of α-TOS. The inhibitors of SDH activity, 3-bromopyruvate (3BP) (Sanborn et al., 1971) and 3-nitropropionic acid (3NPA) (Scallet et al., 2003), were employed as positive controls. To establish the relationship between inhibition of SDH and MTT reduction, initial experiments with 3BP and 3NPA were performed in the absence of added succinate that might otherwise compete for binding to the enzyme, and the results confirmed that 3BP and 3NPA were inhibitors of CII-mediated MTT reduction and that SDH activity and MTT reduction were closely related (Figure 2a).
Figure 2.

The effect of 3NPA, 3BP, TTFA and α-TOS on the ability of NeuTL cells to reduce MTT. (a) MTT reduction in PBS was assessed after a 4 h co-incubation period in the presence of 3NPA or 3BP used at the concentrations (μM) as shown. (b-d) Cells were pre-incubated for 60 min with MitoQ at the concentrations indicated (Ctrl, 2 or 5 μM; insert box) before addition of 3BP (0-100 μM) (b) and assessed for their ability to reduce MTT after a 2 h incubation period. Cells pre-treated with MitoQ were then treated with TTFA (c) and with α-TOS (d) at the concentration shown, and were assessed for their ability to reduce MTT in RPMI containing 20 mM succinic acid (pH 7.4) after a 2 h incubation period. Results are presented as mean reduction (%) of MTT relative to control (untreated) ± S.D. The symbol ‘*’ denotes significant differences with p<0.05.
Thenoyltrifluoroacetone (TTFA) is an inhibitor of CII that blocks the succinate-dependent MTT reduction by targeting the UbQ-binding sites rather than the SDH enzyme catalytic site (Sun et al., 2005). The CII UbQ sites are situated beneath the active site of SDH, in the transmembrane region of the complex. With high succinate levels as the substrate driving CII-mediated MTT reduction in whole cells, treatment with 3BP, α-TOS or TTFA inhibited MTT reduction in a dose-dependent manner (Figure 2b-d). Pre-incubation of cells with MitoQ, a form of UbQ that preferentially localizes to mitochondria where it binds to CII but not CIII (James et al., 2005, 2007; Kelso et al., 2001), overcame the inhibition in MTT reduction by TTFA and α-TOS, but not by 3BP (Figure 2b-d). These results indicate that α-TOS inhibits SDH activity in a similar manner to TTFA.
Verification that α-TOS inhibits mitochondrial respiratory complex II activity
Additional support for the role of α-TOS in inhibiting CII activity was obtained with mitochondrial preparations from rat liver and membrane fractions from P. denitrificans. These experiments employed a mixture of phenazine methosulphate (PMS) and the terminal electron acceptor 2,6-dichlorophenol indophenol (DCPIP) as the readout for SDH activity (Trounce et al., 1996). SDH activity decreased rapidly after treatment with α-TOS (Figure 3). These data strongly indicate that α-TOS, acting directly on the UbQ site(s) of CII, interferes with the electron flow to PMS and DCPIP. In contrast, α-TOS had no effect on NADH dehydrogenase (CI) activity (data not shown), consistent with selective targeting to CII.
Figure 3.

Inhibition of SDH/complex II activity in isolated rat liver mitochondria (a, c), and Paracoccus denitrificans (b, d) by α-TOS. Preparations of mitochondria from rat liver or membranes from P. denitrificans were incubated in a reaction mixture facilitating mitochondrial SDH/CII activity (μmol/min/mg protein) and containing DCPIP+PMS. Samples in a and b contained succinate and were both treated with α-TOS as indicated. Dose-response curves displaying changes in the reaction rate (μmol/min) for DCPIP were measured as absorbance at 600 nm under different concentrations of succinate as indicated in the absence or presence of α-TOS (c, d). Results are represented as mean values ± S.D. (n=3). The symbol ‘*’ indicates values significantly different from the controls with p<0.05.
Next we tested whether α-TOS initiated apoptosis in parental Chinese hamster lung fibroblasts (B1 cells), CI dysfunctional cells (B10 cells) and CII-dysfunctional cells (B9 cells, with a mutation in the gene encoding the succinyl dehydrogenase subunit C (SDHC subunit, CybL). B9 cells were less responsive to α-TOS, with lower levels of ROS accumulation (Figure 4a) and diminished SDH activity (Figure 4b) compared to the parental (B1) or B10 cells. In line with these findings, B9 cells were relatively resistant to apoptosis induced by the VE analogue (Figure 4c). Reconstitution of CII in the CybL-mutant (B9) cells normalised the SDH activity (Figure 4b), and also restored cell sensitivity to α-TOS-induced killing (Figure 4c).
Figure 4.
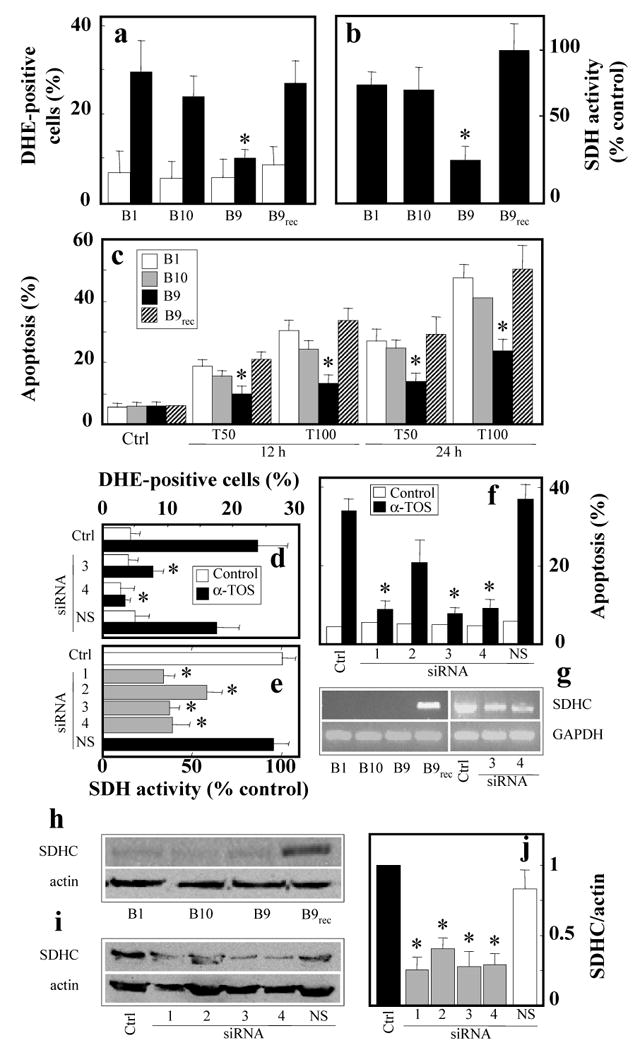
Apoptosis induction by α-TOS is suppressed in CII dysfunctional cells. Parental (B1), CI-dysfunctional (B10), CII-dysfunctional (CybL-mutant; B9), and CybL-mutant cells following complex II reconstitution by transfection with human CybL (B9rec) were exposed to α-TOS 50 μM and for 24 h, unless shown otherwise, harvested and assessed for ROS accumulation (a), SDH activity assessed in whole cells on the basis of MTT reduction with succinate as a substrate (b), and apoptosis (c). MCF7 cells were pre-treated with CybL or non-specific (NS) siRNA, exposed to α-TOS as shown, and assessed for ROS accumulation (d), SDH activity (e), and apoptosis induction (f). Panel g shows results of RT-PCR analysis of B1, B10, B9 or B9rec cells as well as SDHC siRNA-treated MCF7 cells using human SDHC primers. Panel h reveals results of Western blotting of B1, B10, B9 and B9rec cells using monoclonal IgG anti-human SDHC. Western blotting is also shown to document the levels of SDHC in MCF7 cells treated with different SDHC siRNA duplexes and with NS siRNA (i), which was evaluated relative to the actin band (j). Results are represented as mean values ± S.D. (n=3), images are representative of three independent experiments. The symbol ‘*’ indicates values significantly different from the controls with p<0.05.
