

SUMMARY
Loss of the JunB/AP-1 transcription factor induces a myeloproliferative disease (MPD) arising from the hematopoietic stem cell (HSC) compartment. Now we show that JunB inactivation deregulates the cell cycle machinery and increases the proliferation of long-term repopulating HSCs (LT-HSCs) without impairing their self-renewal or regenerative potential in vivo. We found that JunB loss destabilizes a complex network of genes and pathways that normally limit myeloid differentiation, leading to impaired responsiveness to both Notch and TGF-β signaling due, in part, to transcriptional deregulation of the Hes1 gene. These results demonstrate that LT-HSC proliferation and differentiation are uncoupled from self-renewal, and establish some of the mechanisms by which JunB normally limits the production of myeloid progenitors hence preventing initiation of myeloid malignancies.
SIGNIFICANCE
Understanding how clonal dominance occurs in the HSC compartment is a central issue in leukemogenesis. Here, we show that inactivation of the JunB transcription factor causes cell cycle deregulation, disrupts the complex transcriptional network controlling proliferation and differentiation, and decreases responsiveness to Notch and TGF-β signaling. This renders the HSC compartment insensitive to the limits on expansion and myeloid differentiation imposed by those signals. Together, these deregulations lead to the aberrant expansion of myeloid progenitors and MPD development in vivo, without causing LT-HSC exhaustion. Our results provide a mechanism explaining how mutations that increase the proliferation of a single LT-HSC can become dominant and lead to the expansion of an aberrant clone within the otherwise normal HSC compartment.
INTRODUCTION
The mammalian hematopoietic system provides a unique, tractable model for investigating how cancer-associated mutations affect the behavior of specific cell populations and lead to the development of blood cancer or leukemia (Orkin and Zon, 2008). Hematopoietic development is organized hierarchically, starting with a rare population of hematopoietic stem cells (HSCs) that gives rise to a series of committed progenitors and mature cells with particular functional and immunophenotypic properties. HSCs are operationally defined by their ability to provide long-term multilineage reconstitution when transplanted into hematopoietically compromised recipients, and are the only cells that self-renew throughout life. HSCs are found predominantly in the bone marrow (BM) associated with several recently described vascular and endosteal niches (Kiel and Morrison, 2008). A complex balance of cell intrinsic regulators and cell extrinsic factors present in these niches normally maintain HSCs in a state of relative dormancy and regulate their trafficking to and from these BM niches. Under steady-state conditions, HSCs are a largely quiescent, slowly cycling cell population, which, in response to environmental stresses, are capable of dramatic expansion and contraction to ensure proper homeostatic replacement of blood cells (Passegué et al., 2005).
Gene knockout studies in mice have demonstrated that regulation of HSC numbers can be accomplished through direct modulation of HSC proliferative activity, resistance to apoptosis and retention in the BM niches (Orkin and Zon, 2008). Important mediators of these processes include cell cycle regulators such as the D-cyclins and the cyclin-dependant kinase inhibitors (CKIs) p21/CIP1 and p18/INK4c. Recent studies have also highlighted the roles of specific signal transducer (Pten), transcription factors (Gfi1, HoxB4, HoxA9) and extrinsic regulatory pathways (Notch, TGF-β, Wnt) in controlling HSC self-renewal and proliferation (Akala and Clarke, 2006; Blank et al., 2008). However, the precise molecular circuitry controlling HSC fate decisions has yet to be fully elucidated, and the mechanisms by which HSC maintenance, proliferation and differentiation are coordinately regulated to ensure homeostatic production of blood cells remain poorly understood. Recently, it has been suggested that changes in the quiescence status of HSCs (Holyoake et al., 1999) and deregulation of their interaction with BM niches (Jin et al., 2006) could be key events for their leukemic transformation and the development of myeloid leukemia. Still, little is known about the impact of leukemic transformation on HSC biological function and how abnormal HSC-derived leukemia-initiating stem cells (LSCs) differ from normal HSCs.
Originally discovered in leukemia, cancer-initiating stem cells have now been recognized in a variety of solid tumors (Wang and Dick, 2005). They represent a subset of a heterogeneous cancer population and are operationally defined by their ability to drive the formation and growth of a new tumor in transplanted mice. Convincing evidence indicates that LSCs are inefficiently eliminated by current therapeutic treatments and suggests that LSC persistence could be responsible for disease maintenance and/or recurrence (Jordan et al., 2006). Developing therapeutic interventions that specifically target LSCs is an appealing strategy for improving leukemia treatment, which requires an understanding of how LSCs escape normal regulatory mechanisms and become malignant. Few mouse models of human leukemia are currently available in which the LSC population has been identified and can be purified for analysis (Wang and Dick, 2005). This is an essential prerequisite for identifying pathways and molecules available for interventional therapies in patients.
We have previously developed several mouse strains lacking the JunB/AP-1 transcription factor that accurately recapitulate important clinical aspects of human myeloid malignancies, including chronic myelogenous leukemia (CML) (Passegué et al., 2001). We have also identified the LSC population as arising from the HSC compartment during the pre-cancerous myeloproliferative disease (MPD) phase (Passegué et al., 2004). Importantly, JunB inactivation has been observed in a spectrum of human myeloid malignancies, including CML (Yang et al., 2003), and downregulation of junB expression has been found in the HSC compartment of patients with acute myeloid leukemia (Steidl et al., 2006). At present, little is known about the role of JunB in HSC biology and myeloid leukemia development. Here, we have used junB-deficient mice as a model system to understand how JunB normally controls HSC functions and to identify the deregulated mechanisms that are responsible for HSCs transformation into LSCs.
RESULTS
Defects in hematopoietic reconstitution
To evaluate the impact of junB loss on HSC function in hematopoietic reconstitution, we first performed competitive bone marrow (BM) transplantation experiments (Figure 1A). Control and junB-deficient BM cells (1×106 total) were mixed in a 1:1 or 9:1 ratio with GFP-expressing competitor BM cells, and injected into lethally irradiated recipients. Regardless of the donor/competitor ratio used, we found that junB-deficient BM cells consistently displayed ~50% reduced engraftment efficiency compared to control BM cells. We then directly transplanted 1×106 control or junB-deficient BM cells into lethally or sublethally irradiated recipients (Figure 1B and data not shown). While transplantation into lethally irradiated mice gave comparable reconstitution levels (Passegué et al, 2004), transplantation into sublethally irradiated mice revealed extremely variable engraftment levels with junB-deficient BM cells being on average 50% less efficient than control BM cells. To confirm the limited engraftment of junB-deficient cells, we transplanted small numbers of an HSC-enriched population (defined as Lin-/c-Kit+/Sca-1+/Flk2- or Flk2- LSK BM cells, hereafter referred to as hematopoietic stem and progenitor cells or HSPCs) together with 3×105 unfractionated helper BM cells into lethally irradiated recipients (Figure 1C and Table S1). While no engraftment was observed after injection of 50 to 100 junB-deficient HSPCs, transplantation of 250 junB-deficient HSPCs resulted in a 2 to 10-fold decrease in reconstitution levels depending mainly upon the use of Sca-1-depleted (HSC-purged) helper BM cells. In every case where junB-deficient HSPCs engrafted (even at low levels), we observed progressive expansion of the myeloid lineage leading, by 6 to 12 months posttransplantation, to the development of an MPD (Figure 1D) similar to the disease observed in primary junB-deficient mice (Passegué et al., 2004).
Figure 1. Hematopoietic reconstitution potential and homing activity of junB-deficient bone marrow cells.
