

Abstract
Defective hepatitis B virus DNA (dDNA) is reverse-transcribed from spliced hepatitis B virus (HBV) pregenomic messenger RNA (pgRNA) and has been identified in patients with chronic HBV (CH-B). The major 2.2-kb spliced pgRNA encodes a novel HBV gene product, the hepatitis B splice protein (HBSP) via a deletion and frame shift within the polymerase. Although spliced RNA and HBSP expression have been associated with increased HBV DNA levels and liver fibrosis, the role of dDNA in HBV-associated disease is largely undefined. Our aims were to (1) compare the relative proportions of dDNA (% dDNA) in a range of HBV-infected serum samples, including patients with human immunodeficiency virus (HIV)/HBV coinfection and HBV-monoinfected persons with differing severities of liver disease, and (2) determine the effect of mutations associated with drug resistance on defective DNA production. Defective DNA was detected in 90% of persons with CH-B. There was no significant difference in the relative abundance of dDNA between the monoinfected and HIV/HBV-coinfected groups. We also found no association between the%dDNA and alanine aminotransferase, hepatitis B e antigen status, HBV DNA levels, fibrosis levels, compensated or decompensated liver cirrhosis, genotype, or drug treatment. However, the%dDNA was significantly lower in individuals infected with lamivudine-resistant (LMV-R) HBV compared with wild-type HBV (P < 0.0001), indicating that antiviral drug resistance alters the balance between defective and genomic length DNA in circulation. Experiments in vitro using HBV encoding LMV-R mutations confirmed these results.
Conclusion
Our results identified no association between dDNA and parameters associated with disease status and suggested that the relative abundance of dDNA is largely dependent on the integrity of the HBV polymerase and is unrelated to the severity of liver disease.
Hepatitis B (HBV) is a leading cause of cirrhosis and hepatocellular carcinoma. HBV infection is normally not cytopathic, with liver disease resulting from immune-mediated lysis of HBV-infected cells.1 A more aggressive form of hepatitis is associated with human immunodeficiency virus (HIV)/HBV coinfection, with liver mortality rates 14-fold higher than HBV-monoinfected individuals,2 despite these individuals having immune dysfunctions. The reasons for this are unclear, although HBV DNA levels are higher in HIV/HBV-coinfected persons,2,3 and the recent identification of a novel HBV mutant predominantly in coinfected persons suggests that the HBV itself may play a role in disease progression.4
Up to 12 different defective HBV DNAs (dDNA) have been identified in the serum of persons with chronic hepatitis B (CH-B), the most frequently detected being 2 kb in length.5 These defective molecules were more prevalent in the sera of persons with CH-B than in persons who had an acute infection.6 This molecule is reverse transcribed from an RNA transcript produced after excision of a 1.3-kb intron from the 3.5-kb pregenomic messenger RNA (pgRNA).5-9 The interaction between the 2.2kb pgRNA and Pol occurs in trans, as deletion of the 1.3-kb intron removes most of the HBV Pol C-terminus, including the reverse transcriptase (RT) and ribonuclease H encoding domains.7 Spliced pgRNA forms up to 30% of cytoplasmic pgRNA,10 although only 1% to 10% of secreted HBV particles contain spliced dDNA, reflecting the inefficiency of the HBV polymerase activated in trans.6
Removal of the 1.3-kb intron in the major spliced transcript and the resultant frameshift in the Pol reading frame creates a novel open reading frame in the HBV genome, encoding a protein known as the hepatitis B splice protein (HBSP).11 The direct role of HBSP in viral replication and pathogenesis remains unclear, although overexpression of HBSP in vitro led to an increase in caspase3-dependant apoptosis, suggesting a role for HBSP in fibrosis and liver inflammation.11,12 Individuals with CH-B were also more likely to produce anti-HBSP antibodies.13 Anti-HBSP antibodies were associated with higher HBV DNA levels, greater tumor necrosis factor production, and a fourfold increase in the relative risk factor for severe fibrosis.13 However, a recent study showed that the ratio of spliced pgRNA (encoding HBSP) relative to full-length pgRNA was inversely correlated with histology activity, suggesting that HBSP may in fact be a toleragen protecting HBV from immune-mediated clearance.14 Rosmorduc and colleagues6 used polymerase chain reaction (PCR) and Southern blotting to show that dDNA reverse-transcribed from spliced RNA was detected more often in persons with CH-B than in those with acute HBV. Furthermore, persons with higher HBV DNA levels, as determined by semiquantitative PCR, were more likely to have circulating dDNA.6 However, the role of dDNA in disease progression is largely undefined.
We investigated the relationship between defective HBV DNA and disease status, by analyzing the association between dDNA and a number of HBV-associated risk factors. These included high HBV DNA levels, HIV/HBV coinfection,2 high alanine aminotransferase (ALT) levels, hepatitis B e antigen (HBeAg)-negative disease, HBV genotype, or the selection of drug-resistant mutations. In addition, we investigated the relationship of dDNA and disease severity, ranging from compensated liver disease with no evidence of cirrhosis to decompensated cirrhosis. We also investigated the effect of antiviral drug resistance mutations in the HBV polymerase on the relative abundance of dDNA. We hypothesized that mutations that reduce the activity of the HBV polymerase, such as mutations associated with antiviral drug resistance,15,16 would also reduce the relative abundance of defective DNA, because of the inefficiency of the dDNA reverse transcription in trans. To test this hypothesis, we analyzed the effect of drug resistance mutations on the abundance of dDNA in both patient serum and in vitro.