Data obtained with the CII-dysfunctional (B9) and the reconstituted B9 cells were independently verified by treatment of MCF7 cells with four different short-interfering RNAs (siRNA) against CybL. Duplexes 1, 3 and 4 substantially suppressed CII activity (Figure 4d), as well as ROS generation (Figure 4e) and apoptosis induction in response to α-TOS (Figure 4f).
Using a human monoclonal SDHC antibody, we probed the B1, B9 and B10, and the CII-reconstituted Chinese hamster lung fibroblasts (B9rec) for the presence of SDHC. Western blotting analysis (Figure 4h,i) revealed high levels of human SDHC in the B9rec cells and its low level in MCF-7 cells treated with the SDHC siRNAs. The B9 cells with reconstituted CII revealed re-appearance of SDHC. As revealed by densitometric evaluation, siRNA treatment of MCF7 cells lowered the level of the SDHC protein by 50-80% (Figure 4j). These data are consistent with the RT-PCR results showing presence of human SDHC mRNA in CII-reconstituted cells and low levels of the transcript in MCF7 cells treated with SDHC siRNA (Figure 4g). The siRNA approach was specific for SDHC, since we did not observe any changes in the level of the SDHB subunit protein in the SDHC-treated MCF7 cells (data not shown).
Molecular modelling reveals strong binding of α-TOS to UbQ sites on complex II
To rationalize our results, which indicated that α-TOS interacts with CII via the UbQ-binding sites, we undertook a molecular modelling study using AutoDock (Morris et al., 1998). The crystal structure of porcine heart mitochondrial CII has been reported (Sun et al., 2005). It exhibits a high sequence identity with human CII (95-97% for the individual subunits), therefore we used this structure (1ZOY) and the related structure (1ZP0) with the inhibitor TTFA bound as the basis for our modelling study. Structural analysis clearly revealed the proximal UbQ-binding site (QP) and also identified the position of the proposed distal UbQ-binding site (QD). To test the feasibility of using AutoDock for this system, we first simulated the binding of UbQ5 to both the QP and the proposed QD sites. UbQ5 was chosen as it is of similar size to α-TOS (Figure 5e) and contains a similar number of rotatable bonds (16 and 17, respectively). Figure 5a indicates that UbQ5 docked in QP to a deeper position than that observed for the portion of UbQ resolved in the published crystal structure (Sun et al., 2005). UbQ5 was also found to dock into the proposed QD site, with the UbQ ring positioned in front of the binding site and the hydrophobic tail located inside the site (Figure 5c).
Figure 5.
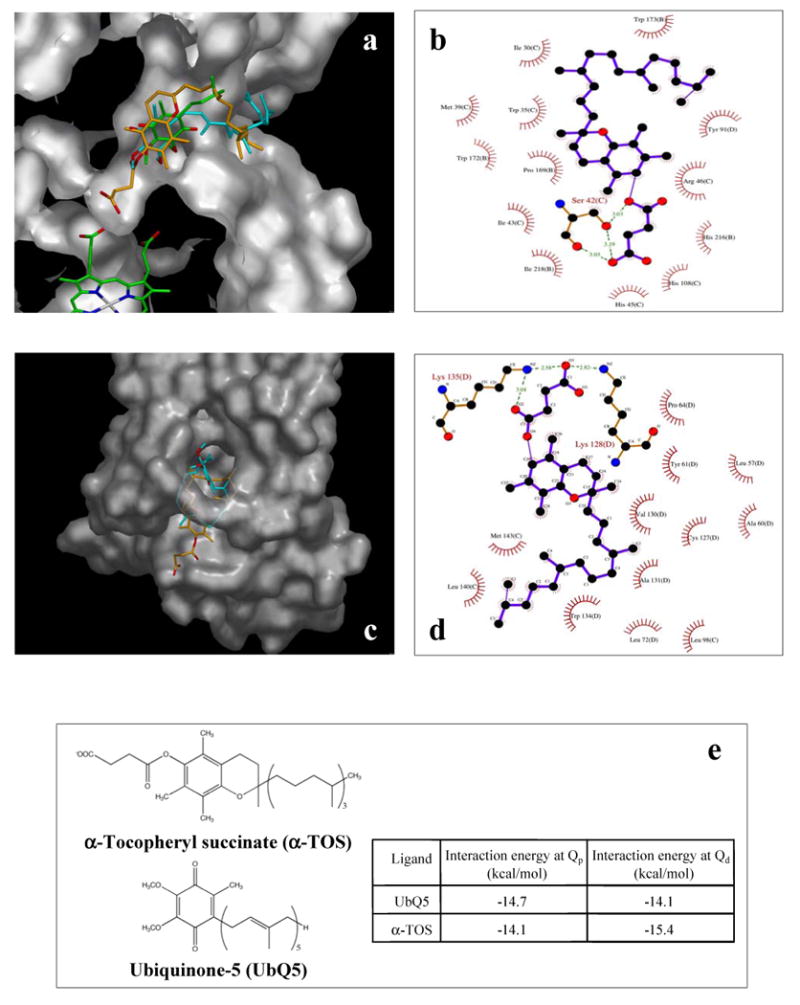
Molecular modelling reveals interaction of α-TOS with UbQ-binding sites in CII. (a) Cut-away view of the QP binding site showing the heme group (bottom left corner) and the position of UbQ (green carbon atoms) as indicated with arrows. The best-fit conformations of UbQ5 (cyan carbons) and α-TOS (orange carbon atoms) are also shown. (b) Ligplot diagram showing the major interactions between the best docked conformation of α-TOS and the QP binding site. (c) View of the proposed QD binding site (with overhanging bridge as a translucent surface) showing the best docked conformations of UbQ5 (cyan carbons) and α-TOS (orange carbons). (d) Ligplot diagram showing the major interactions between the best docked conformation of α-TOS and the QD binding site. (e) Chemical structures of UbQ5 and α-TOS and a table of interaction energies for the best ranked docking conformations for UbQ5 and α-TOS in the QP and QD binding sites. Images in panels a and c were prepared using Astex Viewer (Hartshorn, 2002), while those in panels b and d were prepared using Ligplot (Wallace et al, 1995).