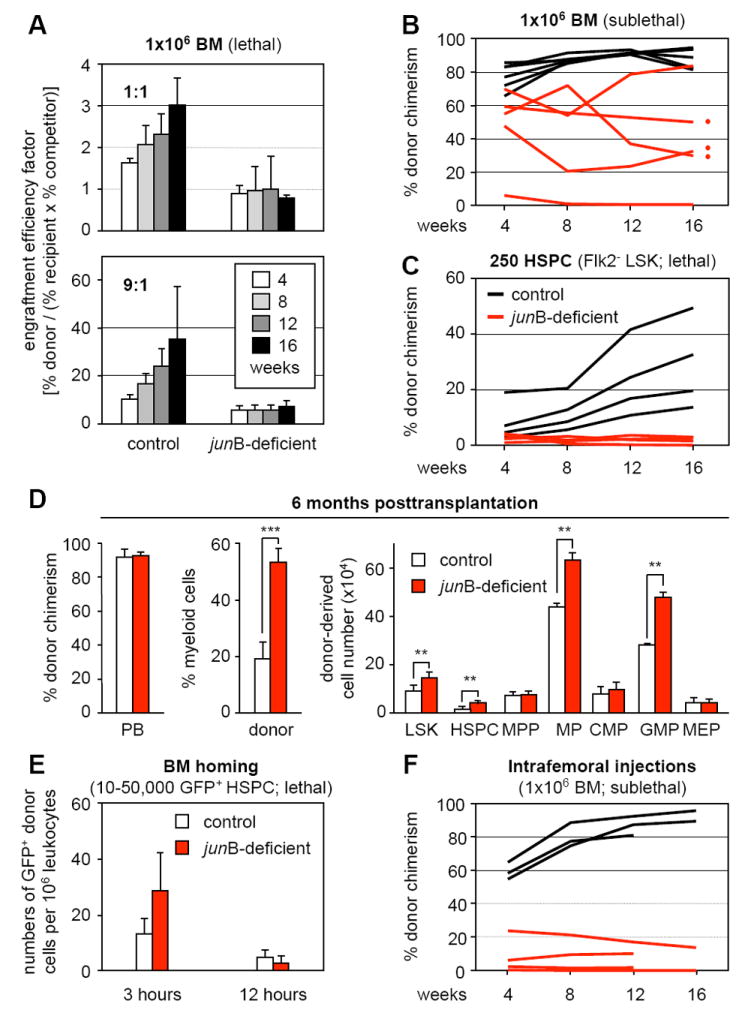
Lethally or sublethally irradiated recipients (CD45.1) were transplanted with the indicated numbers and type of donor control or junB-deficient cells (CD45.2). Mice were bled every 4 weeks and analyzed for % CD45.2 chimerism in PB.
(A) Transplantation of 1×106 unfractionated BM cells (1:1 and 9:1 ratio of donor and competitor GFP+/CD45.1 cells) into lethally irradiated recipients (n = 5 mice per cohort). Engraftment efficiency factor (percentages ± SD) was calculated as [(% CD45.2+ donor cells)/((% CD45.1+ recipient and competitor cells) × (% GFP+ competitor cells))].
(B) Transplantation of 1×106 unfractionated BM cells into sublethally irradiated recipients (n = 5 mice per cohort).
(C) Transplantation of 250 purified HSPCs (Flk2- KLS) together with 3×105 helper CD45.1 BM cells into lethally irradiated recipients (n = 4 mice per cohort).
(D) MPD development in recipients of junB-deficient BM cells (red dots in panel B). The % CD45.2 chimerism and donor-derived myeloid (Gr-1+/Mac-1+) cells in PB, and the total cell numbers for the indicated BM subpopulations is given at 6 months posttransplantation (averages ± SD; n = 3 mice per cohort; **p ≤ 0.01; ***p ≤ 0.001). MPP: multipotent progenitors (Flk2+ LSK); MP: myeloid progenitors (Lin-/Sca-1-/c-Kit+); CMP: common myeloid progenitors; GMP: granulocyte/macrophage progenitors; MEP: megakaryocyte/erythrocyte progenitors.
(E) Short-term in vivo homing assay. Sublethally irradiated recipients (n = 3 mice per cohort) were injected with either 50,000 LSK (3 hours) or 10,000 Flk2- LSK (12 hours) cells isolated from β-actin GFP control and junB-deficient mice. The number (averages ± SD) of transplanted GFP+ cells present in the BM was determined at 3 or 12 hours postinjection.
(F) Intrafemoral injections. Unfractionated 1×106 BM cells were directly injected into the femoral cavity of sublethally irradiated recipients (1 femur injected per mouse, n = 3 to 4 mice per cohort).
The engraftment process requires that transplanted HSCs home to the BM cavity and cross the endothelial layer forming the wall of the blood vessels (Lapidot et al., 2005). To assess whether junB-deficient HSPCs are defective in BM homing, we performed short-term in vivo homing assays by injecting lethally irradiated recipients with deficient GFP+ Flk2- LSK or LSK cells isolated from junB-deficient mice crossed to β-actin GFP transgenic mice. Analysis of the peripheral blood (PB), BM, spleen as well as the hematopoietic component of the liver for the presence of GFP+ cells at 3 or 12 hours postinjection revealed similar numbers of control and junB-deficient HSPCs in all tissues, including BM, at both time points (Figure 1E and data not shown). We also used intrafemoral injections of 1×106 BM cells into sublethally irradiated recipients to confirm that impaired homing did not contribute to the reduced engraftment of junB-deficient cells (Figure 1F and S1). Taken together, these results indicate that despite normal lodging in the BM cavity after transplantation, junB-deficient HSPCs are impaired in their ability to engraft the BM niches and to contribute to hematopoietic reconstitution. This suggests that junB-deficient HSCs are either defective in BM maintenance or that fewer long-term repopulating HSCs (LT-HSCs) are present in the transplanted junB-deficient populations.
Deregulated cell cycle distribution
HSC engraftment following transplantation as well as maintenance during steady-state hematopoiesis critically depends upon their quiescent state (Passegué et al., 2005). We next investigated whether the engraftment defects observed with junB-deficient HSPCs could result from changes in their cell cycle distribution. Analyses performed using either live Hoechst-33342 (H)/Pyronin Y (PY) staining on purified Flk2- LSK or intracellular 7AAD/PY staining on unfractionated BM cells (Figure 2A and Table S2) revealed that junB-deficient HSPCs had on average a 2-fold decrease in the percentage of quiescent G0 cells and a correlative increase in the percentage of cycling G1 and S-G2/M cells. Short-term kinetic analysis of bromodeoxyuridine (BrdU) incorporation indicated increased proliferation rates in junB-deficient HSPCs, with ~13% of BrdU+ cells after 1 hour incorporation reaching 24% after 12 hours compared to ~8% and ~19%, respectively, for control HSPCs (Figure 2B and data not shown). Ki67 and phospho-histone H3 immunofluorescence staining on isolated Flk2- LSK cells further confirmed that junB-deficient HSPCs have a higher proliferative index (Table S2).
Figure 2. Deregulated proliferation and regulation of the cell cycle machinery in junB-deficient HSPCs.
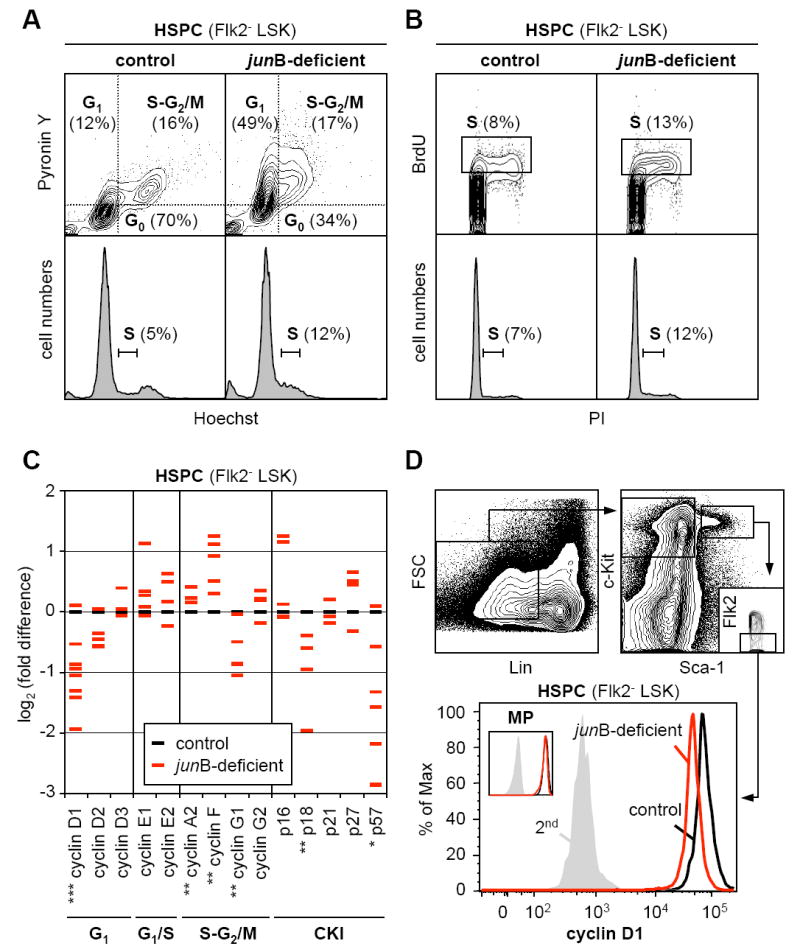
(A) Live Hoechst (H)/Pyronin Y(PY) staining of purified control and junB-deficient HSPCs. Quiescent G0 cells are H2n/PY- while proliferative G1 and S-G2/M cells are H2n/PY+ and H≥2n-4n/PY+, respectively. Bottom histograms indicate DNA contents.
