

Abstract
Chronic microglial activation is an important component of many neurological disorders, and imaging activated microglia in vivo will enable the detection and improved treatment of neuroinflammation. 1-(2-Chlorphenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline-carbox-amide (PK11195), a peripheral benzodiazepine receptor ligand, has been used to image neuroinflammation, but the extent to which PK11195 binding distinguishes activated microglia and reactive astrocytes is unclear. Moreover, PK11195 may lack sufficient sensitivity for detecting mild neuroinflammation. We hypothesized that N-(2,5-dimethoxybenzyl)-N-(4-fluoro-2-phenoxyphenyl) acetamide (DAA1106), a new ligand that binds specifically to peripheral benzodiazepine receptor, binds to activated microglia in human neurological diseases with higher affinity than does PK11195. We therefore compared the pharmacological binding properties of [3H](R)-PK11195 and [3H]DAA1106 in postmortem tissues from patients with cerebral infarcts, amyotrophic lateral sclerosis, Alzheimer disease, frontotemporal dementia, and multiple sclerosis (n = 10 each). In all diseases, [3H]DAA1106 showed a higher binding affinity as reflected by lower dissociation constant (KD) values than that of [3H](R)-PK11195. Moreover, specific binding of both ligands correlated with the presence of activated microglia identified by immunohistochemistry in situ. We conclude that 1) ligands that bind peripheral benzodiazepine receptor mainly label activated microglia in human neurological disorders and that 2) DAA1106 may possess binding characteristics superior to those of PK11195, which may be beneficial for in vivo positron emission tomography imaging.
Keywords: DAA1106, Microglia, Neurological disorders, Peripheral benzodiazepine receptor, PK11195, Positron emission tomography imaging
INTRODUCTION
Microglia undergo activation in response to neuronal injury in a variety of neurological disorders and function as resident brain macrophages in clearing debris from dying neurons. On the other hand, because they are sources of many neurotoxins, chronically activated microglia influence the viability and function of neurons and are thought to exacerbate neuronal injury (1). Imaging activated microglia in vivo using positron emission tomography (PET) would enable the detection of neuroinflammation and the monitoring of therapies that are targeted at modulating inflammatory processes in the CNS.
As pioneered by Dr Richard Banati from the Hammersmith Hospital (UK), [11C](R)-1-(2-chlorphenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline-carbox-amide (PK11195) has been extensively used to detect neuroinflammation using PET in human subjects (2). [11C](R)-PK11195 shows increased brain retention in the comparison of control human subjects to varying extents with patients with Alzheimer disease (AD) (3); amyotrophic lateral sclerosis (ALS) (4); multiple sclerosis (MS) (5–7); Parkinson disease (8, 9); frontotemporal dementia (FTD) (10); corticobasal degeneration (11); Huntington disease (12, 13); and more acute conditions such as stroke (14–17), Rasmussen encephalitis (18), herpes encephalitis (19), and hepatic encephalopathy (20).
Despite extensive use of [11C](R)-PK11195, the specific cell populations bound by PK11195 have been debated. Several studies (mainly in animal models) suggest that [3H](R)-PK11195 binds to activated microglia (5, 21–28). [3H](R)-PK11195 binding to astrocytes has, however, also been reported in various experimental conditions including cell culture systems (29, 30) and in animals injected with neurotoxins (31, 32). Studies in human subjects have generally focused on in vivo PET imaging, where it is not possible to determine cellular correlates of PK11195 binding. To elucidate the relative contributions of activated microglia versus reactive astrocytes to PK11195 binding in human neurological disorders, we used tissues obtained from patients diagnosed postmortem with cerebrovascular infarct, MS, FTD, ALS, and AD. In each of these conditions, we compared [3H](R)-PK11195 binding to the abundance of activated microglia and reactive astrocytosis. Further, we also used a new peripheral benzodiazepine receptor (PBR) ligand, N-(2,5-dimethoxybenzyl)-N-(4-fluoro-2-phenoxyphenyl) acetamide (DAA1106), which binds with higher affinity to PBR compared with [3H](R)-PK11195 (33), to determine if this higher binding affinity translates to human postmortem tissues with neuroinflammatory conditions. In tissues from patients with the neurological diseases assessed, both [3H](R)-PK11195 and [3H]DAA1106 showed increased specific binding compared with that of controls that corresponded mainly to binding of activated microglia. Moreover, [3H]DAA1106 showed a higher binding affinity compared with that of [3H](R)-PK11195, suggesting that DAA1106 may serve as a better ligand for the imaging of activated microglia in neuroinflammatory disorders in patients.
MATERIALS AND METHODS
Archival Human Brain Tissue
Brain tissue was obtained from the University of Pittsburgh and the University of California Los Angles Human Brain and Spinal Fluid Resource Center. Six controls and 10 cases each of patients diagnosed postmortem with cerebrovascular infarcts, MS, FTD, ALS, and AD were identified (Table 1). Tissues from the frontal cortex of patients with AD and FTD, primary motor cortex in patients with ALS, regions containing plaques from patients with MS, and the core of infarcts from patients with cerebrovascular infarction were dissected out and bisected. The anterior half of the tissue from each case was processed to obtain frozen sections for autoradiography and homogenized for filtration binding assays. The posterior halves of the samples were fixed and embedded in paraffin for sectioning for immunohistochemical analysis.
TABLE 1.