Patients and Methods
Detection of Defective DNA in Patient Serum
Patients
Serum was obtained from 286 individuals (536 samples) with chronic HBV infection (Table 1), including 194 persons with HBV monoinfection and 92 persons who were coinfected with HIV. The HBV-monoinfected individuals included 26 persons with decompensated cirrhosis, in addition to 26 patients who had received a liver transplant. Antiviral drug treatment histories were obtained for 77 HBV-monoinfected and 18 HIV/HBV-coinfected persons, respectively. Patients were recruited from multiple sites, including Australia, the United States, and Hong Kong.17 The HIV/HBV-coinfected individuals were recruited from Australia and the United States.4,18 Twenty-six patients were obtained from the “Tenofovir in HIV/HBV Coinfection study,” which was a randomized clinical trial in antiretroviral naïve HIV/HBV-coinfected patients in Thailand. Subjects were required to be HIV-1-positive with evidence of CH-B (HBsAg-positive for longer than 6 months) and naïve to antiviral therapy for both HIV and hepatitis B. All patients analyzed had HBV DNA levels of ≥ 1 × 105 copies/mL and were negative for infection with hepatitis C and delta viruses. Multiple samples for longitudinal analysis were obtained from 107 persons (83 monoinfected and 24 HIV/HBV-coinfected patients), including 40 patients with samples before and following the selection of lamivudine (LMV) resistant virus (LMV-R). Samples were also obtained before (108) and during (85) lamivudine therapy. Liver biopsies were performed for 171 HBV-monoinfected and 18 HIV/HBV-coinfected samples within 3 months of the respective serum sample and were independently scored by the respective pathologists. Childs Pugh scores were obtained for 73 samples. The use of human samples complied with all relevant federal guidelines and relevant institutional policies and ethics committees.
Table 1.
Composition of Groups
| Monoinfected | HIV/HBV-Coinfected | |
|---|---|---|
| Patients | 194 | 92 |
| Samples | 386 | 150 |
| ALT mean, IU/mL | 143 (57) | 217 (21) |
| ALT median | 80 | 71 |
| Viral load mean, copies/mL* | 3.7 × 108 (184) | 2.7 × 109 (37) |
| Viral load median* | 1.79 × 107 | 8.11 × 108 |
| Genotype | ||
| A | 26 | 76 |
| B | 46 | 2 |
| C | 89 | 29 |
| D | 94 | 27 |
| E | 0 | 2 |
| G | 0 | 8 |
| Age mean, years | 52.7 (182) | 47.9 (24) |
| Age median | 50 | 48 |
| Sex (M:F) | 162:30 | 88:4 |
| LMV-R, %† | 25.86% | 38.29% |
| HBeAg positive† | 43.47% | 91.80% |
Bold denotes significance and was determined by the Mann-Whitney U test for independent nonparametric samples. Numbers within parentheses highlight the number of samples used for each detail.
Bold denotes statistical significance determined by chi-square analysis with 2 degrees of freedom.
Viral Load Assays
HBV DNA levels were determined with either the HBV Digene Hybrid Capture II microplate assay (Roche, Branchburg, NJ) or the Versant HBV DNA 3.0 assay (Bayer HealthCare-Diagnostics, Tarrytown, NY), in accordance with the manufacturer's instructions. The limit of detection for the Versant HBV DNA 3.0 assay was 2000 copies/mL.
DNA Extraction, PCR, and Sequencing
DNA was extracted from 200 μL patient serum, using either a Magnapure (Roche Applied Science, Indianapolis, IN) or Qiagen Qiaamp Blood mini kit (Qiagen, Dusseldorf, Germany), according to the manufacturer's protocols. The extracted DNA was resuspended in 50 μL H2O, and stored at -20°C. The HBV polymerase gene was amplified by PCR and sequenced from 299 samples using previously described methods.18 To identify basal core promoter (BCP) and precore mutations, the basal core promoter and precore/Core genes were sequenced from 385 samples.19 To determine whether differences in the abundance of defective DNA were attributable to mutations in splice donor and acceptor sequences, genomic-length sequencing was also completed for 62 samples. 4,19
Quantitative PCR Amplification of Defective DNA
HBV DNA extracted from patient serum was quantified by quantitative polymerase chain reaction (Qpcr) using two primer sets specific for either genomic-length HBV or the spliced defective HBV DNA. 23F (5′CCG CGT CGC AGA AGA TCT3′) and 180R (5′ATG GGA ATA CAG GTG CAA TTT CC3′) primed DNA synthesis across the intron, while 228F (5′TCT AGA CTC GTG GTG GAC TTC TCT C3′) and 404R (5′CAT AGC AGC AGG ATG AAG AGG AA3′) primed DNA synthesis within the intron. Primers were designed with Applied Biosystem's Primer Express software after choosing regions of high conservation both between and within HBV genotypes. Twenty-microliter reactions, consisting of 2 μL sample, 100 pM of each primer, were analyzed using SYBR Green I detection chemistry, with an ABI-Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA). Samples were quantified with a standard curve constructed from plasmid containing either full-length or spliced HBV genome cloned from a genotype D virus.20 Quantification above 500 wild-type copies per well was accepted as valid. Optimization reactions showed no nonspecific amplification of wild-type plasmid with splice primers or vice versa when spiked with less than 109 plasmid copies per well. Intra-assay coefficient of variation was determined to be less than 30% (0.3). The relative abundance of dDNA (% dDNA) in the serum-derived DNA was determined by dividing the amount of dDNA by the (genomic length + dDNA) and multiplying by 100. The ratio of dDNA to total HBV DNA was a preferred measure of dDNA, rather than absolute values, because % dDNA uses total HBV DNA as a loading and purification internal control and (HBV DNA) is correlated with (dDNA) but not % dDNA.
