

Abstract
Strains of Saccharomyces cerevisiae lacking Isw2, the catalytic subunit of the Isw2 chromatin remodeling complex, show the mating type-independent activation of the cell wall integrity (CWI) signaling pathway. Since the CWI pathway activation usually reflects cell wall defects, we searched for the cell wall-related genes changed in expression. The genes DSE1, CTS1, and CHS1 were upregulated as a result of the absence of Isw2, according to previously published gene expression profiles (I. Frydlova, M. Basler, P. Vasicova, I. Malcova, and J. Hasek, Curr. Genet. 52:87-95, 2007). Western blot analyses of double deletion mutants, however, did not indicate the contribution of the chitin metabolism-related genes CTS1 and CHS1 to the CWI pathway activation. Nevertheless, the deletion of the DSE1 gene encoding a daughter cell-specific protein with unknown function suppressed CWI pathway activation in isw2Δ cells. In addition, the deletion of DSE1 also abolished the budding-within-the-birth-scar phenotype of isw2Δ cells. The plasmid-driven overexpression proved that the deregulation of Dse1 synthesis was also responsible for CWI pathway activation and manifestation of the budding-within-the-birth-scar phenotype in wild-type cells. The overproduced Dse1-green fluorescent protein localized to both sides of the septum and persisted in unbudded cells. Although the exact cellular role of this daughter cell-specific protein has to be elucidated, our data point to the involvement of Dse1 in bud site selection in haploid cells.
The yeast cell wall is a crucial extracellular organelle that protects the cell from lysis during environmental stresses and morphogenesis. The cell wall of the budding yeast Saccharomyces cerevisiae is composed of glucans, mannoproteins, and a small amount of chitin (13, 14). To prevent lysis during cell expansion, the cell wall assembly and the cell cycle progression are coordinated by the mitogen-activated protein kinase (MAPK) Slt2 that is a component of the cell wall integrity (CWI) pathway. This pathway is thus periodically activated during budding at the time of polarized growth, but its activation also reflects cell wall damage (15, 35).
The critical point for cell integrity maintenance is the mother-daughter cell separation following cytokinesis when a controlled action of synthetic and degrading enzymes is necessary to prevent cell lysis. Chitinase Cts1 secreted by the daughter cell dissolves the primary chitin septum covered on both sides with a secondary septum and, in collaboration with other cell wall-degrading enzymes, allows the release of the daughter cell, leaving the mother cell with a prominent chitin bud scar and the daughter cell with a much less conspicuous birth scar (2). It has been reported that the birth scar contains little or no chitin (1, 23), although the exact composition of this structure is unknown. In haploid cells, the bud is formed next to the birth scar outwards, and the birth scar forms a zone restricted for budding (4, 29). Besides chitinase, other daughter cell-specific proteins were identified to participate in the cell separation. Dse2 and Scw11 function as glucanases that help to degrade the cell wall from the daughter side of the septum (3, 5). Another protein, Dse1, was suggested to participate in pathways regulating cell wall metabolism, since some diploid strains bearing the dse1 deletion show a cell separation defect and display an increased sensitivity to drugs affecting the cell wall (6). The transcription of DSE1, as well as other daughter cell-specific genes, is Ace2 regulated and peaks in the early G1 phase asymmetrically, in the daughter cells only, due to the localization of the transcription factor Ace2 in the daughter cell nucleus (5). Furthermore, Dse1 interacts in the high-throughput two-hybrid analysis with the bud formation proteins Boi1 and Boi2 (8). However, the precise function of Dse1 is unknown.
Isw2, a member of the ISWI (initiation switch) class of ATPases, was first shown to be a component of a two-subunit (together with Itc1) complex with nucleosome-stimulated ATPase and ATP-dependent nucleosome spacing activities (32). The Isw2 chromatin remodeling complex is involved in the transcriptional regulation of a broad spectrum of genes by influencing the accessibility of chromatin to the transcriptional machinery (32). It was found to participate, e.g., in the repression of the mitotic cyclin-encoding gene CLB2 (24) and MATa-specific genes in MATα cells (20, 31). The absence of either Isw2 or Itc1 in MATα cells results in an inappropriate a-factor production and an autocrine activation of the pheromone response pathway. As a result, the pheromone-inducible genes are upregulated and cells display altered morphology resembling budding “shmoos” (20, 28, 30, 31). However, the level of derepression of MATa-specific genes in isw2Δ MATα cells as a direct effect of the Isw2 absence is very low. Their derepression is potentiated by the activated pheromone response pathway. We proved it by microarray analyses comparing the expression profile of the isw2Δ MATα strain (versus the wild-type strain) with the expression profile of the isw2Δ ste4Δ MATα strain (versus the ste4Δ MATα strain), in which the pheromone response pathway is disrupted (9). The absence of the Isw2 complex also results in the phosphorylation of the Slt2 kinase and the activation of the CWI pathway which does not depend on the mating type (20).
Here, we report on a connection between the daughter cell-specific protein Dse1 and an aberrant budding within the birth scar. We found that the deletion of the DSE1 gene suppressed CWI pathway activation and abolished the aberrant budding of isw2Δ cells. Consistently, the plasmid-driven overexpression of the DSE1 gene in wild-type cells induced the budding-within-the-birth-scar phenotype as well as CWI pathway activation. Our data suggest that a tight temporal and spatial control of the daughter cell-specific Dse1 protein distribution is essential for maintenance of the birth scar as a zone forbidden for budding.