Demographics of human postmortem tissue samples
| Neurological Disease | Number of Cases | Male | Female | Age, mean (SD), Years | PMI, Hours | Disease Classifications (n) |
|---|---|---|---|---|---|---|
| Cerebrovascular infarct | 10 | 7 | 3 | 72 (10) | 19 (6) | NA |
| Multiple sclerosis | 10 | 5 | 5 | 67 (14) | 16 (6) | Relapsing remitting (2), secondary chronic progressive (5), primary progressive (3) |
| Frontotemporal dementia | 10 | 5 | 5 | 77 (10) | 10 (6) | Pick disease (10) |
| Amyotrophic lateral sclerosis | 10 | 7 | 3 | 64 (10) | 15 (4) | NA |
| Alzheimer disease | 10 | 4 | 6 | 82 (5) | 12 (6) | Braak stage VI (5), Braak stage V (5) |
| Control | 6 | 4 | 2 | 66 (8) | 11 (6) | NA |
PMI, postmortem interval; n, number of cases; NA, not applicable.
Immunohistochemistry
Immunostaining and laser confocal microscopic imaging were performed as previously described (26). Paraffin sections from the same regions from the same patients from which frozen tissue was obtained were stained with mouse monoclonal antibody against CD68 (lysosomal marker for activated macrophages that detects activated resident microglia and well as infiltrating macrophages, 1:10,000, clone KP1; DAKO, Carpinteria, CA) and rabbit polyclonal antibody against GFAP (1:500, DAKO). Sections were then incubated with Alexa Flour 488-conjugated goat anti-mouse IgG and Cy3-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Bar Harbor, ME; 1:200, room temperature, 1 hour). Immunostained sections were scanned and quantified on a laser confocal microscope equipped with an argon laser with 458, 477, 488, and 514 nm primary emission lines (LSM 150; Carl Zeiss GmbH, Heidelberg, Germany). Each section was scanned along the z axis to define the middle optical plane used in quantification (262,144 pixels/plane; 1 pixel = 0.25 mm2). Image analysis was performed on a Silicon Graphics computer (Windows NT 4.0 Operating System; Microsoft Corp, Redmond, WA) using the LSM software (version 3.0; Carl Zeiss GmbH). Scanning parameters such as laser power aperture, gain, and photomultiplier tube settings were kept constant for each wavelength.
An individual blinded to the experimental design imaged 10 areas (×40) encompassing 106,100 μm2. For each cell phenotype scanned, the contribution to signal intensity from the autofluorescence was minimized using a threshold that was kept constant. In each area, the average pixel fluorescence and the pixel counts for a given cell phenotype marker that exceeded the threshold were enumerated. The average pixel fluorescence was multiplied by the total number of pixels to measure the total fluorescence for that cell phenotype marker in that area. The total fluorescence values determined from the 10 scanned areas in 1 brain region were averaged to represent a measure of the abundance of the cell phenotype in that brain region.
Saturation Filtration Binding
Brain tissue was weighed and homogenized in ice-cold 50 mmol/L 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic (HEPES, 4°C, pH 7.4). Homogenates were washed 4 times by centrifugation at 40,000×g for 20 minutes at 4°C, and protein concentrations in each sample were estimated using the BCA protein assay kit (Pierce, Rockford, IL). Brain tissue samples (total protein concentration ranging from 150 to 200 μg) were incubated with either 0.5 to 84 nmol/L [3H](R)-PK11195 (sp. act., 89.9 Ci/mmol; custom ordered from NEN Life Sciences Products, Boston, MA) or 0.2 to 25 nmol/L [3H]DAA1106 (specific activity, 80 Ci/mmol; American Radiolabeled Chemical, St. Louis, MO) at 4°C for 2 hours in a final volume of 250 μL of HEPES (4°C, pH 7.4). This was defined as total binding. Nonspecific binding was determined by the inclusion of 10 μM of either PK11195 or DAA11106, respectively. The reaction was terminated by filtration through glass fiber filters (Brandel, Gaithersburg, MD) pre-soaked in 0.3% polyethylenimine by the addition of ice-cold HEPES in a vacuum cell harvester (Brandel). Filter-bound radioactivity was counted in a liquid scintillation spectrometer (Tricarb liquid scintillation counter; Perkin Elmer Life Sciences, Wellesley, MA) after the addition of 6 mL of liquid scintillation fluid (Perkin Elmer Life Sciences). Specific binding at each concentration of 3H-ligand was defined as the difference between total binding and nonspecific binding and ranged from 80% to 90% of total binding. All samples were run in duplicate. Bmax values in femtomoles per milligram protein measuring the maximal number of binding sites for each ligand were taken to reflect the total number of receptors, and KD values, the dissociation constant reported in nanomoles per liter and inversely proportional to the binding affinity of the ligand, were determined for each ligand using PRISM software (GraphPad, San Diego, CA).
Quantitative Film Autoradiography
Brain tissues were sectioned at 15 μm and mounted on Superfrost glass slides (Sigma, St. Louis, MO). Serial brain sections were incubated with 1 nmol/L of either [3H]DAA1106 or [3H](R)-PK11195 for 30 minutes at 37°C and washed for 5 minutes at room temperature in 50 mmol/L HEPES buffer (pH 7.4) to determine total binding. Non-specific binding was determined in adjacent brain sections in the presence of 1 μM DAA1106 or PK11195, respectively. Slides were washed 2 times for 5 minutes each in buffer followed by distilled water, dried, and apposed to Kodak Bio-Max MR films (Eastman Kodak, Rochester, NY) for 3 weeks at room temperature. Reference [3H]-microscales standards (Amersham, IL) were included with each film to enable quantitative analysis of autoradiograms. Densitometric analysis was performed on a Macintosh computer using the public domain National Institutes of Health Image program (developed at the US National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/) v1.62. To evaluate the cellular localization of both [3H](R)-PK11195 and [3H]DAA1106 in brain sections, we combined immunostaining and autoradiography. Unfixed frozen sections were first immunostained with both anti-GFAP and anti-CD68 and then processed for autoradiography and were imaged on the confocal microscope.