Statistical Analysis
To determine whether data were normally distributed, quantitative values were initially tested using a Shapiro-Wilk test. All nonnormal distributions were analyzed with the Mann-Whitney U test for nonparametric populations. Bonferroni correction21 was employed to avoid chance capitalization and resulted in a P value below 0.0083 being significant after correction. Correlations between two continuous variables were tested using linear regression and determination of the Pearson's correlation coefficient. Qualitative comparisons were done using the chi-square test with an α value of 0.05. Cell culture experiments were compared using one-tailed Student t test with an α value of 0.05.
Analysis of HBV dDNA in Cell Culture
Site-Directed Mutagenesis
Point mutations were introduced by site-directed mutagenesis into a 1.3-times genome-length wild-type HBV genotype D clone, as previously described.20 The mutants examined in the genotype D clone included the famciclovir-resistant mutant rtL180M; the lamivudine-resistant mutants rtM204I, rtL180M+rtM204V, and rtV173L+rtL180M+rtM204V; the lamivudine and entecavir-resistant mutants rtI169T+rtV173L+rtL180M+rtM204V, rtI169T+rtV173L+rtL180M+rtM204V+rtM250V, and rtL180M+rtT184G+rtS202I+rtM204V; and the adefovir-resistant mutants rtA181T, rtN236T, and rtA181T+rtN236T. A mutation that inhibits polymerase activity (rtD205A) and a core knockout mutant (coreY6*) were included as controls for plasmid carry-over.
Cell Culture and Transfection
Huh-7 cells were grown in Dulbecco's Modified Eagle's Medium (Gibco BRL) supplemented with 10% (vol/vol) fetal bovine serum at 37°C and 5% CO2. Cells were seeded to semi-confluence in 12-well tissue culture dishes (Greiner, Frickenheim, Germany) and adhered overnight. Transient transfections were achieved using Fugene6 transfection reagent and 2 μg plasmid DNA at a ratio of 8:5 (Roche Diagnostics, Mannheim, Germany) according to the manufacturer's instructions. Media and cell ly-sates were harvested for analysis 5 days after transfection, and the ratio between full-length HBV genomic and dDNA was quantitated, with the amount of full-length genomic HBV being used as an internal control for transfection.
Cell Harvest and Extraction of HBV DNA/RNA
Intracellular HBV core-associated nucleic acids were harvested using the method described.22 HBV DNA or RNA was extracted with the Qiagen Blood DNA kit and viral RNA kits, respectively, as per the manufacturer's instructions. For analysis of extracellular DNA, aliquots of culture medium collected on day 5 posttransfection were clarified by centrifugation, before precipitation of virions at 4°C overnight with 26% polyethylene glycol 8000 -1.4 M NaCl-25 mM ethylenediaminetetraacetic acid. Precipitated virions were collected by centrifugation for 30 minutes at 10,000g and then resuspended in 200 μL 5.5 mM MgCl2-10 mM Tris-HCl (pH 7.5) before extraction of HBV DNA and processing as described.
Reverse Transcription
Core-associated RNA was reverse transcribed using the ABI High capacity RT kit, as per the manufacturer's instructions. Full-length pgRNA and spliced pgRNA were quantified using the PCR technique outlined on the resultant complementary DNA.
Results
Patient Clinical Parameters
Mann-Whitney nonparametric analysis showed that HBV DNA levels were significantly higher in HIV/HBV-coinfected individuals than in HBV monoinfected individuals (P < 0.05), and there was no significant difference in ALT levels between groups (Table 1). Most patients in the study were male, with the greatest proportion of female patients in the HBV-monoinfected group. Genotypes A and D predominated in HIV/HBV-coinfected persons, whereas genotypes C and D were the major genotypes detected in HBV-monoinfected HIV/HBV-coinfected individuals. The prevalence of LMV-R was significantly higher in the coinfected patients (38% of samples) than in the monoinfected samples [26%, P = 0.006, chi-squared, degrees of freedom (df) = 2]. HIV/HBV-coinfected individuals were also more likely to be HBeAg-positive (92%) than HBV-monoinfected persons (44%, P < 0.0001, chi-squared, df = 2).
Analysis of % dDNA
HBV-Related Disease Parameters
Defective DNA was detected in more than 90% of patients, with the % dDNA ranging from 0% to 72% (Table 2). Most samples had less dDNA than full-length DNA, with a mean of 3% of detectable HBV DNA being dDNA. Linear regression analysis showed there was no association between % dDNA and HBV DNA levels (viral load), or ALT levels, with Pearson correlation coefficients of r2 = 0.006 and r2 = 0.001, respectively (data not shown). The % dDNA was not influenced by HBeAg status or HBV genotype (data not shown). Although we did identify an association between total genomic HBV copies/mL and dDNA copies/mL (r2 = 0.36, P < 0.001, df > 100, data not shown), % dDNA was our preferred measure of dDNA, rather than absolute values, because this enabled the total HBV DNA concentration to be used as an internal control and was independent of HBV viral load.
Table 2.