MATERIALS AND METHODS
Strains, plasmids, media, and general methods.
The S. cerevisiae strains used in this study are listed in Table 1 and were derived from either BY4741 and BY4742 (Euroscarf collection) or S288C (yeast green fluorescent protein [GFP] clone collection; Invitrogen). The isw2Δ::HIS3MX strain was prepared by replacing the ISW2 gene in BY4742 with the HIS3MX cassette similarly as described previously (31). Deletion strains expressing specific GFP fusions from the chromosomal locus and double deletion strains were generated by mating, subsequent sporulation on Fowel medium, and spore dissection using the Singer micromanipulator. Escherichia coli DH5α [F− recA1 supE44 endA1 hsdR17(rK− mK+) gyrA96 relA1 thi-1 Δ(lacIZYA-argF)U169deoR φ80dlacZΔM15] was used as the host in cloning procedures.
TABLE 1.
Yeast strains
| Strain | Genotype | Source |
|---|---|---|
| BY4741 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Euroscarf |
| BY4742 | MATahis3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Euroscarf |
| CRY3 | BY4741; MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 isw2Δ::kanMX | Euroscarf |
| CRY4 | BY4742; MATα his3Δ1 leu2Δ0 met15Δ0 lys2Δ0 ura3Δ0 isw2Δ::kanMX | Euroscarf |
| CRY146 | BY4742; MATα his3Δ1 leu2Δ0 met15Δ0 lys2Δ0 ura3Δ0 isw2Δ::kanMX ste4Δ::HIS3MX | 31 |
| CRY555 | S288C; MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 DSE1::GFP::HIS3MX | 11 |
| CRY557 | S288CxBY4742; MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 isw2Δ::kanMX DSE1::GFP::HIS3MX | This study |
| CRY665 | BY4742; MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 isw2Δ::HIS3MX | This study |
| CRY668 | BY4741; MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 dse1Δ::kanMX | Euroscarf |
| CRY870 | BY4741; MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 chs1Δ::kanMX | Euroscarf |
| CRY871 | BY4741; MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 cts1Δ::kanMX | Euroscarf |
| CRY671 | BY4741 × BY4742; MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 dse1Δ::kanMX isw2Δ::HIS3MX | This study |
| CRY900 | BY4741 × BY4742; MATahis3Δ1 leu2Δ0 ura3Δ0 chs1Δ::kanMX isw2Δ::HIS3MX | This study |
| CRY902 | BY4741 × BY4742; MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 cts1Δ::kanMX isw2Δ::HIS3MX | This study |
| CRY826 | BY4741; MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 + pGAL-DSE1-Myc (URA+) | This study |
| CRY827 | BY4741; MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 + pYC2/CT (URA+) | This study |
| CRY979 | BY4741; MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 + pGAL-DSE1-GFP (URA+) | This study |
All the plasmids used in this study carried the galactose-inducible promoter GAL1. The empty vector pYC2/CT was purchased from Invitrogen, and pGAL-DSE1-Myc was constructed and kindly provided by David E. Stone (University of Illinois, Chicago, IL). The plasmid pGAL-DSE1-GFP was constructed as follows. The DSE1 coding region was amplified from the S288C chromosomal DNA using primers Dse1Fwd (5′CGGCCGAGCTCATGCAAGATACCAAATACTA3′) and Dse1Rev (5′GCCGACGTCGACGACAACAGTAGTATCAATAG3′) and Phusion DNA polymerase (Finnzymes, Finland). The purified PCR product was digested sequentially with SacI and SalI and ligated into the SacI/SalI-cut vector pYC2/CT-GFP that we constructed earlier (our unpublished data). The resulting construct was transferred into the BY4741 strain and checked for the production of the Dse1-GFP fusion protein by Western blotting and fluorescence microscopy.
Standard bacterial cultivation media and temperatures were used (21). Yeast cultures were grown at 30°C in either YPD (1% yeast extract, 2% peptone, 2% glucose), SC (0.17% YNB without amino acids and ammonium sulfate, 0.5% ammonium sulfate, 2% glucose, supplemented with a complete mixture of amino acids), or SC lacking uracil to maintain the selection for plasmids. The corresponding solid medium contained 2% agar. For the overexpression analyses, cells were pregrown overnight in SCRaff-Ura (SC with 2% raffinose as the carbon source and lacking uracil) and then shifted for 4 to 6 h to SCGal-Ura (SC with 1% raffinose and 2% galactose and lacking uracil).
Standard methods were used for all DNA manipulations (21), bacterial cells were transformed by electroporation according to the protocol of Dower et al. (7), and yeast transformation was carried out by the lithium acetate method (10).
Cell wall staining.
Cell wall staining was performed according to the protocols described in references 11 and 23. For double labeling of the cell wall structures, yeast cells were washed with potassium phosphate-citric acid (KCP) buffer (pH 5.9). To 20 μl of the cell suspension in KCP buffer (107 cells/ml), an aliquot of calcofluor white (American Cyanamid Co.) stock solution was added (final concentration, 0.5 μg/ml). Further, an aliquot of 5 μl of fluorescein isothiocyanate (FITC)-labeled wheat germ agglutinin (WGA-FITC) stock solution (Sigma) was added (final concentration, 100 μg/ml). The cells were then washed two times with KCP buffer and subjected to microscopic analysis.
Western blot analyses.