(B) BrdU incorporation (1 hour in vivo pulse) in purified control and junB-deficient HSPCs. Cells in S-phase (box) are BrdU+ and have ≥2n DNA content as determined by PI counterstaining (bottom histograms).
(C) Quantitative RT-PCR analysis of cell cycle gene expression in control and junB-deficient HSPCs. Flk2- LSK were isolated from pools of 3 to 5 control mice and from age-matched individual junB-deficient mice. The results shown are averages of triplicate measurements and are expressed as log2(fold difference) compared to the levels found in control HSPCs (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; β-actin normalization). For each gene, between 3 to 9 independent samples are analyzed per genotype (some red dashes are overlapping).
(D) Intracellular FACS analysis of cyclin D1 protein levels in control and junB-deficient HSPCs and myeloid progenitors (MP: Lin-/Sca-1-/c-kit+).
To understand the deregulations occurring at the molecular level, we analyzed by quantitative RT-PCR (qRT-PCR) the expression level of a comprehensive panel of cell cycle genes in Flk2- LSK cells isolated from pools of control mice (3 to 5 mice) or from age-matched individual junB-deficient mice (Figure 2C). Although significant fluctuations in gene expression were observed between the individual junB-deficient HSPC populations (likely reflecting differences in the stage of MPD development in the respective donor mice), we found a consistent decrease in the expression levels of early G1-cyclins (mainly D1) and of two CKIs (p18 and p57), associated with a trend toward increased expression of late G1-cyclins (E1 and E2) and S-G2/M-cyclins (A2 and F). Furthermore, we confirmed a decrease in cyclin D1 protein levels in junB-deficient HSPCs using intracellular FACS analysis (Figure 2D). Taken together, these results provide a molecular signature of the deregulations occurring in the cell cycle regulation of junB-deficient HSPCs and a mechanism for their enhanced proliferation.
Quiescence status and engraftment potential
Increased proliferation and altered ratios of quiescent vs. cycling HSPCs may indicate that the engraftment defect observed with junB-deficient cells results from transplanting fewer numbers of quiescent HSCs. To test this hypothesis, Flk2- LSK cells were isolated, stained with H/PY and re-sorted for the G0 subset after which 250 G0 HSPCs were transplanted into lethally irradiated recipients together with a radio-protective dose of Sca-1-depleted helper BM cells (Figure 3A). Surprisingly, we found that junB-deficient G0 HSPCs still displayed severe defects in hematopoietic reconstitution compared to the same number of control G0 HSPCs. This result prompted our re-evaluation of the cell surface marker combination used to identify engrafting HSCs by performing intracellular 7AAD/PY staining for cell cycle distribution together with additional molecules that enrich for LT-HSC activity: CD34 (Osawa et al., 1996) and the SLAM markers CD150 and CD48 (Kiel et al., 2005). Using the CD34 marker, as expected, we identified a highly quiescent CD34-/Flk2- LSK population in control mice (Figure S2). However, in the junB-deficient mice, the CD34-/Flk2- LSK cells also displayed aberrant cell cycle distribution with massive recruitment into G1 and significant expansion in numbers. In sharp contrast, using the SLAM markers, we identified a highly quiescent CD150+/CD48-/Flk2- LSK population (G0: 92.4 ± 3.5% vs. 89.5 ± 3.8%; G1-S-G2/M: 2.2 ± 2.3 vs. 10.5 ± 4.1; n = 5) that was of similar size in both control and junB-deficient mice (Figure 3B and 3C). This staining strategy also revealed that the increased in size of the junB-deficient HSPCs compartment was almost entirely caused by an increase in CD48+/Flk2- LSK cells (Figure 3B).
Figure 3. Persistence of a highly quiescent LT-HSC population with normal engraftment potential in junB-deficient mice.
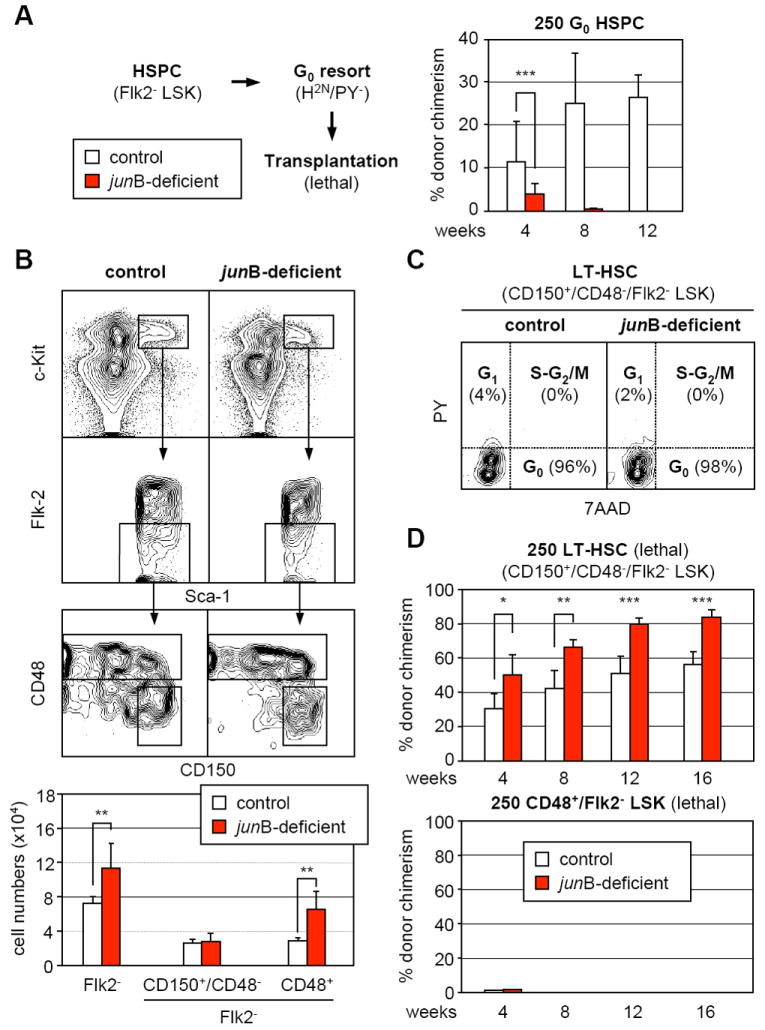
(A) Hematopoietic reconstitution from quiescent HSPCs. Flk2- LSK were first isolated from control and junB-deficient mice (CD45.2), stained with H/PY and re-sorted for the G0 (H2n/PY-) subpopulation. Lethally irradiated recipient mice (CD45.1; n = 3 per cohort) were injected with 250 purified G0 HSPCs together with 3×105 Sca-1-depleted helper CD45.1 BM cells, and analyzed monthly for percentages (± SD) of CD45.2 chimerism in PB (***p ≤ 0.001).
(B) SLAM marker subfractionation of the HSPC compartment in control and junB-deficient mice. The top graphs show examples of CD150 and CD48 expression in the Flk2- LSK compartment and the bottom graph summarizes the total numbers (averages ± SD) of Flk2- LSK and SLAM subsets in control (n = 7) and junB-deficient (n = 5) mice (**p ≤ 0.01).
(C) Cell cycle analysis of CD150+/CD48-/Flk2- LSK in control and junB-deficient mice. BM cells were stained for CD150 expression in the Flk2- LSK compartment (with CD48 included in the lineage cocktail, see Figure S3) in combination with intracellular staining for cell cycle distribution (7AAD/PY). Quiescent G0 cells are 7AAD2n/PY- while proliferative G1 and S-G2/M cells are 7AAD2n/PY+ and 7AAD≥2n-4n/PY+, respectively.
(D) Hematopoietic reconstitution from CD150+/CD48-/Flk2- LSK and CD48+/Flk2- LSK subsets. Cells were isolated from CD45.2 control and junB-deficient mice, and 250 cells for each subset were injected together with 3×105 Sca-1-depleted helper CD45.1 BM cells into lethally irradiated CD45.1 recipients. Mice (n = 5 per cohort) were analyzed every 4 weeks for percentages (± SD) CD45.2 chimerism in PB (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
Subsequently, we transplanted 250 CD150+/CD48-/Flk2- LSK or CD48+/Flk2- LSK cells into lethally irradiated recipients to investigate their engraftment potential (Figure 3D). As expected, neither control nor junB-deficient CD48+/Flk2- LSK cells provided long-term reconstitution (Kiel et al., 2005). In fact, CD48+/Flk2- LSK cells only gave rise to a transient myeloid readout without long lasting lymphoid cell production (data not shown), and their progenies were almost completely absent from the recipient mice at 1 month posttransplantation (Figure 3D). Strikingly, junB-deficient CD150+/CD48-/Flk2- LSK cells displayed quantitatively enhanced and more robust repopulating activity than control cells, which reflects their increased production of mature myeloid cells. By 11 months posttransplantation, robust and sustained engraftment was observed in both cohorts (control: 78 ± 14% chimerism; junB-deficient: 93 ± 2% chimerism; n = 5), accompanied by MPD development in mice transplanted with junB-deficient cells (data not shown). Taken together, these results demonstrate that CD150+/CD48-/Flk2- LSK cells are the true quiescent and engrafting LT-HSCs in both control and junB-deficient mice. They indicate that the reconstitution defects observed upon transplantation of junB-deficient unfractionated BM or purified HSPCs result from dilution of these engrafting LT-HSCs by the expanded, non-engrafting CD48+/Flk2- LSK cells. They also provide an explanation for the puzzling outcome of the G0 HSPC transplantation experiments (Figure 3A) since the transplanted junB-deficient G0 HSPCs contained a much larger fraction of non-engrafting G0 CD48+/Flk2- LSK cells (Figure S3) than control G0 HSPCs, hence again diluting the true engrafting quiescent LT-HSCs.