Statistical Analysis
Data were analyzed using the PRISM software (GraphPad). Student t-tests or one-way analysis of variance tests with posttest Bonferroni correction and 95% confidence intervals were used to analyze data. Data are represented as mean (SD). The number of patients in each disease condition was 10, with 6 controls in each experiment. Nonparametric correlations using 95% confidence intervals were performed to quantify the relationship between ligand binding and immunohistochemical markers. Results from correlational analyses are represented by r, the Spearman coefficient.
RESULTS
Assessment of Neuroinflammation in Postmortem Tissues Derived From Human Neurological Disorders
The extent of microglial activation and of reactive astrocytosis was assessed in postmortem brain tissues from patients with cerebrovascular infarcts, MS, FTD, ALS, and AD (n = 10 each condition) and compared with controls (n = 6). The demographics of the patients and the controls are described in Table 1. Both microglial activation (Figs. 1, 2A) and reactive astrocytosis (Figs. 1, 2B) were greater in all the disease samples compared with those of controls.
FIGURE 1.
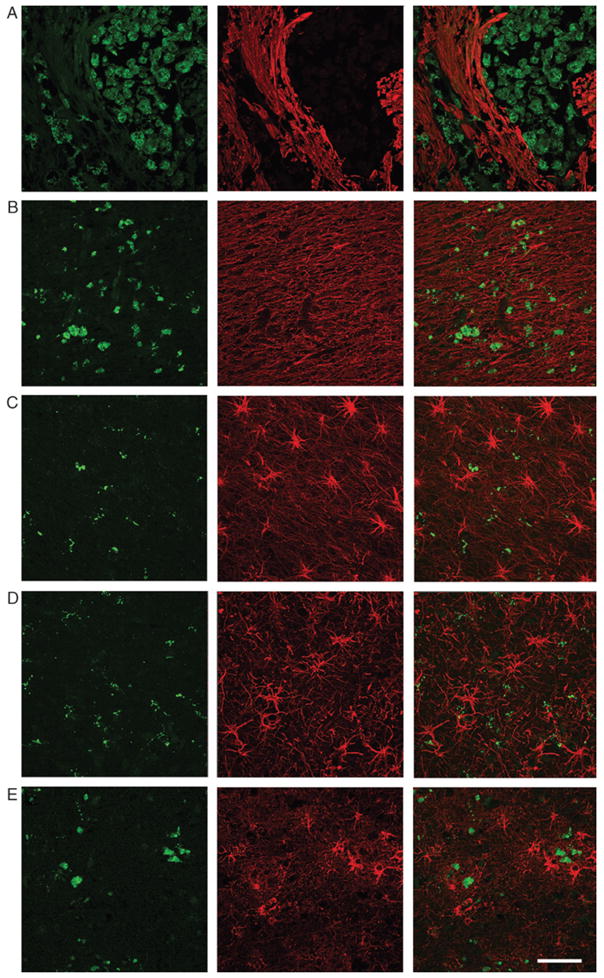
Microglial activation and reactive astrocytosis in various CNS disorders. Activated microglia (CD68, green) and reactive astrocytosis (GFAP, green) were assessed in various CNS diseases. Samples included the core of an acute cerebral infarct core (A), an active multiple sclerosis plaque (B), frontal cortex in a case of frontotemporal dementia (C), primary motor cortex in a case of amyotrophic lateral sclerosis (D), and frontal cortex in a case of Alzheimer disease (E). Individual (left and middle panels) and merged images (right panels) are shown. Scale bar = 50 μm.
FIGURE 2.

Assessment of microglial activation and reactive astrocytosis in human CNS disorders. Activated microglia and reactive astrocytosis were assessed as described in the Materials and Methods section in brain tissue samples from patients diagnosed postmortem with acute cerebral infarcts (Infarct, core of infarct), multiple sclerosis (MS, active plaques), frontotemporal dementia (FTD, frontal cortex), amyotrophic lateral sclerosis (ALS, primary motor cortex), and Alzheimer disease (AD, frontal cortex) and compared with those in controls (Con). Both microglial activation (A) and reactive astrocytosis (B) were higher in patients with the disease conditions assessed compared with those in controls. Data were analyzed using analysis of variance (***, p < 0.0001; **, p < 0.001; *, p < 0.05).
[3H](R)-PK11195 and [3H]DAA1106 Show Higher Bmax Values in Patients With Human Neurological Disorders Compared With Those of Controls
[3H]DAA1106 binding was compared with [3H](R)-PK11195 binding in all the disease conditions and controls using quantitative autoradiographic analysis. Both ligands showed significantly higher binding in patients with the disease conditions assessed compared with that in controls (Fig. 3). On comparing the 2 ligands, [3H]DAA1106 binding was significantly higher in all disease conditions except FTD (Fig. 3). Saturation filtration binding experiments using [3H](R)-PK11195 and [3H]DAA1106 were performed in each condition (curves and Scatchard plots, Fig. 4). The Bmax values (in femtomoles per milligram), reflecting the total number of binding sites, were significantly higher in patients with the disease conditions assessed compared with those in the controls for both [3H](R)-PK11195 (clear bars) and [3H]DAA1106 (black bars, Fig. 5A). Within each condition, Bmax values with [3H]DAA1106 and [3H](R)-PK11195 did not significantly differ (Fig. 5A).
FIGURE 3.