Lysates were prepared according to a modified protocol of Riezman et al. (19). Yeast cells (optical density at 600 nm, 5 to 6) were collected by centrifugation, resuspended in fresh medium with phosphatase and kinase inhibitors (100 mM NaF, 100 mM NaN3, 4 mM sodium orthovanadate, 100 mM β-glycerophosphate), and frozen in liquid nitrogen. Lysis was done by adding an equal volume of 3.7 M NaOH for 10 min on ice. The proteins were precipitated by incubation with trichloracetic acid for 10 min on ice and collected by centrifugation at full speed in a microcentrifuge. The protein pellets were neutralized by 1 M Tris base and dissolved in sodium dodecyl sulfate sample buffer. After being separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels, the proteins were transferred to nitrocellulose membranes. The detection of phosphorylated Slt2 was performed with anti-phospho-p44/42 MAPK (Thr202/Tyr204) antibody (Cell Signaling Technology). The blots were stripped and reprobed with specific anti-Mpk1 (y-244) antibody (Santa Cruz Biotechnology). The pixel densities of the bands were quantified using Adobe Photoshop image software, and the level of Slt2 phosphorylation was normalized for loading using the level of total Slt2p. The statistics represent the results from two independent experiments and at least two different loading amounts for each experiment. The expression of the Dse1-Myc and Dse1-GFP fusion proteins was followed using the anti-Myc tag (Cell Signaling Technology) and anti-GFP (Santa Cruz) antibodies, respectively.
Microscopy.
The cells were inspected after washing with SC medium or KCP buffer, mounting on coverslips, and coating with a slice of 1% agarose in SC medium. The distribution of GFP fusion proteins or fluorescent probes was analyzed with a 100× PlanApochromat objective (numerical aperture, 1.4) using the Olympus IX-71 inverted microscope equipped with a Hammamatsu Orca/ER digital camera and the Cell-R detection and analyzing system Olympus (GFP filter block U-MGFPHQ, excitation maximum 488, emission maximum 507; Calcofluor filter block U-MNUA2, excitation maximum 440, emission maximum 500 to 520). The images were processed and merged using Olympus Cell-R and Adobe CS2 software (Adobe).
RESULTS
Deletion of the DSE1 gene overrode CWI pathway activation in isw2Δ cells.
Among genes whose expression was changed at least twofold in the absence of Isw2, there were three cell wall-related genes, CTS1, CHS1, and DSE1, and all were upregulated (9). We hypothesized that detected changes in the expression of these genes might account for an activation of the CWI pathway reported for isw2Δ cells previously (20).
Here we tested by Western blotting whether the CWI pathway was also activated in the isw2Δ ste4Δ MATα strain, lacking indirect defects induced by an autocrine activation of the pheromone response pathway (31). As shown in Fig. 1A, Slt2 phosphorylation reflecting CWI pathway activation was increased not only in the isw2Δ cells of both mating types but also in the isw2Δ ste4Δ MATα double mutant. To test our notion that the excess of the CHS1, CTS1, and DSE1 gene products might account for CWI pathway activation in the isw2Δ mutants, we prepared double deletion mutants lacking the ISW2 gene and one of the three selected genes and analyzed them for the level of phospho-Slt2. As shown in Fig. 1B, the additional absence of CHS1 or CTS1 genes did not significantly decrease the Slt2 phosphorylation observed in the single isw2 deletant. Quantification of the immunoblots proved that the level of Slt2 phosphorylation in the isw2Δ chs1Δ deletion mutant was about 115% (±6%) of that of the single isw2Δ deletion mutant and about 99% (±15%) for the isw2Δ cts1Δ mutant in comparison with the isw2Δ mutant level. These data showed that the deregulation of the chitin metabolism-related genes CTS1 and CHS1 was not responsible for CWI pathway activation in isw2Δ cells. In the last tested isw2Δ dse1Δ double deletion mutant, the Slt2 kinase was much less phosphorylated (67.7 ± 4%) in comparison with the single isw2Δ mutant (Fig. 1B). This result indicated that the upregulation of the DSE1 gene contributed to the cell wall integrity-signaling phenotype of isw2Δ cells.
FIG. 1.
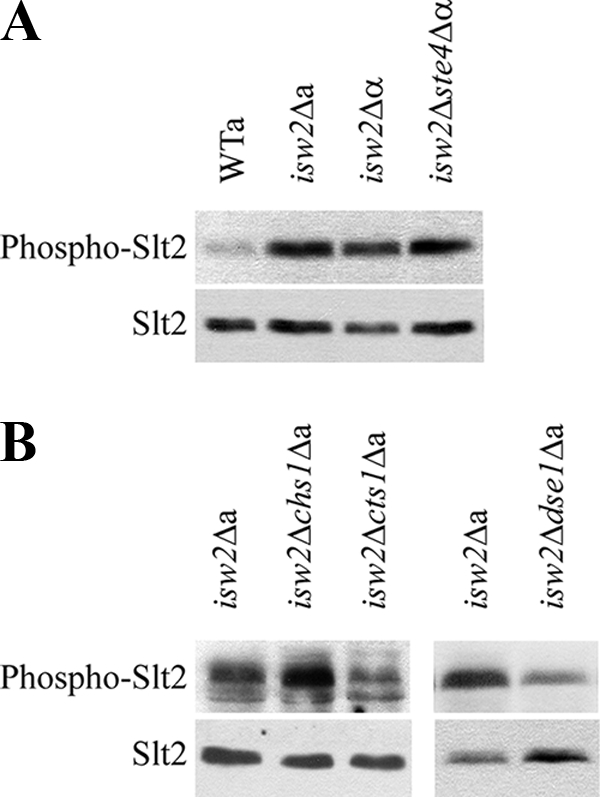
Slt2 phosphorylation indicated CWI pathway activation in cells lacking Isw2. Protein extracts were prepared from the wild-type strain and strains deleted for the ISW2 gene cultivated overnight in YPD medium. Phosphorylated Slt2 (Phospho-Slt2) was detected by the anti-phospho-p44/42 MAPK antibody (top panels). After stripping, we used the specific anti-Mpk1 antibody to quantify the amount of Slt2 protein (bottom panels). (A) The isw2Δ MATa (isw2Δa), isw2Δ MATα (isw2Δα), and isw2Δ ste4Δ MATα (isw2Δste4Δα) strains (strains CRY3, CRY4, and CRY146) displayed increased Slt2 phosphorylation in comparison with that of the wild-type strain (BY4741). (B) In contrast to deletions of the CHS1 and CTS1 genes (isw2Δchs1Δa and isw2Δcts1Δa; strains CRY900 and CRY902), deletion of the DSE1 gene (isw2Δdse1Δa; strain CRY671) lowered the level of Slt2 phosphorylation in the isw2Δ background.