Increased proliferation with normal regenerative potential
Next, we tested whether junB-deficient LT-HSCs, despite maintaining normal numbers in the BM, could have increased proliferation rates by performing long-term BrdU incorporation as recently described (Kiel et al., 2007) (Figure 4A and Table S3). Control and junB-deficient mice were injected intraperitoneally with BrdU (1 mg/6 g body weight) and then fed BrdU (1 mg/ml) in their drinking water for up to 10 days. LT-HSCs (CD150+/CD48-/Flk2- LSK) were isolated and the cells that incorporated BrdU while replicating their DNA were enumerated by immunofluorescence analysis. In addition, we also quantified BrdU incorporation by intracellular FACS analysis (Figure S4). Using both detection methods, we found that junB-deficient LT-HSCs displayed significantly increased BrdU incorporation compared to control LT-HSCs, reaching 60.7% ± 2.5% vs. 42.5% ± 0.7% (n = 4; p ≤ 0.005) after 10 days of BrdU administration. Cell cycle entry rates, calculated by regression on the log of the proportion of unlabelled cells (Cheshier et al., 1999), revealed that ~13.6% junB-deficient LT-HSCs enter the cell cycle each day (with population doubling time of 16.3 days) compared to only ~6.4% per day for control LT-HSCs (with population doubling time of 26.9 days). We also performed qRT-PCR analysis for the expression of quiescence and cell cycle-associated genes in junB-deficient LT-HSCs (Figure 4B). Interestingly, we found a significant increase in the expression level of G0/G1 transition kinase cyclin C together with changes already observed in junB-deficient HSPCs, mainly decreased expression of the CKIs p18, p57 and cyclin D1, and increased expression of the mitotic cyclin A2.
Figure 4. Deregulated turnover rates but normal regeneration potential in junB-deficient LT-HSCs.
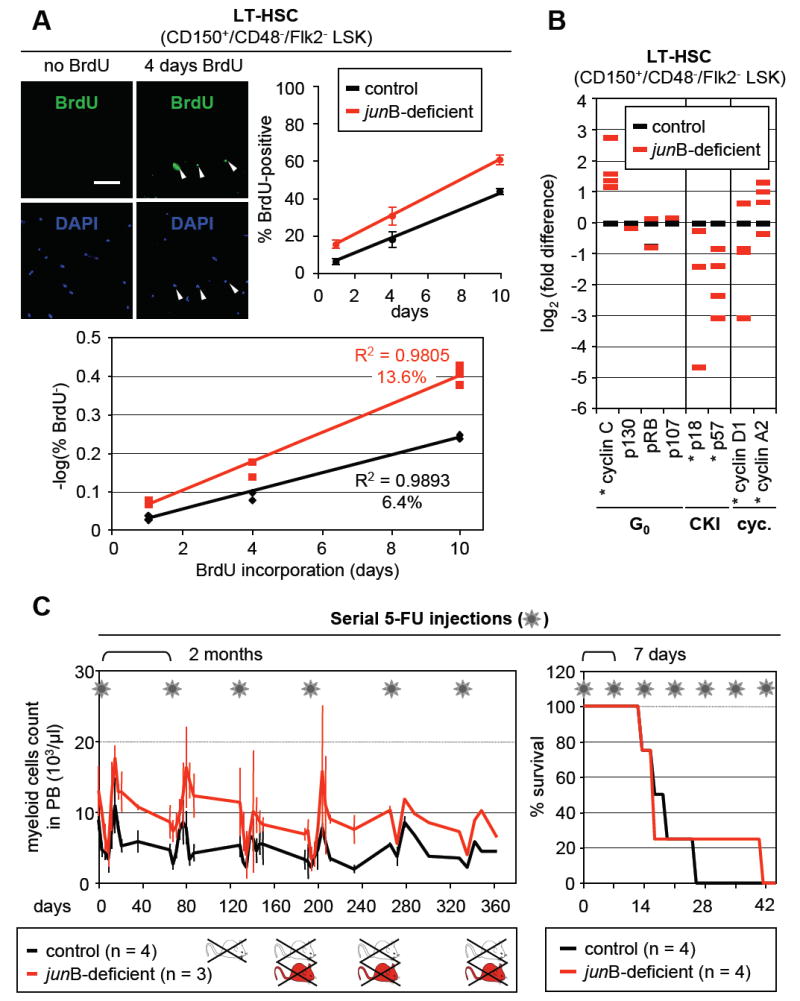
(A) Long-term kinetics of BrdU incorporation in LT-HSCs. CD150+/CD48-/Flk2- LSK were isolated from individual control and junB-deficient mice fed with BrdU for the indicated number of days (n = 2 to 3 mice per time point). The pictures shown representative examples of BrdU staining (scale bar: 100 mm; arrowhead: BrdU+ LT-HSCs). BrdU enumeration upon immunofluorescence detection (top row; averages ± SD) and calculation of the regression on the log of the proportion of unlabelled cells (bottom graph) are shown.
(B) Quantitative RT-PCR analysis of cell cycle gene expression in control and junB-deficient LT-HSCs. CD150+/CD48-/Flk2- LSK were isolated from pools of age-matched control and junB-deficient mice (2 to 3 mice per pool, n = 3). The results shown are averages of triplicate measurements and are expressed as log2(fold difference) compared to the levels found in control LT-HSCs (*p ≤ 0.05; β-actin normalization).
(C) Serial 5-FU injections. Control and junB-deficient mice were injected intraperitoneally with 150 mg/kg 5-FU (asterisk) either every 2 months or every 7 days. For the 2 months protocol (left side), the kinetics of myeloid recovery following 5-FU injection were evaluated. The results (averages ± SD) show the evolution of myeloid cell counts in the peripheral blood as assessed by CBC analysis. For the 7 days protocol (right side), the survival rates postinjection were determined.
The increased proliferation of junB-deficient LT-HSCs raises the possibility that these cells might exhaust faster than normal LT-HSCs under regenerative stress conditions as suggested by other studies using transplantation of more or less purified cell populations (Orford and Scadden, 2008). To address this question without using the transplantation system, we evaluated hematopoietic recovery and survival after serial injections of the myelosuppressive agent 5-fluorouracil (5-FU; 150 mg/kg) (Figure 4C). When 5-FU was injected every second month (to allow complete recovery between injections) both control and junB-deficient mice started dying by the 3rd 5-FU injection without any significant difference between the two groups. As expected, junB-deficient mice always displayed higher restoration levels of the myeloid lineage than control mice indicating constant regeneration of the MPD from junB-deficient LT-HSCs that persisted after 5-FU treatment. When 5-FU was injected every 7 days (to continuously deplete regenerating progenitor populations) both control and junB-deficient mice also displayed similar survival rates, with the majority of deaths occurring by the 3rd or 4th 5-FU injection. These results indicate that junB-deficient LT-HSCs do not exhaust faster and have similar regenerative potential as control LT-HSCs. Taken together with the transplantation data, they demonstrate that the fast cycling junB-deficient LT-HSCs have normal self-renewal activity in vivo.