Differences in dDNA Between Groups
| Mean | Median | Samples | Max | Significance | |
|---|---|---|---|---|---|
| Monoinfected (1 sample/patient) | 3.03% | 0.66% | 194 | 52.34% | |
| HIV-1/HBV-coinfected (1 sample/patient) | 2.52% | 1.16% | 92 | 71.76% | p-MW = 0.3227 |
| Monoinfected | 3.20% | 0.63% | 386 | 52.34% | |
| HIV-1/HBV-coinfected | 2.87% | 0.79% | 150 | 71.76% | p-MW = 0.212 |
| Splice | Full-length | ||||
| Monoinfected (absolute values), copies/mL* | 4.48 × 105 | 7.96 × 107 | |||
| HIV-1/HBV-coinfected (absolute values), copies/mL* | 1.89 × 106 | 2.88 × 108 | |||
| p-MW | 0.00069 | 0.0027 | |||
| Mean | Median | Samples | Max | Significance | |
| HBeAg-positive | 2.37% | 0.60% | 135 | 24.84% | |
| HBeAg-negative | 2.35% | 0.40% | 121 | 49.87% | p-MW = 0.39 |
| Basal core promoter (BCP) mutant | 2.56% | 0.59% | 100 | 49.87% | |
| Basal core promoter (BCP) wt | 2.10% | 0.50% | 285 | 28.80% | p-MW = 0.99 |
| Precore (PC) mutant | 1.74% | 0.53% | 76 | 49.87% | |
| Precore (PC) wt | 2.24% | 0.50% | 309 | 24.84% | p-MW = 0.79 |
| Genotype | |||||
| A | 1.96% | 0.40% | 102 | 49.87% | |
| B | 1.61% | 0.66% | 48 | 11.94% | |
| C | 3.90% | 0.39% | 123 | 71.76% | |
| D | 2.09% | 0.68% | 121 | 43.10% | |
| E | 3.65% | 3.65% | 2 | 4.16% | |
| G | 1.80% | 1.39% | 8 | 4.34% | |
| Antiviral drug-naive | 4.37% | 0.61% | 108 | 71.76% | |
| Antiviral drug-treated | 4.44% | 1.37% | 147 | 52.34% | p-MW = 0.14+ |
| LMV/FTC-treated (monoinfected) | 6.32% | 3.23% | 77 | 52.34% | p-MW = 0.033+ |
| LMV/FTC-treated (HIV/HBV-coinfected) | 0.43% | 0.35% | 8 | 1.02% | p-MW = 0.19+ |
| LMV/FTC-treated (combined) | 5.77% | 2.88% | 85 | 52.34% | p-MW = 0.093+ |
| Tenofovir-treated (HIV/HBV-coinfected) | 2.55% | 0.34% | 10 | 7.47% | p-MW = 0.59+ |
| Decompensated liver disease, CP > A | 2.63% | 0.49% | 26 | 28.80% | |
| Compensated liver disease, CP = A | 2.58% | 0.44% | 47 | 49.87% | p-MW = 0.58 |
| Advanced fibrosis (Metavir F score 3 and 4) | 2.89% | 0.85% | 69 | 29.71% | |
| Limited or no fibrosis (Metavir F score 0, 1, and 2) | 2.21% | 0.38% | 120 | 31.54% | p-MW = 0.18 |
| Single sample per patient | |||||
| Advanced fibrosis (Metavir F score 3 and 4) | 4.61% | 2.62% | 36 | 29.71% | |
| Limited or no fibrosis (Metavir F score 0, 1, and 2) | 2.89% | 0.64% | 79 | 31.54% | p-MW = 0.096 |
Bold denotes significance and was determined by the Mann-Whitney U test for independent nonparametric samples. Attempts to normalize with log transformation failed a Shapiro-Wilk test for an expected normal distribution.
Significance value compared to antiviral drug-naïve.
HBV-Monoinfected Versus HIV/HBV-Coinfected Persons
The % dDNA distributions varied significantly from a normal distribution when examined by a Shapiro-Wilk test (P < 0.0001) and remained nonnormal after log10 transformation or loge transformation (P < 0.0001) (data not shown). Consequently, a nonparametric distribution was assumed for all statistical analysis. Analysis of all samples showed that from HBV-monoinfected persons, the maximum % dDNA observed was 52.3%, and in HIV/HBV-coinfected persons, the maximum was 71.8%. There was no statistical difference in the % dDNA, between HBV-monoinfected and HIV/HBV-coinfected persons (Table 2).
Although % dDNA did not differ between HIV/HBV-coinfected and HBV-monoinfected individuals, the total abundance of defective DNA in circulation was significantly higher in HIV/HBV-coinfected persons (median of 1.9 × 106 copies/mL) when compared with HBV-monoinfected individuals (4.5 × 105 copies/mL, P = 0.0007). This reflects the higher levels of HBV DNA in circulation in HIV/HBV-coinfected persons, compared with HBV-monoinfected persons.
Liver Disease Status
Progressive liver disease was measured using either Metavir fibrosis scores (F) or Child-Pugh scores. No difference in % dDNA was observed between patients with advanced fibrosis (F3 and F4) and those with limited or no detectable fibrosis (F0, F1, and F2, P = 0.096) (Table 2). No difference in % dDNA was observed between patients with compensated cirrhosis (Child-Pugh = A) and those with decompensated cirrhosis (Child-Pugh = B or C, P = 0.58) (Table 2). No difference in % dDNA was observed between patients with cirrhosis and those without (P = 0.36). These results were based on one sample from each patient, to ensure concomitance with the relevant liver biopsy sample. However, no difference in % dDNA was observed between patients with cirrhosis (F3, F4) and those without (F0, F1, F2), or between neighboring F scores (0 versus 1; 1 versus 2; 2 versus 3; 3 versus 4) after the inclusion of multiple samples from individual patients (P = 0.18, Table 2).
Analysis of % dDNA in Patients Infected with LMV-R HBV
Cross-Sectional Analysis
Although no difference was observed between patients taking antiviral drugs and those who were drug-naive, a significant difference in % dDNA was observed between persons infected with wild-type HBV (WT-HBV) and persons infected with HBV harboring LMV-R-HBV (Table 3). LMV resistance was defined by the presence of either rtM204V or rtM204I pol mutations in the dominant HBV species detected by population-based PCR sequencing. In the HBV-monoinfected and the HIV/HBV-coinfected groups, samples with LMV-R-HBV had significantly lower % dDNA compared with persons infected with WT-HBV (P < 0.0001; Table 3). In addition, 70 of 79 samples with greater than 5% dDNA were obtained from patients infected with WT-HBV (P = 0.005, chi-squared, df = 1) (Fig. 1). No mutations were identified in the splice donor or acceptor sites for the 30 LMV-R samples for which full genome sequence was available. Ten of 124 sequences had small deletions (Δ39 nt: 2 samples; Δ6 nt: 8 samples) within the HBV intron; however, all of these patients still had detectable dDNA (data not shown).