isw2Δ cells displayed the budding-within-the-birth-scar phenotype dependent on DSE1.
To analyze the cell wall structure of isw2Δ strains by fluorescence microscopy, we double labeled the cells with calcofluor white and WGA-FITC. Both dyes are routinely used for visualization of the yeast cell wall chitin; however, calcofluor white obviously does not label the residual chitin of the birth scar (17). The isw2Δ MATα cells displayed an aberrant morphology and a complex distribution of the cell wall chitin involving its accumulation at the base of the shmoo-like projections (Fig. 2) as a consequence of an autocrine activation of the pheromone response pathway (31). The pattern of the cell wall labeling of the isw2Δ MATa cells was similar to that of the wild-type cells. No abnormal deposition of chitin was observed, and chitin was detected only in bud and birth scars. However, we came across an intriguing feature of birth scars in these cells. The birth scars that normally expand and fade with age (17) expanded but did not fade in the mutant cells. In the population of isw2Δ MATa cells, we found old cells with enlarged birth scars which were not observed in the wild-type cells. In addition, we discovered an interesting phenomenon that 49.5% of the isw2Δ MATa mother cells in the population formed buds inside the area bordered by the birth scar. No such cells were found in the wild-type population. Observations of the aberrant budding in the isw2Δ ste4Δ MATα cells proved that this phenotype was mating type independent (Fig. 2).
FIG. 2.

Wild-type cells formed buds next to the birth scar, whereas strains lacking Isw2p displayed bud scars within the birth scar. Cells were cultivated in liquid YPD medium overnight and double labeled with calcofluor white and WGA-FITC. The arrows indicate the birth scars in the wild-type (WTa; strain BY4741), isw2Δ MATa (isw2Δa; strain CRY3), and isw2Δ ste4Δ MATα (isw2Δste4Δα; strain CRY146) strains and chitin at the base of the shmoo-like projection in the isw2Δ MATα mutant (isw2Δα; strain CRY4). DIC, differential interference contrast. Bar, 5 μm.
The birth scar normally forms a zone restricted for budding (29). In this respect, we considered that aberrant budding within the birth scar might be the reason for CWI pathway activation. In such a case, deletion of the DSE1 gene in isw2Δ cells that suppressed Slt2 phosphorylation should also prevent the budding phenotype of this mutant. Therefore, we double labeled the isw2Δ dse1Δ cells with calcofluor white and WGA-FITC. Indeed, the budding pattern of these cells was similar to wild-type cells because they displayed neither extremely enlarged birth scars nor the budding-within-the-birth-scar phenotype (Fig. 3). These data indicated that deletion of the DSE1 gene in the isw2Δ genetic background overrode the budding-within-the-birth-scar phenotype and suppressed activation of the CWI pathway.
FIG. 3.

The deletion of the DSE1 gene in the isw2Δ mutant (isw2Δa) background abolished the budding-within-the-birth-scar phenotype. In contrast to the isw2Δ cells (strain CRY3), the isw2Δ dse1Δ mutant (isw2Δdse1Δa; strain CRY671) displayed a common axial budding pattern, i.e., the bud was formed just outside of the birth scar. Cells were cultivated in the liquid YPD medium overnight and double labeled with calcofluor white and WGA-FITC. The arrow indicates the birth scar. DIC, differential interference contrast. Bar, 5 μm.
The ectopic expression of DSE1 in wild-type cells induced the budding-within-the-birth-scar phenotype and CWI pathway activation.
Supposing that the budding-within-the-birth-scar phenotype and the activation of the CWI pathway in isw2Δ cells were caused by an increased expression of the daughter cell-specific gene DSE1, its overexpression from a plasmid should induce the same phenotype. We transformed the wild-type cells with the plasmid pGAL-DSE1-Myc. As confirmed by Western blotting, this strain produced Myc-tagged Dse1 at high levels under inducing conditions in the galactose medium (data not shown).
First, we checked the level of CWI pathway activation. We prepared lysates from cells carrying the plasmid pGAL-DSE1-Myc and control cells carrying the empty vector. Then, we analyzed the level of Slt2 phosphorylation by Western blotting. Indeed, our analysis proved that there was a significant increase in Slt2 phosphorylation in the cells with the induced expression of the DSE1 gene (Fig. 4A).