Broad transcriptional deregulations
To determine how loss of JunB affects HSC fate decisions, we first analyzed junB-deficient HSPCs by qRT-PCR for the expression levels of genes known to play a role in self-renewal, quiescence, proliferation regulation and oncogenic transformation (Figure 5A). Overall, we found a general trend towards decreased gene expression in junB-deficient Flk2- LSK cells, with significant reductions in the levels of Pten, Hoxb4 and Hoxa9 (self-renewal genes), Notch1, Hes1 and Hes5 (Notch pathway) non-significant reductions in n-Myc and c-Myb levels (oncogenes), and no changes in cell cycle regulators and TGF-β pathways components. To exclude measuring changes reflecting only the expansion of the CD48+/Flk2- LSK cells observed in junB-deficient mice, we also analyzed the expression of the self-renewal genes in LT-HSCs (Figure 5B). While Pten, Hoxb4, Hoxa9 and Notch1 did not exhibit significant changes in quiescent junB-deficient LT-HSCs, the expression of Hes1 (a key downstream mediator of both Notch and TGF-β pathways) (Blokzijl et al., 2003) remained significantly decreased. Furthermore, preliminary chromatin immunoprecipitation (ChIP) analysis in wild type HSPCs and detailed promoter studies in 3T3 fibrobasts identified two repeated JunB binding sites in the proximal region of the mouse Hes1 promoter and demonstrated that JunB was a direct transcriptional activator of Hes1 expression (Figure S5).
Figure 5. Broad deregulation of self-renewal and extracellular signaling pathway genes in junB-deficient LT-HSCs.
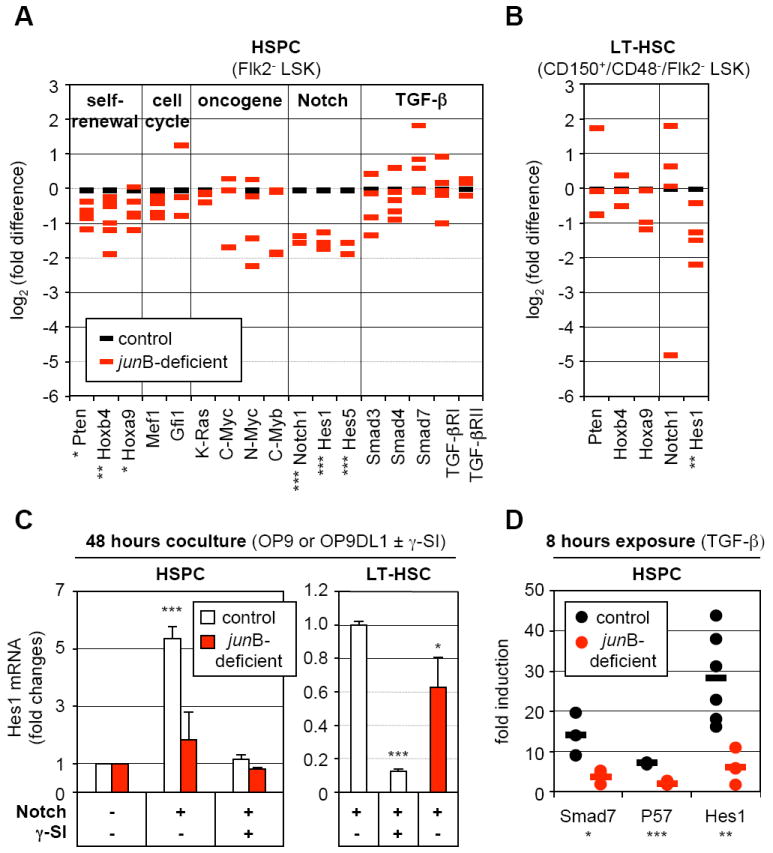
(A) Quantitative RT-PCR analysis of the expression levels of genes and pathways controlling HSC fate decisions in control and junB-deficient HSPCs. The same cDNA samples as in Figure 2C were used for this analysis (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
(B) Quantitative RT-PCR analysis of control and junB-deficient LT-HSCs. The same cDNA samples as in Figure 4B were used for this analysis (*p ≤ 0.05; **p ≤ 0.01).
(C) Control or junB-deficient HSPCs or LT-HSCs (3,000 cells per well) were directly sorted into 96-wells plates containing either OP9 (Notch -) or OP9DL1 (Notch +) cells and incubated for 48 hours with or without γ-secretase inhibitor (γ-SI; 10 μM). The data shown for each population are the averages ± SD of two independent experiments each performed in triplicate. HSPC results are expressed as fold increase compared to Hes1 levels in control or junB-deficient cells cocultured on OP9 cells without γ-SI (arbitrary set to 1). LT-HSC results are expressed as fold decreased compared to Hes1 levels in control cells cocultured on OP9DL1 cells without γ-SI (arbitrary set to 1) (*p ≤ 0.05; ***p ≤ 0.001; β-actin normalization).
(D) Control or junB-deficient HSPCs (3,000 to 20,000 cells per well) were directly sorted into a 96-wells plate, rested overnight and stimulated with 5 ng/ml TGF-β for 8 hours. The results are expressed as fold increase compared to Smad7, Hes1 and p57 levels found in non-stimulated HSPCs incubated for the same length of time. Independent measurements (each performed in triplicate) from 3 to 6 experiments and averages (bars) are shown (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; RL-19 normalization).
To functionally assess the status of the Notch pathway in junB-deficient HSCs, we performed a short-term coculture experiment on OP9 or OP9 stromal cells expressing the Notch ligand delta-1 (OP9DL1). Control and junB-deficient HSPCs were sorted directly into a 96 well plate containing either OP9 or OP9DL1 cells, grown for 48 hours with or without γ-secretase inhibitor (γ-SI) to prevent activation of the Notch pathway, and analyzed by qRT-PCR for Hes1 expression levels (Figure 5C). When cultured on OP9DL1 cells, control HSPCs displayed a strong induction of Hes1 expression that was completely abrogated upon γ-SI treatment. In contrast, Hes1 expression was not significantly induced in junB-deficient HSPCs cultured on OP9DL1 cells, which demonstrate a defective response to Notch stimulation in these cells. We also confirmed defective induction of Hes1 expression in junB-deficient LT-HSCs cocultured for 48 hours on OP9DL1 cells (Figure 5C). To assess the status of the TGF-β pathway, we incubated sorted control and junB-deficient HSPCs for 8 hours with recombinant TGF-β and measured by qRT-PCR the expression levels of three direct transcriptional targets of this pathway (i.e., Smad7, the CKI p57 and Hes1) (Figure 5D). We found significantly reduced induction of all three genes in junB-deficient HSPCs compared to control HSPCs, which indicate that these cells are also defective in their response to TGF-β stimulation. Taken together, these results indicate that loss of JunB destabilizes a broad network of interconnected genes and pathways that regulate HSC fate decisions. They demonstrate that JunB is essential to control the extent to which HSCs respond to Notch and TGF-β stimulation due, in part, to JunB involvement in the transcriptional regulation of the common downstream effecter gene Hes1.
Increased myeloid differentiation in response to impaired Notch and TGF-β regulations
Finally, we asked whether the defective response of junB-deficient HSCs to Notch and TGF-β stimulation could contribute to the increased myelopoiesis observed in junB-deficient mice. To answer this question, control and junB-deficient HSPCs and LT-HSCs were first cocultured for 4 days on OP9 or OP9DL1 cells in the presence or absence of TGF-β (Figure 6A). This initial step was designed to assess growth in response to activation of Notch and TGF-β pathways similar to what is likely provided by the BM niches in vivo. Control cells were also treated with γ-SI and TGF-β inhibitor (T-βI; 5 μM) to mimic the dual impairment observed in junB-deficient HSPCs. After 4 days of coculture, these HSPC- or LT-HSC-derived cells transferred into methylcellulose containing a cocktail of myeloid cytokines and growth factors (but devoid of TGF-β or inhibitors) to assess myeloid colony-forming activity (CFU). This second step allowed us to estimate the numbers of myeloid progenitors generated in each coculture condition. As expected, stimulation with TGF-β and activation of the Notch pathway (alone or in combination) severely limited the growth and myeloid differentiation from control cells compared to untreated cultures (Figure 6A). In contrast, similar treatments did not significantly inhibit junB-deficient HSPCs or LT-HSCs expansion rates. Most strikingly, neither Notch induction nor TGF-β stimulation were able to limit myeloid differentiation from junB-deficient cells, which matched the myeloid differentiation potential obtained in untreated cultures (Figure 6A). Similarly, pharmacological inhibition of Notch and TGF-β signaling in control HSPCs or LT-HSCs partially prevented the growth inhibition and fully relieved the block in myeloid differentiation imposed by these regulatory mechanisms. Taken together, these results indicate that Notch and TGF-β cooperate to limit the rate at which LT-HSCs and early progenitor cells expand (both pathways) and differentiate (mostly TGF-β) to produce myeloid progenitors. They also suggest that lack of response to Notch and TGF-β contribute to the ability of junB-deficient LT-HSCs to constitutively overproduce myeloid progenitors and to induce MPD development in vivo.
Figure 6. Defective Notch and TGF-β pathway responsiveness contributes to the overproduction of myeloid progenitors from junB-deficient LT-HSCs.