In the QP site the ring system of α-TOS sits in the same binding pocket as the UbQ ring but is tilted to one side. The succinate ester moiety extended deeper into the binding site protruding down towards the location of the prosthetic heme group. The carboxyl group fits neatly into this pocket and is involved in a bidentate hydrogen bond with Ser42(C) (Figure 5b). Ser42(C) also interacts with the ester oxygen of α-TOS. As befits the hydrophobic nature of the remainder of the α-TOS molecule, all the other interactions with the protein are hydrophobic. Thus, the hydrocarbon side chain loops around and extends out of the QP site and along the same channel where the isoprenoid side chain of UbQ5 is located.
In the QD site the phenyl ring system of α-TOS sits towards the bottom of the binding site with the succinate ester moiety extending out the bottom of the site in a similar way to that observed for the head group of the phospholipid visible in the original crystal structure (Figure 5c). The succinate moiety hydrogen-bonds to Lys135(D) and Lys128(D) (Figure 5d), while the hydrocarbon side chain loops around the inside of the binding site. The calculated energy of interaction for the docked conformations of α-TOS (Figure 5e) suggested that it can bind at either site. While the binding energy of α-TOS at the QP site is slightly less than that of UbQ5, α-TOS would certainly compete with UbQ5. At the QD site, α-TOS shows a much stronger binding energy than UbQ5 and would be expected to easily displace it from this site (Figure 5e).
We had to use UbQ5 in the modelling study because of the relatively high number of rotatable bonds in α-TOS (17) and UbQ5 (16); adding the full isoprene tail of UbQ10 (with the extra rotatable bonds) makes the AutoDock calculations intractable. Also, data analysis from the published crystal structure only shows electron density for the very start of the isoprenoid chain suggesting that the rest of the tail is not particularly tightly bound at this site (Sun et al., 2005). Therefore, the expected interaction energies of UbQ10 for the two sites will be similar to those calculated for UbQ5.
Apoptosis induced by α-TOS is independent of its BH3 mimetic activity
Shiau et al. (2005) reported that VE analogues possess BH3 mimetic activity, interacting with the BH3 domain of Bcl-2 and Bcl-xL. To ascertain whether the apoptogenic activity of α-TOS is dependent on this activity, we utilized the BH3 mimetic BH3I-2′ (Calbiochem) (Degterev et al., 2001; Milanesi et al., 2006). Cytotoxicity of α-TOS was assessed in MCF7 cells exposed to increasing concentrations of BH3I-2′. We found that levels >5 μM caused mitochondrial destabilization (not shown). Combining BH3I-2′ (5 μM) with α-TOS significantly enhanced the sensitivity of the cells to the VE analogue (Figure 6). These data indicate a synergistic effect of BH3I-2′ and α-TOS, and suggest that the VE analogue induces apoptosis predominantly by other modes than acting as a BH3 mimetic.
Figure 6.

α-TOS causes apoptosis independent of its BH3 mimetic activity. MCF7 cells were treated with 5 μM BH3I-2′ or exposed to 30 μM α-TOS following 10 min pre-treatment with 5 μM BH3I-2′. Cells were then assessed for mitochondrial depolarization (a), ROS generation (b) and apoptosis induction (c). Results are represented as mean values ± S.D. (n=3). The symbol ‘*’ indicates values significantly different from the controls with p<0.05.
α-TOS inhibits tumour growth irrespective of erbB2 status
To demonstrate anti-cancer activity of α-TOS, we first determined whether the VE analogue could suppress tumour growth in an animal model of breast cancer with low erbB2 expression, given that over two thirds of human breast cancers express low levels of the receptor tyrosine kinase (Slamon et al.,1989). Nude mice were xenotransplanted with MCF7 cells and treated with α-TOS. Data in Figure 7a show that tumours progressed to 5-times their initial size in control animals, whereas α-TOS repressed tumour growth with a significant decrease in the resulting tumour size. Next, we tested treatment of erbB2-high breast carcinomas using the transgenic FVB/N c-neu mice (Guy et al., 1992). Compared to the controls, the tumours in the α-TOS-treated mice showed a 30-40% reduction in size over the several weeks of treatment. Collectively, these results suggest a high level of efficacy for α-TOS therapy against breast carcinomas regardless of their erbB-2/HER2 status.
Figure 7.
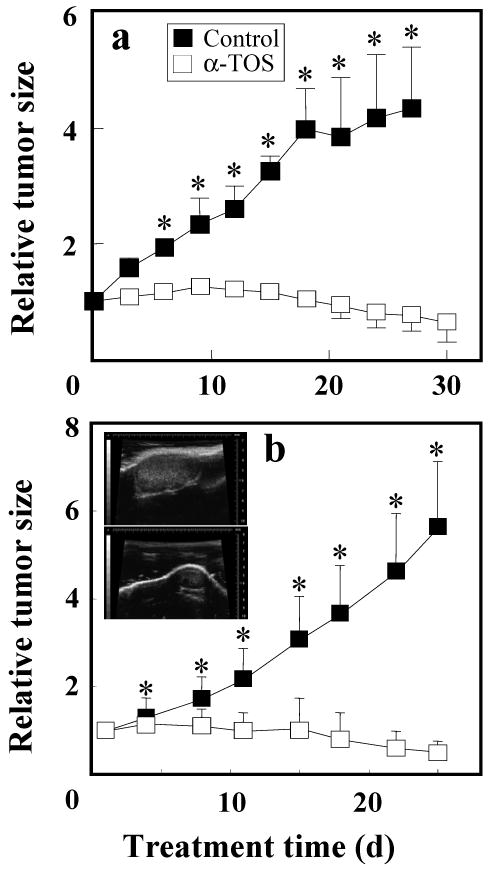
Inhibition of breast cancer in mouse models by α-TOS. (a) Nude mice were inoculated with MCF7 cells and once tumours became established, the animals were treated every 3 d with 10 μmoles per mouse of α-TOS dissolved in DMSO or with DMSO alone, by i.p. injection. Tumour size was measured using callipers and was correlated to the size of the carcinomas at the onset of the therapy. Four animals were used in each group. (b) Female FVB/N c-neu mice with small tumours received either 10 μmoles α-TOS solubilized in corn oil/4% ethanol (n=11) or the excipient alone (control, n=9) by i.p. injection once every 3 d. Tumour size was quantified using USI. Two independent experiments were conducted. The inset in b shows representative images of tumours acquired by USI in the control (upper image) and treated (lower image) FVB/N c-neu mice on day 22 of the experiment. The results shown are mean values ± SEM. The symbol ‘*’ denotes significant differences (p<0.05).