FIG. 4.

The overproduction of Dse1 in wild-type cells (strain BY4741) increased the level of Slt2 phosphorylation and affected the bud site selection. (A) The cells overproducing Dse1 displayed increased Slt2 phosphorylation in comparison with that of the control cells. Protein extracts were prepared from the wild-type (WT) strain carrying either the empty vector pYC2/CT (strain CRY827) or the pGAL-DSE1-Myc plasmid (overexpressing DSE1 [WT+pGAL-DSE1]; strain CRY826) cultivated overnight in the inducing SCGal-Ura medium. Phosphorylated Slt2 (Phospho-Slt2) was detected by the anti-phospho-p44/42 MAPK antibody (top panel). After stripping, we used the specific anti-Mpk1 antibody to quantify the amount of Slt2 protein (bottom panel). (B) Comparison of the budding patterns of wild-type cells with empty vector (WTa+pGAL; strain CRY827 carrying pYC2/CT) and wild-type cells overexpressing DSE1 (WTa+pGAL-DSE1; strain CRY826 carrying the pGAL-DSE1-Myc plasmid). The wild-type cells overexpressing DSE1 showed the enlarging bud or multiple bud scars within the birth scar. The cells were cultivated overnight in the inducing SCGal-Ura medium and double labeled with calcofluor white and WGA-FITC. The arrows indicate the birth scars. DIC, differential interference contrast. Bar, 5 μm.
Further, we examined microscopically the wild-type cells carrying either the empty vector or the plasmid pGAL-DSE1-Myc cultivated in the galactose medium. They were double labeled with calcofluor white and WGA-FITC (Fig. 4B). Whereas the wild-type cells containing empty vector (WTa+pGAL) formed buds adjacent to the birth scars, the cells with overproduced Dse1 (WTa+pGAL-DSE1) displayed enlarged birth scars. In addition, they showed also the budding-within-the-birth-scar phenotype. Moreover, after a serial passage in the galactose medium, Dse1 overproduction induced this phenotype in 97% of the mother cells in the population. We concluded that the overproduction of Dse1 was responsible for the activation of the CWI pathway as well as for the induction of the budding-within-the-birth-scar phenotype in the wild-type cells.
Dse1-GFP produced from the plasmid localized to both sides of the septum.
DSE1 is referred to as a cell cycle-regulated gene that is expressed in the early G1 phase asymmetrically in daughter cells only (5). To analyze the distribution of Dse1-GFP in the wild-type cells (12), the cells were cultivated in YPD overnight, then transferred to fresh SC medium, and further cultivated for 3 h under vigorous shaking. There was no Dse1-GFP-specific fluorescence localized in the unbudded mother cells (Fig. 5A) and the cells with enlarging buds (Fig. 5B and C). The specific signal of the Dse1-GFP fluorescence was observed in separating cells as a ring-like structure associated with the daughter side of the septum (Fig. 5D) and in freshly separated daughters (Fig. 5E).
FIG. 5.

Dse1-GFP was observed only in wild-type daughter cells. It formed a ring at the daughter side of the septum. Wild-type cells (strain CRY555) expressing GFP-tagged Dse1 from the chromosomal locus were cultivated in YPD medium overnight, then transferred to SC medium, and further cultivated under vigorous shaking for 3 h. Cells were stained with calcofluor white and analyzed for Dse1-GFP and calcofluor white fluorescence. DIC, differential interference contrast. Bar, 5 μm.
Since we found DSE1 upregulated in isw2Δ cells, we wanted to analyze Dse1 localization in these cells. We constructed the isw2Δ MATa strain producing the Dse1-GFP fusion from the chromosomal locus. In most of the isw2Δ MATa cells, Dse1-GFP was localized as a ring at the daughter side of the septum (Fig. 6A) similarly to the wild-type cells (as described above; Fig. 5). In addition, approximately 5% of the isw2Δ MATa cells with the localized Dse1-GFP signal displayed Dse1-GFP rings associated also with the mother side of the septum (Fig. 6B). This phenomenon was never found in the wild-type cells. Furthermore, Dse1-GFP frequently persisted at the separation sites (scars) of daughter (birth scar) as well as mother (bud scar) cells in isw2Δ MATa cells (Fig. 6C). Surprisingly, Dse1-GFP was observed also in unbudded mother isw2Δ MATa cells already showing one or several bud scars (Fig. 6D). Our results thus indicated that in isw2Δ MATa mutants, daughter cell-specific Dse1 protein was expressed and localized at the septum not only in daughters but also in mother cells.
FIG. 6.

In separating isw2Δ MATa cells (strain CRY557), Dse1-GFP was observed either as a ring at the daughter side of the septum (A) or forming rings on both sides of the septum (B). Dse1-GFP persisted at the cortical domain of daughter and mother cells after their separation (C). In addition, Dse1-GFP persisted at the cortical domain of a mother cell already showing a bud scar. (D) The isw2Δ MATa cells expressing GFP-tagged Dse1 from the chromosomal site were cultivated in YPD overnight, then transferred to SC medium, and further cultivated under vigorous shaking for 3 h. Cells were stained with calcofluor white and analyzed for Dse1-GFP and calcofluor white fluorescence. The arrows indicate the localization of Dse1. DIC, differential interference contrast. Bar, 5 μm.