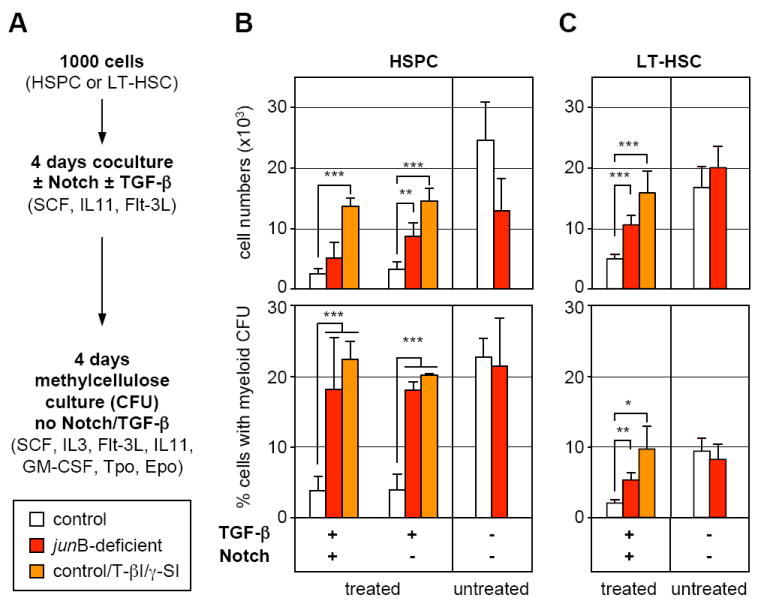
(A) Schematic of the experimental design. Control and junB-deficient HSPCs (B) or LT-HSCs (C) were directly sorted into 96-wells plates (1,000 cells per well) containing OP9 (Notch -) or OP9DL1 (Notch +) cells and incubated for 4 days with or without 5 ng/ml TGF-β in the presence or absence of TGF-β inhibitor (T-βI; 5 μM) and γ-secretase inhibitor (γ-SI; 10 μM). Cells where then counted (top graphs) and replated in methylcellulose to determine the percentage of cells with myeloid-colony forming activity present in each type of culture condition (bottom graph). The results shown are the averages ± SD of four independent experiments performed in duplicate (HSPCs) and two independent experiments performed in triplicate (LT-HSCs) (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
DISCUSSION
In this study, we identified some of the mechanisms by which JunB regulates LT-HSC functions and discovered how inactivation of this key transcription factor can lead to overproduction of myeloid progenitors and MPD development in vivo. We found that JunB normally controls LT-HSC proliferation and limits their rate of production of myeloid progenitors by maintaining appropriate responsiveness to Notch and TGF-β signaling, in part, through transcriptional regulation of Hes1, an important mediator of both pathways. These functions place JunB at the center of the complex network of cell intrinsic and cell extrinsic signals that ensure coordinated regulation of LT-HSC proliferation and differentiation, independently of LT-HSC maintenance and self-renewal activity. Taken together, these results provide a potential mechanism for disease initiation in the range of human myeloid malignancies in which JunB inactivation has already been reported (Yang et al., 2003; Steidl et al., 2006). Moreover, they expand our current understanding of the normal biology of the HSC compartment and the abnormal properties of HSC-derived LSCs.
Re-thinking transplantation assays used to analyze HSCs number and activity
HSCs have been defined and studied for many decades by their ability to engraft and reconstitute the blood system of transplanted mice (Orkin and Zon, 2008). This functional property has allowed for the phenotypic identification and isolation of progressively more enriched HSC populations (Table S4). Hence it has become common practice to enumerate HSCs by performing limit dilution transplantation experiments with unfractionated BM cells. While this technique provides a valid assessment of HSC numbers in normal situations, our results demonstrate that it is a potentially unreliable method to determine HSCs number and function in pathological situations. Hence, when we compared side-by-side the same numbers of control and junB-deficient unfractionated BM cells or enriched, but not pure, HSC populations (Flk2- LSK or HSPCs), we observed engraftment defects from junB-deficient cells, associated with deregulated cell cycle distribution, loss of quiescence and impaired serial transplantability by the third passage (data not shown). Only upon isolation of HSCs to near functional purity (using CD150+/CD48-/Flk2- LSK surface markers, one of the best phenotypic combinations currently available), we uncovered that junB-deficient LT-HSCs had in fact normal engraftment capability as well as normal cell cycle distribution with more than 90% of the BM population quiescent at any given time. By using the same amount of unfractionated BM or partially purified HSPCs for both genotypes, we mistakenly injected fewer of the engrafting LT-HSCs and more of the overproduced non-engrafting differentiating cells for junB-deficient than for control populations (Table S4). This dilution effect was also observed when we transplanted the quiescent fraction of the junB-deficient HSPC compartment, as the expanded CD48+/Flk2- LSK subset also displayed a large portion of quiescent cells (Figure S3). These results have, at least, two important implications. First, they demonstrate that quiescence per se is not a defining characteristic of LT-HSCs and should not be solely used for identifying this cell population even in an enriched subset. In fact, most of the early progenitor cells, including ST-HSCs and MPPs, have ≥50% of their population in the G0 phase of the cell cycle at any given time (Passegué et al., 2005). Second, they indicate that one must transplant a highly purified cell population when assessing LT-HSC numbers and properties in situations where artificially-introduced genetic alterations (in mice) or naturally occurring mutations (in humans) can affect the biological activity of the HSC compartment leading to either overproduction or underproduction of differentiating cells. Incorrect assumptions can otherwise be made regarding the number and self-renewal activity of these mutant LT-HSCs.
Proliferation and HSC maintenance
Several published studies have suggested that increased HSC proliferation may lead to stem cell exhaustion and loss of maintenance in the BM microenvironment (Orford and Scadden, 2008). Here, we show that junB-deficient LT-HSCs incorporate BrdU twice as fast as normal LT-HSCs due to direct or indirect changes in the regulation of their cell cycle machinery (i.e., increased in cyclin C and cyclin A2 expression, decreased in cyclin D1, p18 and p57 CKI levels) but do not display any changes in their BM numbers, quiescence status, engraftment and regenerative potential in vivo. These results clearly demonstrate that HSC maintenance in the BM niches and self-renewal activity is not solely dictated by LT-HSC proliferation rate. One question raised by these observations is how nearly identical numbers of quiescent LT-HSCs can be maintained in the BM microenvironment in both control and junB-deficient mice? One possibility is that junB-deficient LT-HSCs could have shorter cell cycle length leading to increased turnover rates and BrdU accumulation, without an increase in population size or decrease in G0 frequency. Another possibility is that junB-deficient LT-HSCs have increased proliferation (without changes in the length of their cell cycle) but that the overproduced LT-HSCs are displaced from the BM (maybe due to lack of available niches) and released into the periphery where they eventually become exhausted. This would imply the existence of retention mechanisms provided by the BM niches that would specifically maintain a preset number of quiescent LT-HSCs over an excess of cycling LT-HSCs. While such mechanisms are still poorly understood, they are likely to involve cell-cell and cell-matrix adhesion molecules such as α4/β1-VLA4 integrins, CD44 or CXCR4/SDF-1α, which are expressed on HSCs and are important for their retention in the BM. Increased numbers of functional LT-HSCs are found in the PB, liver and spleen of junB-deficient mice (unpublished observations), which also develop pronounced extramedullary hematopoiesis (Passegué et al., 2004), suggesting that displacement of overproduced LT-HSCs might indeed be occurring in these mice as it has already been found in other related mutant mice (Min et al., 2008).
Control of HSC differentiation
Despite being extensively studied, the intricate molecular machinery and signaling mechanisms that coordinately regulate HSC proliferation and differentiation have remained largely elusive (Akala and Clarke, 2006; Blank et al., 2008). Here, we show that at least two developmentally conserved signaling pathways that still play a controversial role in HSCs self-renewal (Notch and TGF-β pathways) are in fact critical for limiting the rates at which HSCs produce myeloid progenitors. First, we found that the MPD-producing junB-deficient LT-HSCs have impaired responsiveness to both Notch activation and TGF-β stimulation as measured by decreased induction of downstream effectors of these pathways (i.e., Smad7, p57 and Hes1). Then, using normal LT-HSCs and pharmacological inhibitors, we show that impairing responsiveness to TGF-β pathway in the presence of attenuated Notch stimulation leads to deregulated production of myeloid progenitors in vitro. Notch signaling has been shown to be necessary for maintaining HSCs in an undifferentiated state in the presence of proliferative signals in vitro (Duncan et al., 2005). Recently, it has been reported that the complete absence of canonical Notch signaling does not affect LT-HSCs maintenance and self-renewal in vivo (Maillard et al., 2008). TGF-β is one of the most potent inhibitors of HSC growth in vitro (Sitnicka et al., 1996) and a negative regulator of myelopoiesis in vivo (Letterio and Roberts, 1998). Similar to Notch, it has been shown that absence of TGF-β signaling does not affect LT-HSC maintenance and self-renewal in vivo (Larsson et al., 2005). Taken together with our findings, these observations suggest that Notch and TGF-β belong to a complex network of signaling mechanisms provided by the BM niches that limit the rates at which LT-HSCs proliferate and produce myeloid progenitors, without affecting their maintenance and self-renewal activity (see model in Figure 7A). In this context, JunB appears essential to maintain appropriate responsiveness to both Notch and TGF-β signaling due to its direct or indirect function in maintaining the expression levels of key downstream effectors of these pathways, including the Hes1 gene. As a consequence, loss of JunB renders the HSC compartment refractory to these negative regulatory mechanisms resulting in increased production of myeloid progenitors and MPD development in vivo.