Table 3.
Significant Differences Between Lamivudine-Sensitive (LMV-S) and Lamivudine-Resistant (LMV-R; rtM204V/I) Samples
| LMV-S |
LMV-R |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | Median | Samples | Max | Mean | Median | Samples | Max | Significance | |
| Monoinfected | 3.69% | 0.84% | 278 | 53.34% | 1.57% | 0.27% | 97 | 28.80% | 0.000085 |
| HIV-1/HBV-coinfected | 4.58% | 1.77% | 87 | 71.76% | 1.07% | 0.46% | 54 | 5.53% | 0.00084 |
| Total | 3.90% | 1.12% | 365 | 71.76% | 1.39% | 0.32% | 151 | 28.80% | 0.0000028 |
Fig. 1.

The relative abundance of defective DNA in persons infected with lamivudine-sensitive (LMV-S) and resistant (LMV-R) HBV. Samples were separated into discrete % dDNA ranges, and the frequency with which each range was represented in the population (y axis) was plotted against the midpoint of the range (x axis). The ranges used were 0%, >0%, and <1%, >1% and <%5, >5% and <10%, and >10%. Eighty-nine percent of samples with >5% dDNA were LMV-S, compared with 70% of samples being LMV-S overall.
Longitudinal Analysis
Longitudinal studies on the relative abundance of defective DNA from 32 HBV-monoinfected and 8 HIV/HBV-coinfected individuals showed that in most cases (38/40) the % dDNA decreased or remained static after selection of LMV-R-HBV (Table 4). Selection of LMV-R-HBV resulted in an increase in % dDNA in only 2 of 40 cases compared with 25 of 62 patients with sequential samples that remained WT (P < 0.0001, chi-squared, df = 3).
Table 4.
Longitudinal Analysis of Changes in % dDNA Between Two Samples from Individual Patients Who Developed LMVr Versus Patient Samples That Remain WT
| Chi-squared, df = 3, P < 0.0001 | % dDNA Increased Between Samples* | % dDNA Decreased Between Samples* | % dDNA Did Not Change Between Samples* |
|---|---|---|---|
| LMV-R developed | 2 | 23 | 15 |
| WT HBV maintained | 25 | 21 | 16 |
Number of individuals that satisfy the category range described. Samples were obtained within 2 years of one another.
Antiviral Therapy
No differences in % dDNA were observed for patients on antiviral therapy, when compared with patients known to be drug naïve (Table 2). Further stratification of the data into patients treated with LMV or Emtricitabine (FTC) also showed no significant difference compared with the drug-naïve samples (P = 0.093). This remained the case when LMV/FTC patients were examined as monoinfected or HIV/HBV-coinfected groups in isolation (P = 0.033, P = 0.19 respectively, (Note: α = 0.0083 for significance)). Finally, a small number of HIV/HBV-coinfected patients were treated with Tenofivir; again, no statistical significance was observed between Tenofovir-treated patients and drug-naïve patients, although numbers were very small (P = 0.59).
In Vitro Analysis of Defective HBV DNA and Spliced pgRNA
To determine whether LMV-R-associated mutations were directly responsible for the reduction in % dDNA, we analyzed the % dDNA and the % spliced pgRNA in Huh-7 cells transiently transfected with infectious plasmid clones of either wild-type or LMV-R mutant HBV genotype D. All fold differences were made relative to wild-type HBV. Consistent with our previous observations in patients, the concentration of genomic-length HBV DNA did not correlate with % dDNA (r2 = 0.26, P > 0.05, df = 11).
Genotype D transient transfections included 12 mutants, six of which encoded LMV-R (Fig. 2). Reduced intracellular and extracellular % dDNA was observed for the LMV-R mutants (rtM204I, rtL180M + M204V, and rtL180M + T184G + S202I + M204V), ranging from a 20% to 50% reduction. Interestingly, the % dDNA for LMV-R mutants (rtM204V) that also harbored the compensatory mutation rtV173L did not differ from wild type but showed a substantial increase over plasmids harboring the M204V/I mutations. LMV-R mutants without rtV173L were compared with all other viable mutants examined and were found to have significantly lower levels of intracellular and extracellular % dDNA (one-tailed Student t test, P = 0.0002 and P = 0.043 respectively). These findings were confirmed in a genotype A genetic background (data not shown), and the combined data strongly support the role of rtM204V/I in reducing the relative abundance of dDNA. The adefovir resistance (ADVr) mutant rtN236T and the double mutant rtA181T+rtN236T also had significantly lower % dDNA relative to wild-type isolated from the cell supernatant, although there was no significant difference in intracellular core-associated % dDNA.
Fig. 2.

The relative abundance (y axis) of defective DNA and spliced pgRNA in HBV nucleocapsids and extracellular virions, harvested 5 days after transient transfection with HBV infectious clones encoding polymerase mutants (x axis). Error bars represent 1 standard deviation, n = 3. *Statistically significant differences from 1.3D WT.
To confirm that HBV DNA detected was the result of reverse transcription and not plasmid carry-over, cells were also transfected with a Pol mutant harboring a YMAD (rtD205A) mutation. As expected, this mutation abolished Pol function, and no HBV DNA, or dDNA, was detected. Similarly, a core mutant with a stop mutation in the core gene (coreY6*) did not produce HBV particles, although pgRNA and spliced RNA were detected, from both mutants, following their transcription from the input plasmid DNA.