Discussion
An ideal anti-cancer drug would be one that specifically targets cancer cells. It is increasingly recognized that mitochondria, which supply much of the cellular energy and are key regulators of apoptosis, are emerging as effective targets that may provide the selectivity needed for potent anti-cancer therapy (Neuzil et al., 2001b; Don and Hogg, 2004). Studies with mitocans have shown exciting potential for cancer therapy since these drugs have limited side-effects, and at least some of these compounds possess specificity for cancer cells (Neuzil et al., 2007).
Evidence from this study and previous work (Yu and Yu, 1983) strongly supports a role for α-TOH and its analogue α-TOS as competitive inhibitors of the UbQ site(s) in CII. Our data reveal that both the QP and QD sites of CII are targets for α-TOS. Binding of the VE analogue at CII promotes ROS generation in cancer cells. The validity of the molecular modelling approach is supported by our identification of several other compounds with known strong interaction energies for the CII UbQ site(s) (results not shown). Of these, pyridoxal phosphate has been previously identified as a potent inhibitor of SDH activity by competing for UbQ (Choudhry et al., 1985, 1986). Presently, we are using this approach to identify other lead compounds that act by displacing UbQ in CII, since they may prove to be efficient anti-cancer agents.
The data we obtained from molecular modelling strongly supports the notion that the binding of α-TOS to CII within the QP and QD sites is a key to the apoptogenic activity of this agent. These observations were verified by experiments with cells where a mutation in CybL disrupts CII function and the ability of cells to survive when growing in the presence of succinate. First, CybL-mutant cells, unlike their CI-deficient and parental counterparts, failed to accumulate substantial levels of ROS and induce apoptosis when exposed to α-TOS, while reconstitution of functional CII reinstated susceptibility to the VE analogue. These findings can be reconciled with a study reporting resistance of CybL-mutant fibroblasts to apoptosis when challenged with the cytotoxic agents paclitaxel, doxorubicin, and cisplatin, or with the Fas ligand (Albayrak et al., 2003). Hence, CII is an important driver of ROS production mediated by cytotoxic drugs. Second, we demonstrate that knocking down CybL rendered cancer cells resistant to α-TOS-triggered ROS accumulation and apoptosis induction (Figure 4). This is important evidence, since the siRNA approach is an acute insult to the cells, while a knockout cell line can undergo adaptive differential expression of genes, potentially obscuring the direct effect of the knockout genotype.
We and others have previously shown α-TOS to be a potent inducer of ROS in cancer cells (Weber et al., 2003; Kang et al., 2004). In addition, the α-TOS drug target we have identified as CII of the respiratory chain has been proposed as an important contributor to cellular ROS production (McLennan and Degli-Esposti, 2000). Addition of MitoQ to cells did not overcome the 3BP-mediated inhibition of MTT reduction suggesting that 3BP acts upstream at the level of the SDH catalytic centre to prevent succinate oxidation. Yet, MitoQ overcame both the TTFA- and α-TOS-mediated inhibition of MTT reduction driven by succinate. While TTFA binds to both QP and QD sites in CII (Sun et al., 2005) like α-TOS, it displays less selective toxicity (Zhang et al., 2001) than the VE analogue for killing cancer cells (Neuzil et al., 2001b). Further studies are required to understand this differential specificity.
Thus, when α-TOS displaces UbQ from its binding site(s) on CII, electrons may no longer be tunneled down the SDH hydrophilic head on to FAD and relayed via the [Fe-S] centres and the heme group to UbQ (Tran et al., 2006; Cheng et al., 2006). Instead, they recombine with molecular oxygen to enhance superoxide anion radical (O2.-) production that ultimately leads to induction of apoptosis in the cancer cell. Although not clearly defined, it is possible that ROS generation occurs via the FAD site (Yankowskaya et al., 2003; Gottlieb and Tomlinson, 2005). It cannot be excluded, though, that electrons in the case of a dysfunctional CII, such as when there are mutations in CybL, interact with dioxygen, which is dissolved within the membrane part of CII (CybL, CybS) (Slane et al., 2006). Another possibility is that electrons move to CI by a process known as reverse electron transport, where they form O2.- (Adam-Vizi and Chinopoulos, 2006). It would appear that blocking UbQ binding promotes ROS generation by removing the opportunity of electrons to react with UbQ (Figure 8). This is strongly supported by studies with the C. elegans mev-1 mutant that can oxidize succinate to fumarate but cannot transfer electrons to ubiquinone, resulting in increased ROS levels and premature aging (Ishii et al., 1998). This observation is consistent with the leakage of electrons as a result of the release from succinate that cannot be accepted by UbQ because of a dysfunctional UbQ-binding site. Further, this can be reconciled with a recent study suggesting that the QP site is important for electron channelling between UbQ and the juxta positioned heme group (Zhao et al., 2006), which in normal cells contributes to stabilization of the transient ubisemiquinone radical (Tran et al., 2006). It is interesting that several human cancers, such as paragangliomas, resulting from genetic disorders due to mutations in CII subunits, including SDHC, may also be induced by increased ROS formation (Gottlieb and Tomlinson, 2005; Niemann and Muller, 2005; Ishii et al., 2005).
Figure 8.

Model for the interference of α-TOS with the mitochondrial electron chain. The scheme shows the proposed effects of α-TOS with the branching of the electron transport chain in its upstream region and clarifies the point of inhibition by α-TOS as specifically interacting with complex II. It also suggests CII as the possible site of superoxide generation, although its precise location within SDH has not been identified. The possible reverse electron transport from CII to CI is also shown.
Our results are not dissimilar to a recent report documenting selective molecular mechanism of apoptosis induction in cancer cells by adaphostin, a compound that binds to the UbQ reduction (Qi) site of CIII, resulting in ROS accumulation (Le et al., 2007). That CII is of importance in ROS formation is further evidenced by a recent report showing that non-steroid anti-inflammatory drugs caused ROS generation in cells, of which the majority was derived from CII (Soller et al., 2007). Moreover, ferulenol, a coumarine derivative, has been reported to affect the succinate ubiquinone reductase by interfering with the UbQ cycle (Lahouel et al., 2007). Given these recent results, more drugs affecting CII are likely to be developed in near future.
Although anti-cancer drugs have been described that may target the UbQ binding sites on the oxidoreductase complexes along the mitochondrial respiratory chain to increase cellular ROS levels and activate apoptosis (Le et al., 2007; Dias and Bailly, 2005; Hall, 2005), α-TOS has an additional feature in that it also disrupts the anti-apoptotic function of Bcl-2 and Bcl-xL by blocking their BH3-binding domain (Shiau et al., 2006). Hence, α-TOS may prove superior for inducing cancer cell death because of its double action on two important targets, QP and QD sites of CII and the Bcl-2 family proteins, promoting the induction of mitochondrially mediated apoptosis. This notion is further corroborated by our data revealing sensitization of cancer cells to α-TOS by the BH3 mimetic BH3I-2′.