Further, we wanted to analyze the distribution of Dse1 protein produced from the plasmid. Microscopic analyses of the cells carrying the plasmid pGAL-DSE1-Myc revealed that Dse1-Myc localized to both sides of the septum (data not shown). To analyze the distribution of overproduced Dse1 in living cells, we constructed the vector pGAL-DSE1-GFP. Whereas the separating wild-type cells showed Dse1-GFP localized only to the daughter side of the septum (see Fig. 5), the separating cells overproducing Dse1-GFP displayed the fusion protein as a double ring at the septum (Fig. 7A). Dse1-GFP persisted at the cell surface after separation (Fig. 7B) and even after the initiation of new budding (Fig. 7C to E). Our data thus showed that the deregulated expression of DSE1 resulted in the persistence of Dse1 in cells after separation which could not be observed in wild-type cells and that the signal for Dse1 localization was present in the mother cells.
FIG. 7.

Overproduced Dse1-GFP localized to both sides of the septum forming a double ring in dividing cells (A), the signal persisted at the same sites during cell separation (B), and it was detected there even after the initiation of new budding (C to E). Cells carrying the pGAL-DSE1-GFP plasmid (strain CRY979) were cultivated overnight in SCRaff-Ura and then shifted to SCGal-Ura for 4 h. DIC, differential interference contrast. Bar, 5 μm.
DISCUSSION
The local remodeling of the cell wall during the separation of S. cerevisiae cells ensured by the balance of cell wall synthetic and degrading activities depends, e.g., on the expression of several genes controlled by the transcription factor Ace2, a member of the CLB2 cluster (26). Ace2 is expressed in the G2 phase of the cell cycle, and then, it is specifically localized by the Cbk1-Mob2 complex to the daughter nucleus only, where it induces the expression of daughter cell-specific genes (5, 36). Their transcript levels peak in the early G1 phase (26). Here we bring evidence that the temporal and spatial deregulation of daughter cell-specific DSE1 gene expression results in CWI pathway activation and the budding-within-the-birth-scar phenotype.
We identified the budding-within-the-birth-scar phenotype of the DSE1 gene deregulation due to our detailed study of the isw2Δ cells showing an upregulation or a derepression of the DSE1 gene (9). Isw2 is a component of a two-subunit (together with Itc1) complex with nucleosome-stimulated ATPase and ATP-dependent nucleosome-spacing activities (32). Sherriff et al. (24) reported that Isw2 cooperates with the Fkh2 and Fkh1 transcription factors to repress the transcription of the B-type cyclin gene CLB2. Cdc28 bound to Clb2 phosphorylates Ace2, the main transcriptional activator of DSE1 gene expression, preventing its transport to the daughter cell nucleus (22), and thus a subsequent DSE1 transcription. If the loss of Isw2 leads to a derepression of CLB2, it should result in an inhibition of DSE1 expression rather than in its activation that was monitored by our DNA microarray analysis (9). A higher level of DSE1 mRNA in isw2Δ cells cannot be a result of its activation by Ace2. Rather, there could be another way for Isw2 to control DSE1 gene expression. Doolin et al. (6) reported that the disruption of ACE2 almost completely abolished the expression of DSE1, but disrupting SWI5 led to an increase in DSE1 expression of at least 25% over wild-type levels in the presence of an intact ACE2 gene. Such a remarkable inhibitory effect of this transcriptional activator suggests that Isw2 might have a direct role as a repressor in the regulation of the DSE1 gene or that it might be a mediator of the action of another yet-unknown repressor. In the DSE1 5′ untranslated region, there are several binding sites not only for Ace2 but also for Fkh1 and Fkh2 transcriptional regulators. It is tempting to speculate that Isw2 can bind to the DSE1 5′ untranslated region, either directly or throughout either Fkh protein (like in the CLB2 regulatory region) or Swi5, and thus influence chromatin structure in this region and, as a consequence, also DSE1 expression. In the absence of Isw2, this region would be remodeled and DSE1 transcription derepressed (like in the absence of Swi5), however, not completely induced like under the action of the activator Ace2.