Quantitative film autoradiography with [3H](R)-1-(2-Chlorphenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline-carbox-amide (PK11195) and [3H]N-(2,5-dimethoxybenzyl)-N-(4-fluoro-2-phenoxyphenyl) acetamide (DAA1106). Quantitative film autoradiography with tritium microscales was performed on frozen brain tissue samples from cases of acute cerebral infarct, multiple sclerosis (MS) plaques, frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), and Alzheimer disease (AD) and compared with that in controls (Con), as described in the Materials and Methods section.(A) Representative images from ALS brain tissue samples incubated with [3H]DAA1106 and [3H](R)-PK11195 (left images). Sections were incubated with DAA1106 and PK11195 in parallel (right images) to assess nonspecific binding. Scale bar = 1 cm. (B) Quantitative film autoradiography was assessed in all the above conditions with [3H](R)-PK11195 (open bars) and [3H]DAA1106 (black bars). Data were analyzed using Student t-test (*, p < 0.05).
FIGURE 4.

Saturation binding curves and representative Scatchard plots with [3H](R)-1-(2-Chlorphenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline-carbox-amide (PK11195) and [3H]N-(2,5-dimethoxybenzyl)-N-(4-fluoro-2-phenoxyphenyl) acetamide (DAA1106). Saturation binding curves with [3H]DAA1106 (A, B) and [3H](R)-PK11195 (C, D) (insets show representative Scatchard plots with x intercepts representing Bmax values and slopes representing KD values) in brain tissues derived from patients diagnosed postmortem with amyotrophic lateral sclerosis (ALS, black squares), acute cerebral infarct core (Infarct, gray squares), frontotemporal dementia (FTD) cortex (clear squares), multiple sclerosis (MS) active plaque (black circles), Alzheimer disease (AD, gray circles), and controls (Con, clear circles).
FIGURE 5.

[3H](R)-1-(2-Chlorphenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline-carbox-amide (PK11195) and [3H]N-(2,5-dimethoxybenzyl)-N-(4-fluoro-2-phenoxyphenyl) acetamide (DAA1106) show higher Bmax values compared with those in controls (Con), with [3H]DAA1106 binding with higher affinity in human neurological disorders. Saturation filtration binding experiments using [3H](R)-PK11195 and [3H]DAA1106 were performed on acute cerebral infarct core, multiple sclerosis (MS) active plaque, frontotemporal dementia (FTD) frontal cortex, amyotrophic lateral sclerosis (ALS) primary motor cortex, and Alzheimer disease (AD) frontal cortex samples and compared with those performed on the Con samples. (A) The Bmax values (in femtomoles per milligram), which reflect the total number of binding sites, were significantly higher in patients with the disease conditions assessed compared with those in Con with both [3H](R)-PK11195 (clear bars) and [3H]DAA1106 (black bars). Bmax values with [3H]DAA1106 and [3H](R)-PK11195 did not differ significantly within the same disease condition. (B) The KD values (in nanomoles per liter) of [3H]DAA1106 were significantly lower than those of [3H]-(R)-PK11195 in each disease condition, indicating higher ligand binding affinities. The KD values did not differ from the Con with either [3H]DAA1106 or [3H]-(R)-PK11195. Data were analyzed using Student t-test (***, p < 0.0001; **, p < 0.001; *, p < 0.05).
[3H]DAA1106 Shows Lower KD Values Compared With [3H](R)-PK11195
Within each condition, KD values (in nanomoles per liter; which inversely correlate with ligand binding affinity) of [3H]DAA1106 were significantly lower than those of [3H]-(R)-PK11195 (Fig. 5B). KD values did not differ in patients from each disease condition when compared with those in controls with either [3H]DAA1106 or [3H]-(R)-PK11195 (Fig. 5B).
[3H]DAA1106 and [3H]-(R)-PK11195 Binding Corresponds to Activated Microglia in Human Neurological Disorders
To determine the relative contributions of astrocytes and activated microglia to [3H]DAA1106 binding, we combined immunostaining for astrocytes (GFAP) and activated microglia (CD68) with [3H]-(R)-PK11195 or [3H]DAA1106 autoradiography on frozen brain samples from each of the disease conditions. [3H]DAA1106 binding overlapped most prominently with CD68-labeled activated microglia compared with GFAP-labeled astrocytes in all the conditions (Fig. 6).
FIGURE 6.

[3H](R)-1-(2-Chlorphenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline-carbox-amide (PK11195) and [3H]N-(2,5-dimethoxybenzyl)-N-(4-fluoro-2-phenoxyphenyl) acetamide (DAA1106) autoradiography overlaps mainly with CD68-labeled cells. Combined autoradiography and immunostaining were performed on brain tissue from acute infarcts (INFARCT, A), multiple sclerosis (MS, B), frontotemporal dementia (FTD, C), amyotrophic lateral sclerosis (ALS, D), and Alzheimer disease (AD, E) samples. Autoradiograms with [3H](R)-PK11195 (PK) and [3H]DAA1106 (DAA) are shown in the column on the left; the white boxes indicate the regions from which high-resolution images were taken (scale bar = 1 cm). Immunostaining was performed for astrocytes (GFAP, red) and activated microglia (CD68, green). [3H](R)-PK11195 (PK)- and [3H]DAA1106 (DAA)-specific binding ([3H]-, black grains) of both ligands overlapped mainly with CD68-labeled activated microglia (merge). Scale bar = 100 μM.