We also show here that α-TOS strongly suppresses tumour progression in mouse models of breast cancer regardless of their erbB2 status, prolonging animal survival. This could be explained by α-TOS blocking the CII UbQ-binding sites, thereby causing ROS generation with ensuing induction of apoptosis, acting in a dominant manner downstream of any anti-apoptotic pro-survival activity resulting from the erbB2 receptor tyrosine kinase signalling. This makes the VE analogue a potentially useful anti-cancer agent when dealing with the challenge of breast cancers with high levels of expression of erbB2/HER2 that are rather recalcitrant to other forms of therapy (Slamon et al., 1989). Similarly, we recently demonstrated that a peptide conjugate of α-TOS (with relatively high affinity for the erbB2 receptor) effectively kills breast cancer cells (Wang et al., 2007).
To conclude, we present the molecular targets of the intriguing anti-cancer agent α-TOS with low toxicity to non-cancerous tissues (Weber et al., 2002), a compound that acts by selectively affecting mitochondria, organelles that are essential both for life and for unleashing the apoptosis machinery to induce cancer cell death (Newmeyer and Ferguson-Miller, 2005). We identify the UbQ-binding sites of the mitochondrial CII as a new target for anti-cancer drugs, and propose that these results will not only lead to the establishment of VE analogues as efficient anti-cancer agents but also will increase the interest in mitochondrial components as targets that are yet to be fully exploited for selective cancer therapy. Work is presently underway, using in silico modelling, to identify lead compounds that induce apoptosis by interfering with the UbQ-binding site(s) of CII and develop them into efficient and selective anti-cancer drugs. Our results on the molecular mechanism of apoptosis induced in cancer cells by α-TOS may be reconciled with our clinical outcome from a mesothelioma patient who has been treated with the VE analogue. The data reveals a significant clinical benefit with α-TOS therapy, causing a reduction in tumour volume and improved the well-being of our subject who had a lethal type of neoplastic pathology (Robinson et al., 2005). We are currently preparing to set up a larger clinical trial in which a cohort of mesothelioma patients will be treated with the mitocan, α-TOS.
Materials and methods
Cell culture and treatment
Human breast cancer cells MCF7 with low and MDA-MB-453 with high levels of erbB2 expression, and the murine breast cancer erbB2-high NeuTL cells derived from the c-neu transgenic mice were cultured in DMEM with 10% FCS and antibiotics. Jurkat T lymphoma cells were cultured in RPMI-1640 medium with 10% FCS and antibiotics. MCF7 cells deficient in mtDNA (ρ0) were prepared and used as described (Weber et al., 2003). CI-dysfunctional (B10 cells) (Seo et al., 1997), CII-dysfunctional (B9 cells with mutant CybL (Oostveen et al., 1995), and the parental Chinese hamster lung fibroblasts (B1 cells) (Oostveen et al., 1995) were grown in DMEM with 10% FCS, antibiotics, 10 mg/ml glucose and non-essential aminoacids.
Assessment of apoptosis, ROS accumulation, and membrane potential
Apoptosis was quantified using the annexin V-FITC kit (PharMingen) as described (Weber et al., 2003). Cellular ROS were detected with the probe dihydroethidium (DHE) (Molecular Probes) by flow cytometry (Weber et al., 2003), or by trapping with 5,5-dimethyl-1-pyrroline N-oxide (DMPO; Sigma) using electron paramagnetic resonance (EPR) spectroscopy (Weber et al., 2003). In some studies, the cells were pre-treated for 1 h with 2 μM MitoQ (James et al., 2005) or co-incubated with SOD (PEG-SOD, 750 units/ml; Sigma). The mitochondrial inner transmembrane potential (ΔΨm,i) was assessed using the polychromatic probe 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidoazolyl-carbocyanino iodide (JC-1; Molecular Probes) as specified elsewhere (Weber et al., 2003).
MTT assay (SDH activity)
MTT solutions were prepared immediately before use by dissolving 2.5 mg/ml 3-(4,5-dimethyl-2,5-diphenyl 2H-tetrazolium bromide (MTT) (Sigma) in PBS alone or phenol red-free RPMI media with 20 mM succinic acid, pH 7.4. Solutions of 3NPA, α-TOS and MitoQ in ethanol, TTFA in DMSO or 3BP in PBS were prepared and added to cells simultaneously with MTT. The drugs were tested on cells cultured in exponential growth phase in 96-well plates using 4-8 replicates per dilution. In some experiments, cells were pre-incubated for 60 min with 2 or 5 μM MitoQ. Final concentrations of ethanol or DMSO in cultures were <0.1% (v/v). Treated and control cells were exposed to MTT for 2 or 4 h at 37°C and 5% CO2. Supernatants were then removed, except for 30 μl, before adding 150 μl DMSO to dissolve the formazan crystals, and absorbance measured at 570 nm.
Preparation of mitochondrial particles
Rat liver mitochondria were freshly prepared as described (Rice and Lindsay, 1997) and stored at -20°C until used. P. denitrificans CCM982 (NCIB 8944) was grown anaerobically at 30°C in a medium containing 50 mM succinate as the carbon source and 10 mM nitrate as the terminal acceptor. Membranes were prepared as published (Burnell et al., 1975) and stored at -20°C until used.
Measurement of mitochondrial complex I and II activity
Reduction of the CII substrate DCPIP in the presence of cells or liver mitochondrial preparations was measured at 600 nm. Reaction mixtures contained 0.5 mM NADH, 5 mM succinate, 10 mM KCN, 50 μM DCPIP, and 50 μM PMS. For each assay point, 0.5 mg sample protein was used and α-TOS added as indicated. When measuring the CI (NADH dehydrogenase) activity, PMS was omitted.
RNA interference, cell transfection, Western blotting and RT-PCR
The siGENOME ON-TARGETplus set of four duplexes of siRNA oligonucleotides (Dharmacon) targeting the CybL (SDHC) subunit of CII were used. The sequences were: siRNA1: sense GCA CUG GUA UUG CUU UGA GUU, antisense 5′-P.CUC AAA GCA AUA CCA GUG CUU; siRNA2: sense GAA AAG UUC UCC UUA UUU GUU, antisense 5′-P.CAA AUA AGG AGA ACU UUU CUU; siRNA3: sense CAU CAU CUU CCU ACA CAU UUU, antisense 5′-P.AAU GUG UAG GAA GAU GAU GUU; siRNA4: sense AGA UAA AGA GGG CUA GUU AUU, antisense 5′-P.UAA CUA GCC CUC UUU AUC UUU. Non-specific siRNA was used as a negative control. Transfections of MCF7 cells with siRNA were performed as detailed elsewhere (Stapelberg et al., 2005). B9 cells were transfected using the Topo pCR3.1 Uni plasmid harbouring the CybL gene (Slane et al., 2006) and selected as described (Weber et al., 2003). Stably transfected and siRNA-treated cells were assessed for their SDH activity and expression of SDHC. Western blotting was performed as described (Wang et al., 2007) using anti-SDHC IgG (clone 3E2; Novus Biologicals) with anti-β-actin IgG (Santa Cruz) as a loading control. RT-PCR was performed using a standard protocol. The primers used were as follows: human CybL primers (Slane et al., 2006) - sense 5′-CAC TTC CGT CCA GAC CGG AAC-3′, antisense 5′-ATG CTG GGA GCC TCC TTT CTT CA-3′; Chinese hamster GAP-DH primers (Sever et al., 2004) - sense 5′-GCA AGT TCA AAG GCA CAG TCA A-3′; antisense 5′-CGC TCC TGG AAG ATG GTG AT-3′.