Figure 7. JunB is a key transcriptional regulator of the HSC compartment that control LT-HSC proliferation and differentiation without affecting self-renewal.
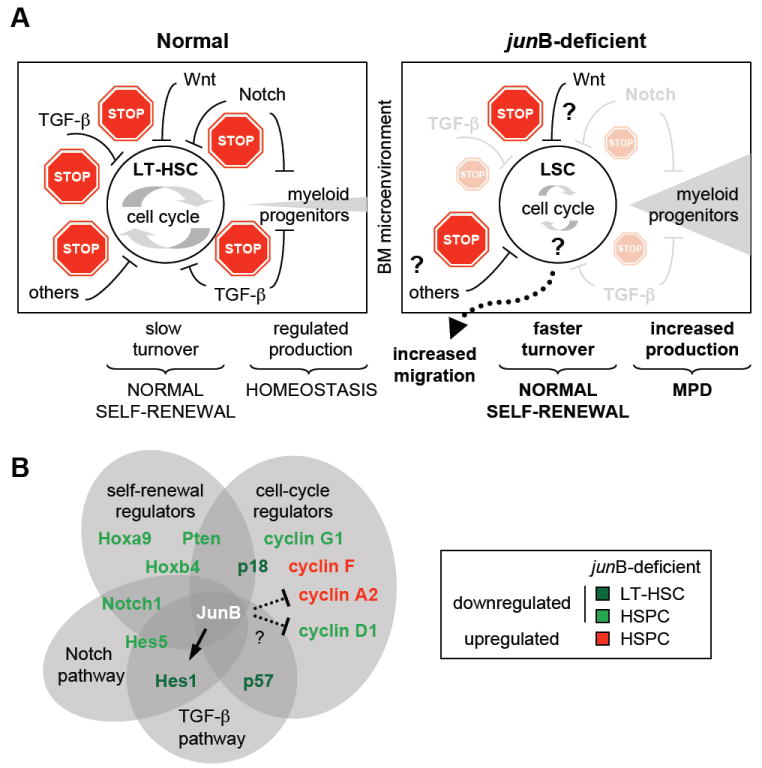
(A) Model summarizing the defective regulation of junB-deficient LT-HSCs. Normal LT-HSCs (left side) in their BM microenvironment are maintained as slowly cycling, mostly quiescent cells due to the concerted action of a complex network of extrinsic regulatory pathways that provide multiple stop signals to limit proliferation and rates of myeloid progenitor production. In absence of junB expression, LT-HSCs lose responsiveness to at least two of these key negative regulatory pathways (Notch and TGF-β) leading to a dampening of the stop signals provided by the BM microenvironment. As a result, junB-deficient LT-HSCs display increased turnover rates without overt expansion in the BM niches (most likely due to increase migration into the periphery) and overproduce myeloid progenitors, hence leading to MPD development in vivo. Importantly, junB-deficient LT-HSCs have normal self-renewal potential indicating that the mechanisms controlling LT-HSC maintenance are unaffected in these LSCs.
(B) JunB transcriptional network in hematopoietic stem and early progenitor cells. Lines (arrow = activation; blunted = inhibition) represent confirmed (plain) or un-confirmed (dotted) direct JunB transcriptional targets.
Deconstructing the JunB transcriptional network in HSCs
Here, we demonstrate that JunB is a core transcriptional regulator of HSC functions that control a vast network of interconnected genes and pathways involved in numerous fate decisions including proliferation and early myeloid differentiation (see model in Figure 7B). We also identify Hes1 as a direct JunB transcriptional target that could be involved in mediating part of these effects. However, direct or indirect deregulation of other effecter genes including (but not limited) to Hes5, Notch1, Pten, HoxB4 and HoxA9 are likely to play a role in establishing or maintaining the aberrant biological functions displayed by junB-deficient HSPCs. Furthermore, it remains to be determined how loss of junB results in the activation of LT-HSCs cell cycle machinery. JunB is a known transcriptional activator of p16 (Passegué et al., 2000) and a transcriptional repressor of both cyclin D1 and cyclin A (Bakiri et al., 2000; Andrecht et al., 2002). While we did observe the expected increase in cyclin A2 in junB-deficient HSPCs, neither p16 (unchanged) nor cyclin D1 (decreased) behave as predicted. It is possible that still uncharted changes in other transcriptional regulators and/or signaling pathways could counterbalance the direct effect of loss of JunB on the expression level of those genes. Alternatively, differences in JunB-mediated transcriptional regulation in distinct cells types (fibroblasts vs. HSCs) or other confounding factors, such as the age of the mice, could explain these discrepancies. In fact, decreased p16 expression is observed in junB-deficient HSPCs isolated from older MPD mice (Passegué et al., 2004), consistent with an enhanced role for p16 in controlling the biology of aging HSCs (Janzen et al., 2006). Genome-wide ChIP analysis is now required to gain a better understanding of these complex regulations and further delineate the JunB transcription network in HSCs.
LSC transformation
Our results indicate that deregulating the network of signaling mechanisms that control the balance between HSC proliferation and differentiation can lead to aberrant myeloid progenitor expansion and MPD development, without causing LT-HSC exhaustion. These findings have important implications for understanding the development of myeloid malignancies in humans. They provide a mechanism to explain how mutations that increase the proliferation of a single LT-HSC can become dominant and lead to the expansion of an aberrant clone within the otherwise normal HSC compartment. They also explain how human LSCs emerging from the HSC compartment, such as in CML, can be maintained as a quiescent, self-renewing, non-expanding population that overproduces myeloid progenitors and mature cells (Holyoake et al., 1999) and can resist most of the current anti-leukemia treatments (Jordan et al., 2006). Similar to junB-deficient LT-HSCs escaping 5-FU killing and regenerating the MPD in treated mice, CML LSCs might escape therapeutic treatment by taking advantage of the intact HSC-associated protection mechanisms provided by the BM niches. It is also tempting to speculate that BCR/ABL (the fusion oncoprotein encoded by the t(9:22) translocation, the hallmark of CML) might act very similarly to junB-inactivation by deregulating differentiation without affecting LT-HSC self-renewal mechanisms. It will be interesting to use the spectrum of deregulation observed in junB-deficient HSCs as a road map to investigate BCR/ABL-expressing HSCs and determine whether similar pathways are affected. This type of comparison could uncover common deregulated mechanisms used by different oncogenic events and identify shared targets available for therapeutic intervention aimed at limiting LSCs aberrant functional properties or preventing HSC transformation into LSCs.
EXPERIMENTAL PROCEDURES
Mice
Congenic C57BL/6-CD45.1 mice were used as donors (4 to 6 week-old) for purification of wild type cells and as recipients (8 to 12 week-old) for transplantation experiments. Control and junB-deficient (junBfl/flMORE-Cre) C57BL/6-CD45.2 mice have been described previously (Passegué et al., 2004). β-actin-GFP C57BL/6-CD45.1 transgenic mice (Forsberg et al., 2006) were used as donors (4 to 6 week-old) for purification of GFP expressing competitor cells and to obtain, upon crossing, GFP expressing control and junB-deficient mice. 5-Fluorouracil (150 mg/kg) was administrated by intraperitoneal injection. All animal experiments were performed in accordance with UCSF Institutional Animal Care and Use Committee approved protocols.
Flow cytometry
Cell preparation, staining, analysis and isolation procedures were performed as previously described (Forsberg et al., 2006; Kiel et al., 2005). Additional information can be found in Table S5. After staining, cells were resuspended in Hank’s buffered saline solution (HBSS) containing 2% heat-inactivated FBS (Hyclone) and 1 μg/ml propidium iodide (PI) for dead cell exclusion, and were sorted on a FACS Aria or analyzed on a LSRII (Becton Dickinson). Each subpopulation was double sorted and re-analyzed to ensure maximum purity.