Although we observed significant variation in the levels of dDNA for the different Pol mutants, packaged RNA levels did not vary, with the exception of adefovir resistance (ADVr) mutant rtN236T, which showed a 40% reduction in percent splice RNA compared with wild-type.
Retrospective Analysis of Patient Samples Indicated That rtV173L Rescued dDNA Replication in the rtM204V/I Background
Our in vitro data indicated that the reduced % dDNA observed in the presence of rtM204V/I could be restored to near wild-type levels by the compensatory mutation rtV173L. This hypothesis was tested against the clinical patient data. Twelve LMV-R isolates with the rtV173L mutation were compared with the remaining LMV-R isolates, which lacked the rtV173L (Table 5). Although numbers were limited and statistical significance was not obtained for the continuous data, a significant difference was evident when the data were categorized into samples that had less than or greater than 2% dDNA. This arbitrary value was chosen as the discriminator because it represented 1 standard deviation above the mean for LMV-R-HBV. A higher proportion of patients infected with HBV encoding the rtM204V/I mutations alone had less than 2% dDNA than patients who were also infected with HBV encoding the compensatory rtV173L mutation (P = 0.014, chi-squared, df = 1). This is further illustrated in Fig. 3, which shows that although a small number of outliers with high levels of % dDNA were detected in patients infected with HBV encoding the rtM204V/I mutations alone (0.0557, 25th percentile; 0.314, median; 1.147, 75th percentile), the median % dDNA in those who harbored LMV-R-HBV with the compensatory rtV173L mutation was higher (0.156, 25th percentile; 0.578, median; 3.155, 75th percentile). Unfortunately longitudinal samples were not available from individual patients before and after acquisition of the rtV173L, although this will form the basis of future studies.
Table 5.
rtV173L Increased the Proportion of Samples with Greater Than 2% dDNA
| Chi-squared, df = 1, P = 0.0135 | <2% dDNA | >2% dDNA | n |
|---|---|---|---|
| LMV-R | 84% | 16% | 140 |
| LMV-R+rtV173L | 58% | 42% | 12 |
Fig. 3.
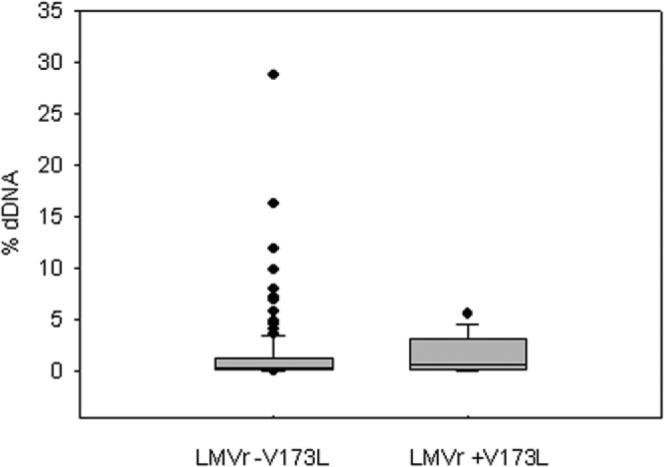
Box and whisker plot of % dDNA (y axis) distributions for all LMVr samples without the rtV173L mutation compared with a population of LMVr samples with the rtV173L compensatory mutation. The horizontal line inside the box represents the median values, with the box enclosing values between the 25% percentile value and the 75% percentile value. Error bars enclose 90% of values, and dot points represent outliers beyond the 90th percentile.
Only seven patients harbored the rtN236T mutation, and although statistical differences in the % dDNA were observed for this mutation in vitro, no difference was found in patients, possibly because of the limited numbers.
Discussion
Defective DNA was detected in sera from 90% of patients with CH-B, which is consistent with previous findings6; however, no association was identified between the relative abundance of dDNA and HBV risk factors, including ALT levels, HBeAg status, HBV DNA levels, fibrosis levels, compensated or decompensated liver cirrhosis, drug treatment, or HIV coinfection.
Although previous studies have suggested an association between HBSP expression and disease progression,6,13 the role of dDNA reverse transcribed from the HBSP RNA template had not been defined. Rosmorduc and colleagues6 showed that dDNA encoding the HBSP was detected more often in serum of persons with CH-B than in those with acute hepatitis B; however, they did not investigate the relationship between dDNA and disease severity/progression. We also identified dDNA in serum of a large proportion of subjects with CH-B; however, sensitive quantitative analysis of dDNA from 500-plus samples from nearly 300 patients showed there was no relationship between the relative abundance of dDNA and disease severity or progression. Soussan and colleagues13 showed that HBSP antibody positivity resulted in a fourfold increase in the relative risk factor for severe fibrosis; however, there is no evidence that the level of serum dDNA reflects HBSP expression. The HBSP is translated directly from spliced messenger RNA; however, it is possible that the level of intracellular dDNA reverse-transcribed from the spliced messenger RNA also alters HBSP expression. This is dependent on whether intracellular dDNA undergoes a typical “HBV replication cycle,” in which relaxed circular DNA is converted into a nuclear covalently closed circular DNA template enabling additional HBSP expression. Although defective dDNA lacks the recently characterized hM cis acting region, which may be required for synthesis of relaxed circular DNA and covalently closed circular DNA,23 Rosmorduc et al.6 identified both linear and double-stranded dDNA after replication of dDNA in vitro. It remains to be determined whether intracellular dDNA is a source of covalently closed circular DNA that could result in additional HBSP expression.