[3H]-(R)-PK11195 and [3H]DAA1106 Binding Correlates With Activated Microglia in Human Neurological Disorders
We next tested whether [3H]-(R)-PK11195 and [3H]DAA1106 binding (Bmax values) in homogenized brain tissue correlated with the abundance of reactive astrogliosis versus activated microglia in each of the disease conditions. Astrocytes and activated microglia were labeled with GFAP and CD68, respectively, and quantified in brain tissues using laser confocal microscopy conducted by an individual blinded to the experimental design. These values were correlated with Bmax values in each of these cases in the same area. In each of the disorders, [3H]-(R)-PK11195 and [3H]DAA1106 correlated with the abundance of activated microglia (Table 2). When data from all disease conditions were pooled, Bmax values correlated with the abundance of CD68-labeled activated microglia assessed in the same area in these same cases with both [3H](R)-PK11195 (r = 0.8213, p < 0.0001, Fig. 7A) and [3H]DAA1106 (r = 0.8259, p < 0.0001, Fig. 8A). [3H](R)-PK11195 Bmax values did not correlate significantly with the extent of reactive astrocytosis with either [3H](R)-PK11195 (r = 0.2666, p = 0.0839, Fig. 7B) or [3H]DAA1106 (r = 0.1822, p = 0.2480, Fig. 8B).
TABLE 2.
[3H](R)-PK11195 and [3H]DAA1106 Binding Correlates Mainly With Activated Microglia in Human Neurological Disorders
| [3H]DAA1106 |
[3H](R)-PK11195 |
||||
|---|---|---|---|---|---|
| Neurological Disease | Statistical Parameter | Microglia (CD68) | Astrocytes (GFAP) | Microglia (CD68) | Astrocytes (GFAP) |
| Cerebrovascular infarct | r value | 0.8447 | −0.0146 | 0.7503 | −0.4524 |
| p value | 0.0168 | 0.9751 | 0.0502 | 0.3081 | |
| Multiple sclerosis | r value | 0.8870 | −0.2664 | 0.7130 | −0.1249 |
| p value | 0.0033 | 0.5237 | 0.0471 | 0.7683 | |
| Frontotemporal dementia | r value | 0.7338 | 0.3724 | 0.7735 | 0.3781 |
| p value | 0.0383 | 0.3636 | 0.0243 | 0.3558 | |
| Amyotrophic lateral sclerosis | r value | 0.7578 | 0.1690 | 0.7310 | 0.1593 |
| p value | 0.0294 | 0.6890 | 0.0394 | 0.7064 | |
| Alzheimer disease | r value | 0.8904 | 0.5998 | 0.7933 | 0.6510 |
| p value | 0.0002 | 0.0877 | 0.0062 | 0.0804 | |
[3H](R)-1-(2-Chlorphenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline-carbox-amide (PK11195) and [3H]N-(2,5-dimethoxybenzyl)-N-(4-fluoro-2-phenoxyphenyl) acetamide (DAA1106) Bmax values in brain tissues derived from patients diagnosed postmortem with acute cerebral infarcts (core of infarct), multiple sclerosis (active plaques), frontotemporal dementia (frontal cortex), amyotrophic lateral sclerosis (primary motor cortex), and Alzheimer disease (frontal cortex) were correlated with the abundance of CD68-labeled activated microglia or GFAP-labeled reactive astrocytes. Increases in both [3H](R)-PK11195 and [3H]DAA1106 binding correlated mainly with activated microglia.
FIGURE 7.

[3H](R)-1-(2-Chlorphenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline-carbox-amide (PK11195) binding in human CNS disorders correlates with CD68-labeled but not GFAP-labeled cells. (A) [3H](R)-PK11195 Bmax values (y axis, in femtomoles per milligram) derived from tissues from patients with the disease conditions assessed and controls correlated with the abundance of CD68-labeled activated microglia (x axis, CD68 units) assessed in the same area in these same patients (r = 0.8213, p < 0.0001). (B) [3H](R)-PK11195 Bmax values (y axis, in femtomoles per milligram) did not correlate significantly with the extent of reactive astrocytosis (r = 0.2666, p = 0.0839).
FIGURE 8.

[3H]N-(2,5-dimethoxybenzyl)-N-(4-fluoro-2-phenoxyphenyl) acetamide (DAA1106) binding in human CNS disorders correlates with CD68-labeled but not GFAP-labeled cells. (A) [3H]DAA1106 Bmax values (y axis, in femtomoles per milligram) derived from tissues from patients with the disease conditions assessed and controls correlated with the abundance of CD68-labeled activated microglia (x axis, CD68 units) assessed in the same area in these same patients (r = 0.8259, p < 0.0001). (B) [3H]DAA1106 Bmax values (y axis, in femtomoles per milligram) did not correlate significantly with the extent of reactive astrocytosis (r = 0.1822, p = 0.2480).
DISCUSSION
Microglia undergo activation in response to neuronal injury, but when they are chronically activated, they are also sources of neurotoxins that may enhance neuronal and synaptic damage (1). By imaging activated microglia in vivo, the development of neuroinflammation could be detected and its progression monitored, thereby enabling assessment of therapies that modulate CNS inflammation. [11C](R)-PK11195, a ligand that binds to PBR expressed in low levels in the normal brain (mainly on astrocytes and resting microglia), has been extensively used to image neuroinflammation in vivo (2, 34). Although it is hypothesized that increases in PK11195 binding are specific to activated microglia in neurological disorders, the relative contributions to PK11195 binding from reactive astrocytes and activated microglia have not been elucidated in human neurological disorders due to the inability to make such assessments in human subjects in vivo.
Here, we used postmortem tissues obtained from patients with various CNS diseases to address this question by comparing 10 patients of each disease condition to 6 controls. In all of the pathological conditions, increases in [3H](R)-PK11195 corresponded mainly to activated microglia (Table 2; Figs. 6, 7). [3H]DAA1106, a new PBR ligand, showed increased binding in patients with the disease conditions assessed compared with that in controls, and this binding corresponded mainly to activated microglia (Table 2; Figs. 6, 7). Further, the KD values with [3H]DAA1106 were significantly lower (~10-fold) compared with those of [3H](R)-PK11195, suggesting that DAA1106 bound to activated microglia with higher affinity (Fig. 5).