The budding-within-the-birth scar is an unusual phenotype, since the area inside the birth scar is believed to be an exclusion zone for budding. In wild-type cells, the first bud scar can very rarely (∼1%) overlap the birth scar; however, the bud scar has never been observed completely within the birth scar (4, 29). Under a deregulation of DSE1, the area of the birth scar loses restriction, and there is a feasibility to form buds in this zone of inhibition. Data on the budding-within-the-birth-scar phenotype are very rare in the literature. In this respect, during the search for genes affecting bipolar budding, deletion mutants with mutations in two genes involved in splicing, IST3 and BUD13, were reported to display individual bud sites adjacent to, overlapping, or within the birth scar (16). However, the mechanism behind this defect is unknown. Recently, the budding-within-the-old-division-site phenotype was found in cells lacking Rga1p (29). Rga1p is a GTPase-activating protein for Cdc42 that specifically prevents Cdc42 activation at the previous division site, thus establishing an exclusion zone that blocks subsequent polarization within that site. In rga1Δ cells, Cdc42 remains in its GTP-bound state at the same place and new buds are nearly always formed at the old division site, thus displaying concentric bud scars at one pole of the cell. In contrast, cells with deregulated Dse1 (isw2Δ cells and cells overexpressing DSE1) displayed one or several bud scars within the birth scar region, and they had a normal localization of Rga1-GFP (our unpublished data). This suggests that the Dse1-related budding-within-the-birth-scar phenotype is not a result of the absence of Rga1. More likely, it might be a consequence of problems in cell polarity signaling, since the protein Dse1 that we identified as being responsible for this phenotype was found to interact in high-throughput two-hybrid assays (not verified) with the Boi1, Boi2, and Zds2 proteins implicated in polar growth (8). Purevdorj-Gage et al. (18) showed that S. cerevisiae cells under the influence of low-shear microgravity exhibit problems in polarity establishment, random budding, and increased aggregation accompanied with the increased expression of BUD5 and the decreased expression of daughter cell-specific genes DSE1, DSE2, and EGT2. A connection of Dse1 with the cell polarity machinery might also be behind the detected activation of the CWI pathway in isw2Δ cells, since the Dse1-interacting protein Zds2 was found to interact in high-throughput two-hybrid assays (8, 33) with Pkc1, a central component of the CWI pathway. Another indication of links between Dse1 and protein kinase C comes from the recent work of Sprowl et al. (27). They found that expression of bovine protein kinase C α in S. cerevisiae resulted in the growth inhibition of G2/M cells with defects in septum formation and decreased the expression of daughter cell-specific genes CTS1, DSE1, and DSE2. The ability of Dse1 to suppress an osmosensitive phenotype of the cdc37-34 mutant (34), most probably by adaptive changes of the cell wall, further suggests that Dse1 might have a signaling role in cell wall metabolism. Hypothetical WD40 repeats enhancing protein-protein interactions were identified by computer analysis in the Dse1 molecule and also support the idea that Dse1 might have a function in signal transduction.
Here, we demonstrate an interesting effect of the deregulation of the DSE1 gene leading to the budding-within-the-birth-scar phenotype and the activation of the CWI pathway. The elucidation of Dse1 links to potential interacting proteins and to other proteins localized to similar areas of daughter cells, e.g., Cwp1 protein that is specifically targeted to the birth scar area (25), is an important task to be undertaken in the near future. It may help to uncover the precise function of the Dse1 protein and its role in cell morphogenesis.
Acknowledgments
We are grateful to Michael Breitenbach and Miroslav Patek for critical reading of the manuscript and helpful comments. The technical assistance of J. Serbouskova, L. Novakova, and D. Janoskova is also gratefully acknowledged. The plasmid pGAL-DSE1-Myc was a kind gift from David E. Stone (University of Illinois, Chicago, IL).
This work was supported by grants GAAV A5020409 and LC545 (MSMT) and also by institutional research concept no. AV0Z50200510.
Footnotes
Published ahead of print on 27 February 2009.
REFERENCES
- 1.Beran, K., Z. Holan, and J. Baldrian. 1972. The chitin-glucan complex in Saccharomyces cerevisiae. I. IR and X-ray observations. Folia Microbiol. (Praha) 17322-330. [DOI] [PubMed] [Google Scholar]
- 2.Cabib, E. 2004. The septation apparatus, a chitin-requiring machine in budding yeast. Arch. Biochem. Biophys. 426201-207. [DOI] [PubMed] [Google Scholar]
- 3.Cappellaro, C., V. Mrsa, and W. Tanner. 1998. New potential cell wall glucanases of Saccharomyces cerevisiae and their involvement in mating. J. Bacteriol. 1805030-5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chant, J., and J. R. Pringle. 1995. Patterns of bud-site selection in the yeast Saccharomyces cerevisiae. J. Cell Biol. 129751-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colman-Lerner, A., T. E. Chin, and R. Brent. 2001. Yeast Cbk1 and Mob2 activate daughter-specific genetic programs to induce asymmetric cell fates. Cell 107739-750. [DOI] [PubMed] [Google Scholar]
- 6.Doolin, M. T., A. L. Johnson, L. H. Johnston, and G. Butler. 2001. Overlapping and distinct roles of the duplicated yeast transcription factors Ace2p and Swi5p. Mol. Microbiol. 40422-432. [DOI] [PubMed] [Google Scholar]
- 7.Dower, W. J., J. F. Miller, and C. W. Ragsdale. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 166127-6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drees, B. L., B. Sundin, E. Brazeau, J. P. Caviston, G. C. Chen, W. Guo, K. G. Kozminski, M. W. Lau, J. J. Moskow, A. Tong, L. R. Schenkman, A. McKenzie III, P. Brennwald, M. Longtine, E. Bi, C. Chan, P. Novick, C. Boone, J. R. Pringle, T. N. Davis, S. Fields, and D. G. Drubin. 2001. A protein interaction map for cell polarity development. J. Cell Biol. 154549-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frydlova, I., M. Basler, P. Vasicova, I. Malcova, and J. Hasek. 2007. Special type of pheromone-induced invasive growth in Saccharomyces cerevisiae. Curr. Genet. 5287-95. [DOI] [PubMed] [Google Scholar]