Molecular modelling of α-TOS interaction with UbQ binding in complex II
For details, see the Supplementary material.
Animal experiments
Balb/c nu/nu mice were inoculated s.c. with MCF7 cells (2×106 cells/mouse). After tumours developed, mice were injected with 10 μmoles α-TOS in DMSO i.p. every 3 d. Control mice were injected with an equal volume (100 μl) of DMSO. Tumour size was estimated with digital callipers.
Transgenic FVB/N c-neu mice carrying the rat HER2/neu proto-oncogene driven by the MMTV promoter on the H-2q FVB/N background (Guy et al., 1992) were used with ∼70% of the female mice developing spontaneous ductal mammary carcinomas with the latency time of ∼7 months. Tumours were quantified by ultrasound imaging (USI) using the Vevo770 device fitted with the RMV704 scan-head (VisualSonics) operated at 60 MHz and featuring 40 μm resolution (Wang et al., 2007; Dong et al., in press. Mice received treatment with 10 μmoles α-TOS in corn oil/4% ethanol or the vehicle administered i.p. every 3 d. α-TOS therapy of the animals commenced when the tumour volume was approximately 40 mm3. Animal studies were performed according to the guidelines of the Australian and New Zealand Council for the Care and Use of Animals in Research and Teaching and were approved by the local Animal Ethics Committee.
Statistical analyses
Between-group comparisons were made using mean±SD and the unpaired Student t test. Differences in the mean relative tumour size (±SEM) was examined using analyses of covariance (ANCOVA) with days as the covariate. Statistical analyses were performed using SPSS® 10.0 analytical software. Statistical significance was accepted at p<0.05.
Acknowledgments
The authors would like to thank Prof. R.A.J. Smith for providing MitoQ, and Prof. R.K. Ralph and Prof. J.W. Eaton for critical reading of the manuscript. This work was supported in part by grants from the National Breast Cancer Foundation, the Queensland Cancer Fund and the Australian Research Council (Discovery Grant DP0453372 to JN and Fellowship DP0343325 to PKW), and a grants from the Academy of Sciences of the Czech Republic(IAA500520702 and KAN2005207203 to J.N), the Institutional Research Concept Funds of the Academy of Sciences of the Czech Republic AV0Z50520701 (to J.N.), and by a grant from the Ministry of Agriculture of the Czech Republic (MZE 0002716201 to J.T.). D.R.S. and F.E.D. were supported by NIH grants RO1-CA100045 (D.R.S.) and NIH RO1-CA73612 (F.E.D.)
References
- Accelrys. InsightII. Accelrys Inc; San Diego: 2001. [Google Scholar]
- Adam-Vizi V, Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci. 2006;27:639–645. doi: 10.1016/j.tips.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Albayrak T, Scherhammer V, Schoenfeld N, Braziulis E, Mund T, Bauer MK, et al. The tumor suppressor cybL, a component of the respiratory chain, mediates apoptosis induction. Mol Biol Cell. 2003;14:3082–3096. doi: 10.1091/mbc.E02-10-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr B. 2002;5:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- Allen RG, Balin AK. Effects of oxygen on the antioxidant responses of normal and transformed cells. Exp Cell Res. 2003;289:307–316. doi: 10.1016/s0014-4827(03)00279-9. [DOI] [PubMed] [Google Scholar]
- Alleva R, Tomasetti M, Andera L, Gellert N, Borghi B, Weber C, et al. Coenzyme Q blocks chemical but not receptor-mediated apoptosis by increasing mitochondrial antioxidant protection. FEBS Lett. 2001;503:46–50. doi: 10.1016/s0014-5793(01)02694-1. [DOI] [PubMed] [Google Scholar]
- Berridge MV, Tan AS. Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch Biochem Biophys. 1993;303:474–478. doi: 10.1006/abbi.1993.1311. [DOI] [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Burnell JN, John P, Whatley FR. The reversibility of active sulphate transport in membrane vesicles of Paracoccus denitrificans. Biochem J. 1975;150:527–536. doi: 10.1042/bj1500527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Pearlman DA, Caldwell JW. Amber 7. University of California; San Francisco: 2002. [Google Scholar]
- Cheng VW, Ma E, Zhao Z, Rothery RA, Weiner JH. The iron-sulfur clusters in Escherichia coli succinate dehydrogenase direct electron flow. J Biol Chem. 2006;281:27662–27668. doi: 10.1074/jbc.M604900200. [DOI] [PubMed] [Google Scholar]
- Choudhry ZM, Gavrikova EV, Kotlyar AB, Tushurashvili PR, Vinogradov AD. Pyridoxal phosphate-induced dissociation of the succinate: ubiquinone reductase. FEBS Lett. 1985;182:171–175. doi: 10.1016/0014-5793(85)81177-7. [DOI] [PubMed] [Google Scholar]
- Choudhry ZM, Kotlyar AB, Vinogradov AD. Studies on the succinate dehydrogenating system. Interaction of the mitochondrial succinate-ubiquinone reductase with pyridoxal phosphate. Biochim Biophys Acta. 1986;850:131–138. doi: 10.1016/0005-2728(86)90017-4. [DOI] [PubMed] [Google Scholar]
- Church SL, Grant JW, Ridnour LA, Oberley LW, Swanson PE, Meltzer PS, Trent JM. Increased manganese superoxide dismutase expression suppresses the malignant phenotype of human melanoma cells. Proc Natl Acad Sci USA. 1993;90:3113–3117. doi: 10.1073/pnas.90.7.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- Dias N, Bailly C. Drugs targeting mitochondrial functions to control tumor cell growth. Biochem Pharmacol. 2005;70:1–12. doi: 10.1016/j.bcp.2005.03.021. [DOI] [PubMed] [Google Scholar]
- Don AS, Hogg PJ. Mitochondria as cancer drug targets. Trends Mol Med. 2004;10:372–378. doi: 10.1016/j.molmed.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Dong LF, Swettenham E, Eliasson J, Wang XF, Gold M, Medunic Y, et al. Vitamin E analogs inhibit angiogenesis by selective apoptosis induction in proliferating endothelial cells: The role of oxidative stress. Cancer Res. doi: 10.1158/0008-5472.CAN-07-3034. in press. [DOI] [PubMed] [Google Scholar]
- Fariss MW, Nicholls-Grzemski FA, Tirmerstein MA, Zhang JG. Enhanced antioxidant and cytoprotective abilities of vitamin E succinate is associated with a rapid uptake advantage in rat hepatocytes and mitochondria. Free Radic Biol Med. 2001;31:530–541. doi: 10.1016/s0891-5849(01)00615-3. [DOI] [PubMed] [Google Scholar]
- Geschwind JF, Ko YH, Torbenson MS, Magee C, Pedersen PL. Novel therapy for liver cancer: direct intraarterial injection of a potent inhibitor of ATP production. Cancer Res. 2002;62:3909–3913. [PubMed] [Google Scholar]
- Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5:857–866. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Natl Acad Sci USA. 1992;89:10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall N. Mitochondria: A novel target for the chemoprevention of cancer. Apoptosis. 2005;10:687–705. doi: 10.1007/s10495-005-0792-8. [DOI] [PubMed] [Google Scholar]
- Hartshorn MJ. AstexViewer™: An aid for structure-based drug design. J Computer Aided Mol Des. 2002;16:871–881. doi: 10.1023/a:1023813504011. [DOI] [PubMed] [Google Scholar]
- Huang LS, Sun G, Cobessi D, Wang AC, Shen JT, Tung EY, et al. 3-Nitropropionic acid is a suicide inhibitor of mitochondrial respiration that, upon oxidation by complex II, forms a covalent adduct with a catalytic base arginine in the active site of the enzyme. J Biol Chem. 2006;281:5965–5972. doi: 10.1074/jbc.M511270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, et al. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and aging in nematodes. Nature. 1998;349:694–697. doi: 10.1038/29331. [DOI] [PubMed] [Google Scholar]
- Ishii N, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005;65:203–209. [PubMed] [Google Scholar]
- James EM, Cocheme HM, Smith RA, Murphy MP. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J Biol Chem. 2005;280:21295–21312. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]
- James AM, Sharpley MS, Manas AB, Frerman FE, Hirst J, Smith RAJ, Murphy MP. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem. 2007;282:14708–14718. doi: 10.1074/jbc.M611463200. [DOI] [PubMed] [Google Scholar]
- Kang YH, Lee E, Choi MK, Ku JL, Kim SH, Park YG, Lim SJ. Role of reactive oxygen species in the induction of apoptosis by α-tocopheryl succinate. Int J Cancer. 2004;112:385–392. doi: 10.1002/ijc.20424. [DOI] [PubMed] [Google Scholar]
- Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, et al. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- Ko YH, Pedersen PL, Geschwind JF. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: characterization and targeting hexokinase. Cancer Lett. 2001;173:83–91. doi: 10.1016/s0304-3835(01)00667-x. [DOI] [PubMed] [Google Scholar]
- Ko YH, Smith BL, Wang Y, Pomper MG, Rini DA, Torbenson MS, et al. Advanced cancers: eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem Biophys Res Commun. 2004;324:269–275. doi: 10.1016/j.bbrc.2004.09.047. [DOI] [PubMed] [Google Scholar]
- Kogure K, Hama S, Manabe S, Tokumura A, Fukuzawa K. High cytotoxicity of α-tocopheryl hemisuccinate to cancer cells is due to failure of their antioxidative defense systems. Cancer Lett. 2002;186:151–156. doi: 10.1016/s0304-3835(02)00344-0. [DOI] [PubMed] [Google Scholar]
- Lahouel M, Zini R, Zellagui A, Rhouati S, Carrupt PA, Morin D. Ferulenol specifically inhibits succinate ubiquinone reductase at the level of the ubiquinone cycle. Biochem Biophys Res Commun. 2007;355:252–257. doi: 10.1016/j.bbrc.2007.01.145. [DOI] [PubMed] [Google Scholar]
- Le SB, Hailer MK, Buhrow S, Wang Q, Flatten K, Pediaditakis P, et al. Inhibition of mitochondrial respiration as a source of adaphostin-induced reactive oxygen species and cytotoxicity. J Biol Chem. 2007;282:8860–8872. doi: 10.1074/jbc.M611777200. [DOI] [PubMed] [Google Scholar]
- Maehara Y, Kusumoto T, Kusumoto H, Anai H, Sugimachi K. Sodium succinate enhances the colorimetric reaction of the in vitro chemosensitivity test: MTT assay. Oncology. 1988;5:434–436. doi: 10.1159/000226660. [DOI] [PubMed] [Google Scholar]
- McLennan HR, Degli-Esposti M. The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J Bioenerg Biomembr. 2000;32:153–162. doi: 10.1023/a:1005507913372. [DOI] [PubMed] [Google Scholar]
- Milanesi E, Costantini P, Gambalunga A, Colonna R, Petronilli V, Cabrelle A, et al. The mitochondrial effects of small organic ligands of BCL-2: sensitization of BCL-2-overexpressing cells to apoptosis by a pyrimidine-2,4,6- trione derivative. J Biol Chem. 2006;281:10066–10072. doi: 10.1074/jbc.M513708200. [DOI] [PubMed] [Google Scholar]
- Morris GM, Goodsell DS, Halliday RS. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J Comp Chem. 1998;19:1639–1662. [Google Scholar]
- Neuzil J, Massa H. Hepatic processing determines dual activity of vitamin E succinate. Biochem Biophys Res Commun. 2005;327:1024–1027. doi: 10.1016/j.bbrc.2004.12.115. [DOI] [PubMed] [Google Scholar]
- Neuzil J, Tomasetti M, Zhao Y, Dong LF, Birringer M, Wang XF, et al. Vitamin E analogs, a novel group of ‘mitocans’, as anti-cancer agents: The importance of being redox-silent. Mol Pharmacol. 2007;71:1185–1199. doi: 10.1124/mol.106.030122. [DOI] [PubMed] [Google Scholar]
- Neuzil J, Wang XF, Dong LF, Low P, Ralph SJ. Molecular mechanism of ‘mitocan’-induced apoptosis in cancer cells epitomizes the multiple roles of reactive oxygen species and Bcl-2 family proteins. FEBS Lett. 2006;580:5125–5129. doi: 10.1016/j.febslet.2006.05.072. [DOI] [PubMed] [Google Scholar]
- Neuzil J, Weber T, Gellert N, Weber C. Selective cancer cell killing by α-tocopheryl succinate. Br J Cancer. 2001a;84:87–89. doi: 10.1054/bjoc.2000.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuzil J, Weber T, Schröder A, Lu M, Ostermann G, Gellert N, et al. Induction of apoptosis in cancer cells by α-tocopheryl succinate: Molecular pathways and structural requirements. FASEB J. 2001b;15:403–415. doi: 10.1096/fj.00-0251com. [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Ferguson-Miller S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–490. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- Oostveen FG, Au HC, Meijer PJ, Scheffler IE. A Chinese hamster mutant cell line with a defect in the integral membrane protein CII-3 of complex II of the mitochondrial electron transport chain. J Biol Chem. 1995;270:26104–26108. doi: 10.1074/jbc.270.44.26104. [DOI] [PubMed] [Google Scholar]
- Ralph SJ, Dyason JC, Freeman R, Dong LF, Prochazka L, Wang XF, et al. Mitocans as anti-cancer agents targeting mitochondria: Lessons from studies with vitamin E analogues, inhibitors of complex II. J Bioenerg Biomembr. 2007;39:65–72. doi: 10.1007/s10863-006-9060-z. [DOI] [PubMed] [Google Scholar]