Transplantations
Congenic recipient mice were irradiated using a cesium source irradiator with lethal (1200 Rad) or sublethal (950 Rad) doses delivered in split dose 3 hours apart and given antibiotic-containing water for at least 6 weeks postirradiation. For in vivo homing experiments, mice were irradiated one day prior to injections. For all other transplantation experiments, mice were injected immediately after irradiation. For intravenous injections, cells were resuspended in a volume of 100 μl and injected into the retro-orbital plexus. For intrafemoral injections, cells were resuspended in a volume of 30 μl and injected through the knee into the femoral cavity of anesthetized mice. Donor and recipient cells were distinguished by expression of GFP or different allelic forms of CD45 (CD45.1 vs. CD45.2).
Cell cycle and intracellular staining
Short and long-term BrdU kinetics as well as live Hoechst 33342 (H)/Pyronin Y (PY) staining were performed as previously reported (Passegué et al., 2005; Kiel et al., 2007) (Table S5). Intracellular 7-Amino-actinomycin D (7-AAD)/PY and cyclin D1 staining of unfractionated BM cells were performed as described in Table S5.
Gene expression analysis
Total RNA was isolated using Trizol reagent (Invitrogen), digested with DNaseI and used for reverse-transcription according to the manufacturer’s instructions (SuperScript III™ kit, Invitrogen). QRT-PCR primers were designed using Primer Express software (Applied Biosystems) (Table S6). All reactions were performed in an ABI-7300 sequence detection system using SYBR® Green PCR Core reagents (Applied Biosystems) and cDNA equivalent of 200 cells per reaction as previously described (Forsberg et al., 2006). Expression levels of β-actin or ribosomal protein L-19 (RL-19) genes were used to normalize the amount of the investigated transcript.
Cell cultures
Hematopoietic (co)cultures were performed in Iscove’s modified Dulbecco’s media (IMDM) containing 5% FBS, 1x penicillin/streptomycin, 2 mM GlutaMax-1, 0.1 mM non-essential amino acid, 1 mM sodium pyruvate, 50 μM 2-mercaptoethanol and supplemented with 25 ng/ml of SCF, Flt3-L and IL11. For coculture experiments, HSCs were sorted directly over a monolayer of OP9 and OP9DL1 cells plated in a 96 well plate (5,000 to 20,000 cells per well) the day before and grown for up to 4 days (with medium addition every 2 days) with or without 5 ng/ml recombinant human TGF-β 1 (R&D System), 10μM γ-secretase inhibitor (XXI; Calbiochem) and/or 5 μM TGF-β inhibitor (SB431542; Sigma). Following the culture period, HSC-derived cells were collected, transferred in a 96 well plate, incubated for 20 min at 37°C to deplete for contaminating stromal cells, re-collected, rinsed once with HBSS and then either resuspended in Trizol reagent for RNA isolation or counted, diluted (1:3 to 1:50) and replated in methylcellulose. Myeloid colony-forming activity was evaluated after 4 days of culture in IMDM-based methylcellulose media (M3231; Stem Cell) supplemented with SCF (25 ng/ml), Flt3-L (25 ng/ml), IL11 (25 ng/ml), IL3 (10 ng/ml), Tpo (25 ng/ml), Epo (4 U/ml) and GM-CSF (10 ng/ml).
Statistical analysis
P values were calculated by using the unpaired student t test.
Acknowledgments
We thank Amy Wagers, Diana Laird and Robert Blelloch for critical reading of the manuscript, Joshua Stuart for bioinformatics assistance and Monique Dail for help with the Notch experiments. M.S. is supported by a fellowship from FRSQ and K.S. by a Rubicon grant from NWO and a fellowship from KWF. E.C.F. is the recipient of a CIRM New Investigator Award, E.P. and B.S.B. of ASH Scholar Awards. This work was supported by research grants from CONCERN Foundation and UCSF REAC, a shared V Foundation Translational Award and NIH grant HL092471 to E.P.
Footnotes
The authors have no financial interests to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akala OO, Clarke MF. Hematopoietic stem cell self-renewal. Curr Opin Genet Dev. 2006;16:496–501. doi: 10.1016/j.gde.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Andrecht S, Kolbus A, Hartenstein B, Angel P, Schorpp-Kistner M. Cell cycle promoting activity of JunB through cyclin A activation. J Biol Chem. 2002;277:35961–35968. doi: 10.1074/jbc.M202847200. [DOI] [PubMed] [Google Scholar]
- Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 2000;19:2056–2068. doi: 10.1093/emboj/19.9.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank U, Karlsson G, Karlsson S. Signaling pathways governing stem-cell fate. Blood. 2008;111:492–503. doi: 10.1182/blood-2007-07-075168. [DOI] [PubMed] [Google Scholar]
- Blokzijl A, Dahlqvist C, Reissmann E, Falk A, Moliner A, Lendahl U, Ibáñez CF. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J Cell Biol. 2003;163:723–728. doi: 10.1083/jcb.200305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheshier S, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci USA. 1999;96:3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C, Yoon K, Cook JM, Willert K, Gaiano N, et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol. 2005;6:314–322. doi: 10.1038/ni1164. [DOI] [PubMed] [Google Scholar]
- Forsberg EC, Serwold TC, Kogan S, Weissman IL, Passegué E. New evidence supporting megakaryocyte-erythrocyte potential of Flk2/Flt3+ multipotent hematopoietic progenitors. Cell. 2006;26:415–426. doi: 10.1016/j.cell.2006.06.037. [DOI] [PubMed] [Google Scholar]
- Holyoake T, Jiang X, Eaves C, Eaves A. Isolation of highly quiescent subpopulation of primitive leukemic cells in chronic myelogenous leukemia. Blood. 1999;94:2056–2064. [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MY, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependant kinase inhibitor p16/INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Eng J Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Kiel MJ, He S, Ashkenazi R, Gentry SN, Teta M, Kushner JA, Jackson TL, Morrison SJ. Haematopoietic stem cells do not asymetrically segregate chromosomes or retain BrdU. Nature. 2007;449:238–242. doi: 10.1038/nature06115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol. 2008;8:290–301. doi: 10.1038/nri2279. [DOI] [PubMed] [Google Scholar]
- Lapidot T, Dar A, Kollet O. How do stem cells find their way home? Blood. 2005;106:1901–1910. doi: 10.1182/blood-2005-04-1417. [DOI] [PubMed] [Google Scholar]
- Larsson J, Blank U, Klintman J, Magnusson M, Karlsson S. Quiescence of hematopoietic stem cells and maintenance of the stem cell pool is not dependent on TGF-beta signaling in vivo. Exp Hematol. 2005;33:592–596. doi: 10.1016/j.exphem.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Maillard I, Koch U, Dumortier A, Shestova O, Xu L, Sai H, Pross SE, Aster JC, Bhandoola A, Radtke F, et al. Canonical Notch signaling is dispensable for the maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2008;2:356–366. doi: 10.1016/j.stem.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min IM, Pietramaggiori G, Kim FS, Passegué E, Stevenson KE, Wagers AJ. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell. 2008;2:380–391. doi: 10.1016/j.stem.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat rev Genet. 2008;9:115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa M, Hanada K, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273:242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- Passegué E, Wagner EF. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 2000;19:2969–2979. doi: 10.1093/emboj/19.12.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegué E, Jochum W, Schorpp-Kistner M, Möhle-Steinlein U, Wagner EF. Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking JunB expression in the myeloid lineage. Cell. 2001;104:21–32. doi: 10.1016/s0092-8674(01)00188-x. [DOI] [PubMed] [Google Scholar]
- Passegué E, Wagner EF, Weissman IL. JunB-deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell. 2004;119:431–443. doi: 10.1016/j.cell.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Passegué E, Wagers AJ, Guiriato S, Anderson W, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steidl U, Rosenbauer F, Verhaak R, Gu X, Otu HU, Bruns I, Steidl C, Costa DB, Klippel S, Wagner K, et al. Essential role of Jun family transcription factors in PU.1-induced leukemic stem cells. Nat Genetics. 2006;38:1269–1277. doi: 10.1038/ng1898. [DOI] [PubMed] [Google Scholar]
- Sitnicka E, Ruscetti FW, Priestley GV, Wolf NS, Bartelmez SH. Transforming growth factor beta 1 directly and reversibly inhibits the initial cell divisions of long-term repopulating hematopoietic stem cells. Blood. 1996;88:82–88. [PubMed] [Google Scholar]
- Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Yang MY, Liu TC, Chang JG, Lin PM, Lin SF. JunB gene expression is inactivated by methylation in chronic myeloid leukemia. Blood. 2003;101:3205–3211. doi: 10.1182/blood-2002-05-1598. [DOI] [PubMed] [Google Scholar]