We also observed a significant reduction in HBV dDNA on the selection of HBV encoding lamivudine resistance mutations. Cross-sectional analysis showed that the LMV-R mutations, rtM204V and rtM204I, were associated with a reduction in % dDNA detected in patient serum and suggested that the compensatory mutation rtV173L had higher % dDNA compared with other LMV-R samples. This was supported by our in vitro analysis, which showed wild-type levels of intracellular and extracellular dDNA in cells transfected with the rtV173L. Our in vitro data also showed that the presence of the LMV-R mutations rtM204V/I reduced the % dDNA without altering the % splice pgRNA, suggesting a reduction in catalytic efficiency of reverse transcription. Our results suggest that the action of the rtM204V/I alters either the relative rate at which spliced pgRNA is reverse transcribed when compared with genomic-length DNA, or the likelihood that spliced pgRNA is reverse transcribed at all. Together, these results suggest that the relative abundance of dDNA is directly related to RT processivity, although this requires experimental verification. It has been shown that the rtM204V and rtM204I mutations increase the fidelity and reduces the processivity of the Pol protein,24 and this reduction in processivity also reduces the promiscuity of the Pol protein.15,16 The pgRNA that encodes Pol is almost exclusively reverse transcribed in cis.25,26 It is possible that the rtM204V and rtM204I mutations further bias this natural preference toward in cis RT processing, further reducing in trans RT processing. This would explain the reduction in % dDNA we observe in patients and in vitro for HBV harboring the rtM204V and rtM204I mutations. This hypothesis is strengthened by the observation that the compensatory mutation rtV173L, in addition to restoring Pol processivity, also restores the ratio at which dDNA is reverse transcribed.27
Our findings show that although dDNA was detected at varying levels in serum of most patients with CH-B, it was unsuitable as a marker for disease status and severity. Experiments are in progress to determine the “life cycle” of dDNA, and whether intracellular dDNA, spliced RNA, or HBSP expression are more suitable markers of disease progression in persons living with CH-B.
Acknowledgment
The authors thank Dr. Heath Kelly for assistance with statistical analysis and Dr. Eric Seaberg for providing clinical data from the Multicenter AIDS Cohort Study.
Supported by NIH grant no. AI060449. Some of the data presented in this article were collected by the Multicenter AIDS Cohort Study (United States), which is funded by the National Institute of Allergy and Infectious Diseases, the National Cancer Institute, and the National Heart, Lung, and Blood Institute. The majority of this analysis was performed at the Victorian Infectious Diseases Reference Laboratory, a Melbourne Health facility.
Abbreviations
- % dDNA
relative proportions of defective hepatitis B virus DNA
- ALT
alanine aminotransferase
- CH-B
chronic hepatitis B virus
- dDNA
defective hepatitis B virus DNA
- df
degrees of freedom
- F
Metavir fibrosis score
- HBeAg
hepatitis B e antigen
- HBSP
hepatitis B splice protein
- HBV
hepatitis B virus
- HIV
human immunodeficiency virus
- LMV
lamivudine
- LMV-R
lamivudine-resistant
- LMV-S
lamivudine-sensitive
- PCR
polymerase chain reaction
- pgRNA
pregenomic messenger RNA
- RT
reverse transcription
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1.Guidotti LG, Rochford R, Chung J, Shapiro M, Purcell R, Chisari FV. Viral clearance without destruction of infected cells during acute HBV infection. Science. 1999;284:825–829. doi: 10.1126/science.284.5415.825. [DOI] [PubMed] [Google Scholar]
- 2.Thio CL, Seaberg EC, Skolasky R, Jr, Phair J, Visscher B, Munoz A, et al. HIV-1, hepatitis B virus, and risk of liver-related mortality in the Multicenter Cohort Study (MACS) Lancet. 2002;360:1921–1926. doi: 10.1016/s0140-6736(02)11913-1. [DOI] [PubMed] [Google Scholar]
- 3.Bica I, McGovern B, Dhar R, Stone D, McGowan K, Scheib R, et al. Increasing mortality due to end-stage liver disease in patients with human immunodeficiency virus infection. Clin Infect Dis. 2001;32:492–497. doi: 10.1086/318501. [DOI] [PubMed] [Google Scholar]
- 4.Revill PA, Littlejohn M, Ayres A, Yuen L, Colledge D, Bartholomeusz A, et al. Identification of a novel hepatitis B virus precore/core deletion mutant in HIV/hepatitis B virus co-infected individuals. AIDS. 2007;21:1701–1710. doi: 10.1097/QAD.0b013e32826fb305. [DOI] [PubMed] [Google Scholar]
- 5.Gunther S, Sommer G, Iwanska A, Will H. Heterogeneity and common features of defective hepatitis B virus genomes derived from spliced pregenomic RNA. Virology. 1997;238:363–371. doi: 10.1006/viro.1997.8863. [DOI] [PubMed] [Google Scholar]
- 6.Rosmorduc O, Petit MA, Pol S, Capel F, Bortolotti F, Berthelot P, et al. In vivo and in vitro expression of defective hepatitis B virus particles generated by spliced hepatitis B virus RNA. HEPATOLOGY. 1995;22:10–19. [PubMed] [Google Scholar]