- Rice JE, Lindsay JG. Subcellular fractionation of mitochondria. In: Graham JM, Rickwood D, editors. Subcellular Fractionation. A Practical Approach Series. Oxford University Press; 1997. pp. 107–142. [Google Scholar]
- Robinson BW, Musk AW, Lake RA. Malignant mesothelioma. Lancet. 2005;366:397–408. doi: 10.1016/S0140-6736(05)67025-0. [DOI] [PubMed] [Google Scholar]
- Safford SE, Oberley TD, Urano M, St Clair DK. Suppression of fibrosarcoma metastasis by elevated expression of manganese superoxide dismutase. Cancer Res. 1994;54:4261–4265. [PubMed] [Google Scholar]
- Sanborn BM, Felberg NT, Hollocher TC. The inactivation of succinate dehydrogenase by bromopyruvate. Biochim Biophys Acta. 1971;227:219–231. doi: 10.1016/0005-2744(71)90055-6. [DOI] [PubMed] [Google Scholar]
- Sanner MF. Python: a programming language for software integration and development. J Mol Graphic Mod. 1999;17:57–61. [PubMed] [Google Scholar]
- Scallet AC, Haley RL, Scallet DM, Duhart HM, Binieda ZK. 3-Nitropropionic acid inhibition of succinate dehydrogenase (complex II) activity in cultured Chinese hamster ovary cells: antagonism by L-carnitine. Ann NY Acad Sci. 2003;993:305–312. doi: 10.1111/j.1749-6632.2003.tb07538.x. [DOI] [PubMed] [Google Scholar]
- Seo BB, Kitajima-Ihara T, Chan EK, Scheffler IE, Matsuno-Yagi A, Yagi T. Molecular remedy of complex I defects: rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc Natl Acad Sci USA. 1997;95:9167–9171. doi: 10.1073/pnas.95.16.9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever N, Lee PC, Song BL, Rawson RB, Debose-Boyd RA. Isolation of mutant cells lacking Insig-1 through selection with SR-12813, an agent that stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J Biol Chem. 2004;279:43136–43147. doi: 10.1074/jbc.M406406200. [DOI] [PubMed] [Google Scholar]
- Shiau CW, Huang JW, Wang DS, Weng JR, Yang CC, Lin CH, et al. α-Tocopheryl succinate induces apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 function. J Biol Chem. 2006;281:11819–11825. doi: 10.1074/jbc.M511015200. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- Slane BG, Aykin-Burns N, Smith BJ, Kalen AL, Goswami PC, Domann FE, Spitz DR. Mutation of succinate dehydrogenase subunit C results in increased oxidative stress, and genomic instability. Cancer Res. 2006;66:7615–7620. doi: 10.1158/0008-5472.CAN-06-0833. [DOI] [PubMed] [Google Scholar]
- Soller M, Drose S, Brandt U, Brune B, von Knethen A. Mechanism of thiazolidinedione-dependent cell death in Jurkat T cells. Mol Pharmacol. 2007;71:1535–1544. doi: 10.1124/mol.107.034371. [DOI] [PubMed] [Google Scholar]
- Spencer TL. The transport and oxidation of succinate by Ehrlich ascites-tumour cells. Biochem J. 1977;160:121–123. doi: 10.1042/bj1600121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapelberg M, Gellert N, Swettenham E, Tomasetti M, Witting PK, Procopio A, Neuzil J. α-Tocopheryl succinate inhibits malignant mesothelioma by disruption of the FGF autocrine signaling loop: Mechanism and the role of oxidative stress. J Biol Chem. 2005;280:25369–25376. doi: 10.1074/jbc.M414498200. [DOI] [PubMed] [Google Scholar]
- Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, et al. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005;121:1043–1057. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- Swettenham E, Witting PK, Salvatore BA, Neuzil J. α-Tocopheryl succinate selectively induces apoptosis in neuroblastoma cells: Potential therapy of malignancies of the nervous system? J Neurochem. 2005;94:1448–1456. doi: 10.1111/j.1471-4159.2005.03298.x. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Tran QM, Rothery RA, Maklashina E, Cecchini G, Weiner JH. The quinone binding site in Escherichia coli succinate dehydrogenase is required for electron transfer to the heme b. J Biol Chem. 2006;281:32310–32317. doi: 10.1074/jbc.M607476200. [DOI] [PubMed] [Google Scholar]
- Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Meth Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- Wang XF, Birringer M, Dong LF, Veprek P, Low P, Swettenham E, et al. A peptide adduct of vitamin E succinate targets breast cancer cells with high erbB2 expression. Cancer Res. 2007;67:3337–3344. doi: 10.1158/0008-5472.CAN-06-2480. [DOI] [PubMed] [Google Scholar]
- Wang XF, Witting PK, Salvatore BA, Neuzil J. α-Tocopheryl succinate induces apoptosis in HER2/erbB2-overexpressing breast cancer cells by signalling via the mitochondrial pathway. Biochem Biophys Res Commun. 2005;326:282–289. doi: 10.1016/j.bbrc.2004.11.028. [DOI] [PubMed] [Google Scholar]
- Weber T, Dalen H, Andera L, Nègre-Salvayre A, Augé N, Sticha M, et al. Mitochondria play a central role in apoptosis induced by α-tocopheryl succinate, an agent with anticancer activity. Biochemistry. 2003;42:4277–4291. doi: 10.1021/bi020527j. [DOI] [PubMed] [Google Scholar]
- Weber T, Lu M, Andera L, Lahm H, Gellert N, Fariss MW, et al. Vitamin E succinate is a potent novel anti-neoplastic agent with high tumor selectivity and cooperativity with tumor necrosis factor-related apoptosis-inducing ligand (TRAIL, Apo2L) in vivo. Clin Cancer Res. 2002;8:863–869. [PubMed] [Google Scholar]
- Word JM, Lovell SC, Richardson JS, Richardson DC. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, et al. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613–621. [PubMed] [Google Scholar]
- Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, et al. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- Yu L, Yu CA. Inhibitory effect of α-tocopherol and its derivatives on bovine heart succinate-cytochrome c reductase. Biochim Biophys Acta. 1983;723:139–149. doi: 10.1016/0005-2728(83)90113-5. [DOI] [PubMed] [Google Scholar]
- Zhang JG, Tirmerstein MA, Nicholls-Grzemski FA, Fariss MW. Mitochondrial electron transport inhibitors cause lipid peroxidation-dependent and -independent cell death: protective role of antioxidants. Arch Biochem Biophys. 2001;393:87–96. doi: 10.1006/abbi.2001.2486. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Rothery RA, Weiner JH. Effects of site-directed mutations in Escherichia coli succinate dehydrogenase on the enzyme activity and production of superoxide radicals. Biochem Cell Biol. 2006;84:1013–1021. doi: 10.1139/o06-188. [DOI] [PubMed] [Google Scholar]