Our working definition of neuroinflammation included astrogliosis (GFAP-labeled cells) and activation of microglia (defined by an increase in CD68-labeled cells). CD68 is a lysosomal marker that labels CNS-resident activated microglia and macrophages that infiltrate into the brain from the periphery. Although immunohistochemical markers that can differentiate between these 2 cell populations in human brain tissues are not available, we have previously shown that PBR ligand binding does not differ in primary human activated microglia and macrophages (35). Overall, the present data suggest that both PBR ligands show increased binding corresponding mainly to activated microglia in human neurological disorders but that DAA1106 may be more useful for PET imaging.
Whereas [11C](R)-PK11195 has been extensively used to image activated microglia, the specific binding signal with [11C](R)-PK11195 is generally very low and difficult to quantify because of typical PET image noise levels (5, 36–38). Moreover, [11C](R)-PK11195 may not detect milder forms of neuroinflammation. For example, no differences were seen in [11C](R)-PK11195 brain retention in individuals with human immunodeficiency virus infection with or without neurological deficits (39, 40). Similarly, [11C](R)-PK11195 retention in the brain did not differ in normal subjects when compared with that in patients with minor cognitive impairment, a condition thought to progress to AD in approximately 50% of the subjects (41). These concerns highlight the need for developing newer ligands that bind with more specificity and sensitivity to activated microglia for PET imaging in neurological diseases. The higher binding affinity of [3H]DAA1106 (reflected by lower KD values) compared with that of [3H](R)-PK11195 suggests that DAA1106 may be a better ligand than PK11195 to image activated microglia in vivo using PET. It is possible, however, that filtration binding experiments in brain tissues may not entirely reflect in vivo properties of the ligand.
Various parameters such as the ability of a given ligand to cross the blood-brain barrier, in vivo binding kinetics to PBR, in vivo nonspecific binding, specific activity of the ligand, regional delivery, and the rate of metabolism of the ligand may determine the use of a ligand as a PET tracer. We are currently conducting in vivo experiments using [11C]DAA1106 to address these issues. Nevertheless, the assertion that a PET radioligand with potential clinical use binds to the intended cellular component (i.e. activated microglia) and that changes in measured Bmax values determined using a standardized assay are observed in a manner that is consistent with the known disease pathology is an important validation that is often overlooked or bypassed in the progression to human investigational studies. Further studies in human subjects with various neurological disorders are required to fully characterize the potential enhancements of DAA1106.
The importance of ascertaining the cellular source of ligands that bind PBR in neuropathologic conditions is important for interpreting PET imaging. Because PBRs are thought to be expressed by both astrocytes and microglia, the finding that [11C](R)-PK11195 is increased in a given neurological disorder is more useful when the cell type responsible for this increase is identified. In this context, PK11195 binding to reactive astrocytes and activated microglia has been the subject of continuous debate. Increased PK11195 binding to activated microglia is seen in animal models of ischemia (21, 22, 42), nerve axotomy (23, 28), MS (5, 24), brain trauma (25), quinolonate injection (27), and simian immunodeficiency virus encephalitis (26). In cell culture systems, PK11195 binding to astrocytes increases under different experimental conditions (29, 30). In addition, rodents treated with the neurotoxin trimethyltin and cuprizone show increased PK11195 binding to astrocytes after an initial increase in activated microglia in brain tissues (31, 32). In human neurological disorders, the data are largely from in vivo PET studies in which the cellular source of PK11195 binding cannot be determined. We addressed this by using postmortem tissues obtained from patients from various conditions, and although PK11195 may bind to astrocytes, our data suggest that increases in [3H](R)-PK11195 binding correlate mainly with increases in activated microglia. Reactive astrocytes certainly bind [3H](R)-PK11195, but increases in the abundance of GFAP-labeled astrocytes did not correlate with increases in [3H](R)-PK11195 binding. There is appreciable binding of both [3H](R)-PK11195 and [3H]DAA1106 in control tissue (Figs. 2, 3), consistent with previous reports in nonpathological human brain tissues (43). The tissue components bound are unclear and may represent the low level of receptor expression in the nonpathological brain. Although activated microglia have been reported in the aging brain (all controls used in this study were more than 64 years old) (44), we were unable to determine any definite relationship between [3H](R)-PK11195 and [3H]DAA1106 and microglial activation or reactive astrocytosis in control brain tissues. Future studies of aging, microglial activation, and PBR ligand binding may address this issue.
Overall, the present data support the hypothesis that ligands that bind PBR show increased binding that corresponds mainly to activated microglia in human neurological disorders. Our data support the use of ligands that bind PBR as tools in detecting activated microglia in vivo using PET.
Acknowledgments
This work was supported by the National Institutes of Health Grant Nos. RO1 MH64921 and K24 MH01717 (C.A.W.).
The authors thank James Kasenchak and Susan Slagel, MS, for technical help; Brian Lopresti, MS, for helpful comments; Dr Ronald L. Hamilton, MD, and Jonette Werley, BA, the University of Pittsburgh brain bank and the University of California Los Angles Human Brain and Spinal Fluid Resource Center, for providing human postmortem tissues.