- 7.Terre S, Petit MA, Brechot C. Defective hepatitis B virus particles are generated by packaging and reverse transcription of spliced viral RNAs in vivo. J Virol. 1991;65:5539–5543. doi: 10.1128/jvi.65.10.5539-5543.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schranz P, Zentgraf H, Loncarevic IF, Niepmann M, Schroder CH. Defective replication units of hepatitis B virus. J Virol. 1990;64:1851–1854. doi: 10.1128/jvi.64.4.1851-1854.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schranz P, Zentgraf H, Schroder CH. Integrated defective replication units of hepatitis B virus. Virus Genes. 1990;4:367–374. doi: 10.1007/BF00570031. [DOI] [PubMed] [Google Scholar]
- 10.Wu HL, Chen PJ, Tu SJ, Lin MH, Lai MY, Chen DS. Characterization and genetic analysis of alternatively spliced transcripts of hepatitis B virus in infected human liver tissues and transfected HepG2 cells. J Virol. 1991;65:1680–1686. doi: 10.1128/jvi.65.4.1680-1686.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soussan P, Garreau F, Zylberberg H, Ferray C, Brechot C, Kremsdorf D. In vivo expression of a new hepatitis B virus protein encoded by a spliced RNA. J Clin Invest. 2000;105:55–60. doi: 10.1172/JCI8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu YW, Tan TL, Chan V, Chen WN. The HBSP gene is expressed during HBV replication, and its coded BH3-containing spliced viral protein induces apoptosis in HepG2 cells. Biochem Biophys Res Commun. 2006;351:64–70. doi: 10.1016/j.bbrc.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Soussan P, Tuveri R, Nalpas B, Garreau F, Zavala F, Masson A, et al. The expression of hepatitis B spliced protein (HBSP) encoded by a spliced hepatitis B virus RNA is associated with viral replication and liver fibrosis. J Hepatol. 2003;38:343–348. doi: 10.1016/s0168-8278(02)00422-1. [DOI] [PubMed] [Google Scholar]
- 14.Sheen IS, Tsou YK, Lin SM, Lin CJ, Lin CC, Hsu CW, et al. Nuclear HBcAg and histology activity index as independent predictors of the expression of singly spliced HBV-RNA. J Viral Hepatol. 2007;14:70–74. doi: 10.1111/j.1365-2893.2006.00781.x. [DOI] [PubMed] [Google Scholar]
- 15.Gaillard RK, Barnard J, Lopez V, Hodges P, Bourne E, Johnson L, et al. Kinetic analysis of wild-type and YMDD mutant hepatitis B virus polymerases and effects of deoxyribonucleotide concentrations on polymerase activity. Antimicrob Agents Chemother. 2002;46:1005–1013. doi: 10.1128/AAC.46.4.1005-1013.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong YB, Choi Y, Jung G. Increased DNA polymerase fidelity of the Lamivudine resistant variants of human hepatitis B virus DNA polymerase. J Biochem Mol Biol. 2004;37:167–176. doi: 10.5483/bmbrep.2004.37.2.167. [DOI] [PubMed] [Google Scholar]
- 17.Lau GK, Cooksley H, Ribeiro RM, Powers KA, Shudo E, Bowden S, et al. Impact of early viral kinetics on T-cell reactivity during antiviral therapy in chronic hepatitis B. Antivir Ther. 2007;12:705–718. [PubMed] [Google Scholar]
- 18.Matthews GV, Bartholomeusz A, Locarnini S, Ayres A, Sasaduesz J, Seaberg E, et al. Characteristics of drug resistant HBV in an international collaborative study of HIV-HBV-infected individuals on extended lamivudine therapy. AIDS. 2006;20:863–870. doi: 10.1097/01.aids.0000218550.85081.59. [DOI] [PubMed] [Google Scholar]
- 19.Ayres A, Bartholomeusz A, Lau G, Lam KC, Lee JY, Locarnini S. Lamivudine and Famciclovir resistant hepatitis B virus associated with fatal hepatic failure. J Clin Virol. 2003;27:111–116. doi: 10.1016/s1386-6532(02)00167-1. [DOI] [PubMed] [Google Scholar]
- 20.Chen RY, Edwards R, Shaw T, Colledge D, Delaney WEt, Isom H, et al. Effect of the G1896A precore mutation on drug sensitivity and replication yield of lamivudine-resistant HBV in vitro. HEPATOLOGY. 2003;37:27–35. doi: 10.1053/jhep.2003.50012. [DOI] [PubMed] [Google Scholar]
- 21.Shaffer JP. Multiple hypothesis testing. Annu Rev Psychol. 1995:561–584. [Google Scholar]
- 22.Chin R, Shaw T, Torresi J, Sozzi V, Trautwein C, Bock T, et al. In vitro susceptibilities of wild-type or drug-resistant hepatitis B virus to (-)-beta-D-2,6-diaminopurine dioxolane and 2′-fluoro-5-methyl-beta-L-arabino-furanosyluracil. Antimicrob Agents Chemother. 2001;45:2495–2501. doi: 10.1128/AAC.45.9.2495-2501.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewellyn EB, Loeb DD. Base pairing between cis-acting sequences contributes to template switching during plus-strand DNA synthesis in human hepatitis B virus. J Virol. 2007;81:6207–6215. doi: 10.1128/JVI.00210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Torresi J, Earnest-Silveira L, Civitico G, Walters TE, Lewin SR, Fyfe J, et al. Restoration of replication phenotype of lamivudine-resistant hepatitis B virus mutants by compensatory changes in the “fingers” subdomain of the viral polymerase selected as a consequence of mutations in the overlapping S gene. Virology. 2002;299:88–99. doi: 10.1006/viro.2002.1448. [DOI] [PubMed] [Google Scholar]
- 25.Bartenschlager R, Junker-Niepmann M, Schaller H. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J Virol. 1990;64:5324–5332. doi: 10.1128/jvi.64.11.5324-5332.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirsch RC, Lavine JE, Chang LJ, Varmus HE, Ganem D. Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as well as for reverse transcription. Nature. 1990;344:552–555. doi: 10.1038/344552a0. [DOI] [PubMed] [Google Scholar]
- 27.Delaney WEt, Yang H, Westland CE, Das K, Arnold E, Gibbs CS, et al. The hepatitis B virus polymerase mutation rtV173L is selected during lamivudine therapy and enhances viral replication in vitro. J Virol. 2003;77:11833–11841. doi: 10.1128/JVI.77.21.11833-11841.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]