- 10.Gietz, D., A. St. Jean, R. A. Woods, and R. H. Schiestl. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 201425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hasek, J. 2006. Yeast fluorescence microscopy. Methods Mol. Biol. 31385-96. [DOI] [PubMed] [Google Scholar]
- 12.Huh, W. K., J. V. Falvo, L. C. Gerke, A. S. Carroll, R. W. Howson, J. S. Weissman, and E. K. O'Shea. 2003. Global analysis of protein localization in budding yeast. Nature 425686-691. [DOI] [PubMed] [Google Scholar]
- 13.Klis, F. M., A. Boorsma, and P. W. De Groot. 2006. Cell wall construction in Saccharomyces cerevisiae. Yeast 23185-202. [DOI] [PubMed] [Google Scholar]
- 14.Lesage, G., and H. Bussey. 2006. Cell wall assembly in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 70317-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levin, D. E. 2005. Cell wall integrity signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 69262-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ni, L., and M. Snyder. 2001. A genomic study of the bipolar bud site selection pattern in Saccharomyces cerevisiae. Mol. Biol. Cell 122147-2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Powell, C. D., D. E. Quain, and K. A. Smart. 2003. Chitin scar breaks in aged Saccharomyces cerevisiae. Microbiology 1493129-3137. [DOI] [PubMed] [Google Scholar]
- 18.Purevdorj-Gage, B., K. B. Sheehan, and L. E. Hyman. 2006. Effects of low-shear modeled microgravity on cell function, gene expression, and phenotype in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 724569-4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riezman, H., T. Hase, A. P. van Loon, L. A. Grivell, K. Suda, and G. Schatz. 1983. Import of proteins into mitochondria: a 70 kilodalton outer membrane protein with a large carboxy-terminal deletion is still transported to the outer membrane. EMBO J. 22161-2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruiz, C., V. Escribano, E. Morgado, M. Molina, and M. J. Mazon. 2003. Cell-type-dependent repression of yeast a-specific genes requires Itc1p, a subunit of the Isw2p-Itc1p chromatin remodelling complex. Microbiology 149341-351. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 22.Sbia, M., E. J. Parnell, Y. Yu, A. E. Olsen, K. L. Kretschmann, W. P. Voth, and D. J. Stillman. 2008. Regulation of the yeast Ace2 transcription factor during the cell cycle. J. Biol. Chem. 2511135-11145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaw, J. A., P. C. Mol, B. Bowers, S. J. Silverman, M. H. Valdivieso, A. Duran, and E. Cabib. 1991. The function of chitin synthases 2 and 3 in the Saccharomyces cerevisiae cell cycle. J. Cell Biol. 114111-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherriff, J. A., N. A. Kent, and J. Mellor. 2007. The Isw2 chromatin-remodeling ATPase cooperates with the Fkh2 transcription factor to repress transcription of the B-type cyclin gene CLB2. Mol. Cell. Biol. 272848-2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smits, G. J., L. R. Schenkman, S. Brul, J. R. Pringle, and F. M. Klis. 2006. Role of cell cycle-regulated expression in the localized incorporation of cell wall proteins in yeast. Mol. Biol. Cell 173267-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spellman, P. T., G. Sherlock, M. Q. Zhang, V. R. Iyer, K. Anders, M. B. Eisen, P. O. Brown, D. Botstein, and B. Futcher. 1998. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 93273-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sprowl, J. A., D. J. Villeneuve, B. Guo, A. J. Young, S. L. Hembruff, and A. M. Parissenti. 2007. Changes in expression of cell wall turnover genes accompany inhibition of chromosome segregation by bovine protein kinase C alpha expression in Saccharomyces cerevisiae. Cell Biol. Int. 311160-1172. [DOI] [PubMed] [Google Scholar]
- 28.Sugiyama, M., and J. Nikawa. 2001. The Saccharomyces cerevisiae Isw2p-Itc1p complex represses INO1 expression and maintains cell morphology. J. Bacteriol. 1834985-4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tong, Z., X. D. Gao, A. S. Howell, I. Bose, D. J. Lew, and E. Bi. 2007. Adjacent positioning of cellular structures enabled by a Cdc42 GTPase-activating protein-mediated zone of inhibition. J. Cell Biol. 1791375-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trachtulcova, P., I. Frydlova, I. Janatova, A. Dorosh, and J. Hasek. 2003. The W303 genetic background affects the isw2 delta mutant phenotype in Saccharomyces cerevisiae. Folia Microbiol. (Praha) 48745-753. [DOI] [PubMed] [Google Scholar]
- 31.Trachtulcova, P., I. Frydlova, I. Janatova, and J. Hasek. 2004. The absence of the Isw2p-Itc1p chromatin-remodelling complex induces mating type-specific and Flo11p-independent invasive growth of Saccharomyces cerevisiae. Yeast 21389-401. [DOI] [PubMed] [Google Scholar]
- 32.Tsukiyama, T., J. Palmer, C. C. Landel, J. Shiloach, and C. Wu. 1999. Characterization of the imitation switch subfamily of ATP-dependent chromatin-remodeling factors in Saccharomyces cerevisiae. Genes Dev. 13686-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uetz, P., and R. E. Hughes. 2000. Systematic and large-scale two-hybrid screens. Curr. Opin. Microbiol. 3303-308. [DOI] [PubMed] [Google Scholar]
- 34.Yang, X. X., P. Hawle, J. P. Bebelman, A. Meenhuis, M. Siderius, and S. M. van der Vies. 2007. Cdc37p is involved in osmoadaptation and controls high osmolarity-induced cross-talk via the MAP kinase Kss1p. FEMS Yeast Res. 7796-807. [DOI] [PubMed] [Google Scholar]
- 35.Zarzov, P., C. Mazzoni, and C. Mann. 1996. The SLT2(MPK1) MAP kinase is activated during periods of polarized cell growth in yeast. EMBO J. 1583-91. [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu, G., P. T. Spellman, T. Volpe, P. O. Brown, D. Botstein, T. N. Davis, and B. Futcher. 2000. Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature 40690-94. [DOI] [PubMed] [Google Scholar]