References
- 1.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 2.Banati RB. Visualising microglial activation in vivo. Glia. 2002;40:206–17. doi: 10.1002/glia.10144. [DOI] [PubMed] [Google Scholar]
- 3.Cagnin A, Brooks DJ, Kennedy AM, et al. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–67. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 4.Turner MR, Cagnin A, Turkheimer FE, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15:601–9. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 5.Banati RB, Newcombe J, Gunn RN, et al. The peripheral benzodiazepine binding site in the brain in multiple sclerosis: Quantitative in vivo imaging of microglia as a measure of disease activity. Brain. 2000;123:2321–37. doi: 10.1093/brain/123.11.2321. [DOI] [PubMed] [Google Scholar]
- 6.Debruyne JC, Versijpt J, Van Laere KJ, et al. PET visualization of microglia in multiple sclerosis patients using [11C]PK11195. Eur J Neurol. 2003;10:257–64. doi: 10.1046/j.1468-1331.2003.00571.x. [DOI] [PubMed] [Google Scholar]
- 7.Versijpt J, Debruyne JC, Van Laere KJ, et al. Microglial imaging with positron emission tomography and atrophy measurements with magnetic resonance imaging in multiple sclerosis: A correlative study. Mult Scler. 2005;11:127–34. doi: 10.1191/1352458505ms1140oa. [DOI] [PubMed] [Google Scholar]
- 8.Ouchi Y, Yoshikawa E, Sekine Y, et al. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol. 2005;57:168–75. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- 9.Gerhard A, Pavese N, Hotton G, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21:404–12. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Cagnin A, Rossor M, Sampson EL, Mackinnon T, Banati RB. In vivo detection of microglial activation in frontotemporal dementia. Ann Neurol. 2004;56:894–97. doi: 10.1002/ana.20332. [DOI] [PubMed] [Google Scholar]
- 11.Gerhard A, Watts J, Trender-Gerhard I, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in corticobasal degeneration. Mov Disord. 2004;19:1221–26. doi: 10.1002/mds.20162. [DOI] [PubMed] [Google Scholar]
- 12.Pavese N, Gerhard A, Tai YF, et al. Microglial activation correlates with severity in Huntington disease: A clinical and PET study. Neurology. 2006;66:1638–43. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- 13.Tai YF, Pavese N, Gerhard A, et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130:1759–66. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- 14.Ramsay SC, Weiller C, Myers R, et al. Monitoring by PET of macrophage accumulation in brain after ischaemic stroke. Lancet. 1992;339:1054–55. doi: 10.1016/0140-6736(92)90576-o. [DOI] [PubMed] [Google Scholar]
- 15.Gerhard A, Neumaier B, Elitok E, et al. In vivo imaging of activated microglia using [11C]PK11195 and positron emission tomography in patients after ischemic stroke. Neuroreport. 2000;11:2957–60. doi: 10.1097/00001756-200009110-00025. [DOI] [PubMed] [Google Scholar]
- 16.Gerhard A, Schwarz J, Myers R, Wise R, Banati RB. Evolution of microglial activation in patients after ischemic stroke: A [11C](R)-PK11195 PET study. Neuroimage. 2005;24:591–95. doi: 10.1016/j.neuroimage.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 17.Price CJ, Wang D, Menon DK, et al. Intrinsic activated microglia map to the peri-infarct zone in the subacute phase of ischemic stroke. Stroke. 2006;37:1749–53. doi: 10.1161/01.STR.0000226980.95389.0b. [DOI] [PubMed] [Google Scholar]
- 18.Banati RB, Goerres GW, Myers R, et al. [11C](R)-PK11195 positron emission tomography imaging of activated microglia in vivo in Rasmussen’s encephalitis. Neurology. 1999;53:2199–203. doi: 10.1212/wnl.53.9.2199. [DOI] [PubMed] [Google Scholar]
- 19.Cagnin A, Myers R, Gunn RN, et al. In vivo visualization of activated glia by [11C] (R)-PK11195-PET following herpes encephalitis reveals projected neuronal damage beyond the primary focal lesion. Brain. 2001;124:2014–27. doi: 10.1093/brain/124.10.2014. [DOI] [PubMed] [Google Scholar]
- 20.Cagnin A, Taylor-Robinson SD, Forton DM, Banati RB. In vivo imaging of cerebral “peripheral benzodiazepine binding sites” in patients with hepatic encephalopathy. Gut. 2006;55:547–53. doi: 10.1136/gut.2005.075051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myers R, Manjil LG, Cullen BM, et al. Macrophage and astrocyte populations in relation to [3H]PK 11195 binding in rat cerebral cortex following a local ischaemic lesion. J Cereb Blood Flow Metab. 1991;11:314–22. doi: 10.1038/jcbfm.1991.64. [DOI] [PubMed] [Google Scholar]
- 22.Stephenson DT, Schober DA, Smalstig EB, et al. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J Neurosci. 1995;15:5263–74. doi: 10.1523/JNEUROSCI.15-07-05263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banati RB, Myers R, Kreutzberg GW. PK (‘peripheral benzodiazepine’)-binding sites in the CNS indicate early and discrete brain lesions: Microautoradiographic detection of [3H]PK11195 binding to activated microglia. J Neurocytol. 1997;26:77–82. doi: 10.1023/a:1018567510105. [DOI] [PubMed] [Google Scholar]
- 24.Vowinckel E, Reutens D, Becher B, et al. PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurosci Res. 1997;50:345–53. doi: 10.1002/(SICI)1097-4547(19971015)50:2<345::AID-JNR22>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 25.Raghavendra Rao VL, Dogan A, Bowen KK, Dempsey RJ. Traumatic brain injury leads to increased expression of peripheral-type benzodiazepine receptors, neuronal death, and activation of astrocytes and microglia in rat thalamus. Exp Neurol. 2000;161:102–14. doi: 10.1006/exnr.1999.7269. [DOI] [PubMed] [Google Scholar]
- 26.Venneti S, Lopresti BJ, Wang G, et al. PET imaging of brain macrophages using the peripheral benzodiazepine receptor in a macaque model of neuroAIDS. J Clin Invest. 2004;113:981–89. doi: 10.1172/JCI20227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryu JK, Choi HB, McLarnon JG. Peripheral benzodiazepine receptor ligand PK11195 reduces microglial activation and neuronal death in quinolinic acid-injected rat striatum. Neurobiol Dis. 2005;20:550–61. doi: 10.1016/j.nbd.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Pedersen MD, Minuzzi L, Wirenfeldt M, et al. Up-regulation of PK11195 binding in areas of axonal degeneration coincides with early microglial activation in mouse brain. Eur J Neurosci. 2006;24:991–1000. doi: 10.1111/j.1460-9568.2006.04975.x. [DOI] [PubMed] [Google Scholar]
- 29.Itzhak Y, Norenberg MD. Ammonia-induced upregulation of peripheral-type benzodiazepine receptors in cultured astrocytes labeled with [3H]PK 11195. Neurosci Lett. 1994;177:35–38. doi: 10.1016/0304-3940(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 30.Hazell AS, Desjardins P, Butterworth RF. Chronic exposure of rat primary astrocyte cultures to manganese results in increased binding sites for the ‘peripheral-type’ benzodiazepine receptor ligand 3H-PK 11195. Neurosci Lett. 1999;271:5–8. doi: 10.1016/s0304-3940(99)00489-9. [DOI] [PubMed] [Google Scholar]
- 31.Kuhlmann AC, Guilarte TR. Cellular and subcellular localization of peripheral benzodiazepine receptors after trimethyltin neurotoxicity. J Neurochem. 2000;74:1694–1704. doi: 10.1046/j.1471-4159.2000.0741694.x. [DOI] [PubMed] [Google Scholar]
- 32.Chen MK, Baidoo K, Verina T, Guilarte TR. Peripheral benzodiazepine receptor imaging in CNS demyelination: Functional implications of anatomical and cellular localization. Brain. 2004;127:1379–92. doi: 10.1093/brain/awh161. [DOI] [PubMed] [Google Scholar]
- 33.Chaki S, Funakoshi T, Yoshikawa R, et al. Binding characteristics of [3H]DAA1106, a novel and selective ligand for peripheral benzodiazepine receptors. Eur J Pharmacol. 1999;371:197–204. doi: 10.1016/s0014-2999(99)00118-1. [DOI] [PubMed] [Google Scholar]
- 34.Venneti S, Lopresti BJ, Wiley CA. The peripheral benzodiazepine receptor (Translocator protein 18 kDa) in microglia: From pathology to imaging. Prog Neurobiol. 2006;80:308–22. doi: 10.1016/j.pneurobio.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venneti S, Wang G, Wiley CA. The high affinity peripheral benzodiazepine receptor ligand DAA1106 binds to activated and infected brain macrophages in areas of synaptic degeneration: Implications for PET imaging of neuroinflammation in lentiviral encephalitis. Neurobiol Dis. 2008;29:232–41. doi: 10.1016/j.nbd.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petit-Taboue MC, Baron JC, Barre L, et al. Brain kinetics and specific binding of [11C]PK11195 to omega 3 sites in baboons: Positron emission tomography study. Eur J Pharmacol. 1991;200:347–51. doi: 10.1016/0014-2999(91)90594-g. [DOI] [PubMed] [Google Scholar]
- 37.Groom GN, Junck L, Foster NL, Frey KA, Kuhl DE. PET of peripheral benzodiazepine binding sites in the microgliosis of Alzheimer’s disease. J Nucl Med. 1995;36:2207–10. [PubMed] [Google Scholar]
- 38.Pappata S, Levasseur M, Gunn RN, et al. Thalamic microglial activation in ischemic stroke detected in vivo by PET and [11C]PK11195. Neurology. 2000;55:1052–54. doi: 10.1212/wnl.55.7.1052. [DOI] [PubMed] [Google Scholar]
- 39.Hammoud DA, Endres CJ, Chander AR, et al. Imaging glial cell activation with [11C]-R-PK11195 in patients with AIDS. J Neurovirol. 2005;11:346–55. doi: 10.1080/13550280500187351. [DOI] [PubMed] [Google Scholar]
- 40.Wiley CA, Lopresti BJ, Becker JT, et al. Positron emission tomography imaging of peripheral benzodiazepine receptor binding in human immunodeficiency virus-infected subjects with and without cognitive impairment. J Neurovirol. 2006;12:262–71. doi: 10.1080/13550280600873868. [DOI] [PubMed] [Google Scholar]
- 41.Schuitemaker A, Van Berckel BN, Boellaard R, et al. Assessment of microglial activation in mild cognitive impairment using [11C](R)-PK11195 and PET. Neuroimage. 2006;31:T159. [Google Scholar]
- 42.Conway EL, Gundlach AL, Craven JA. Temporal changes in glial fibrillary acidic protein messenger RNA and [3H]PK11195 binding in relation to imidazoline-I2-receptor and alpha 2-adrenoceptor binding in the hippocampus following transient global forebrain ischaemia in the rat. Neuroscience. 1998;82:805–17. doi: 10.1016/s0306-4522(97)00321-7. [DOI] [PubMed] [Google Scholar]
- 43.Rao VL, Butterworth RF. Characterization of binding sites for the omega3 receptor ligands [3H]PK11195 and [3H]RO5-4864 in human brain. Eur J Pharmacol. 1997;340:89–99. doi: 10.1016/s0014-2999(97)01395-2. [DOI] [PubMed] [Google Scholar]
- 44.Conde JR, Streit WJ. Microglia in the aging brain. J Neuropathol Exp Neurol. 2006;65:199–203. doi: 10.1097/01.jnen.0000202887.22082.63. [DOI] [PubMed] [Google Scholar]