

Abstract
High-throughput screening of a library of diverse molecules has identified the 1,4-naphthoquinone scaffold as a new class of Hsp90 inhibitors. The synthesis and evaluation of a rationally–designed series of analogues containing the naphthoquinone core scaffold has provided key structure–activity relationships for these compounds. The most active inhibitors exhibited potent in vitro activity with low micromolar IC50 values in anti-proliferation and Her2 degradation assays. In addition, 3g, 12, and 13a induced the degradation of oncogenic Hsp90 client proteins, a hallmark of Hsp90 inhibition. The identification of these naphthoquinones as Hsp90 inhibitors provides a new scaffold upon which improved Hsp90 inhibitors can be developed.
Keywords: Hsp90, Cancer, Naphthoquinone, MCF-7, Anti-proliferation, High-Throughput Screening
Introduction
The 90 kDa family of heat shock proteins (Hsp90) has become a validated target for the treatment of cancer because of their role as molecular chaperones responsible for the folding of nascent polypeptides into conformationally active three-dimensional structures.1 Client proteins of Hsp90 have been implicated in all six hallmarks of cancer and inhibition of Hsp90 can provide a multifaceted attack on cancer and malignant cell growth. Studies have revealed two nucleotide binding pockets in the Hsp90 protein, one at the N-terminus and the other in the C-terminal domain.2,3 Only the N-terminal ATP-binding pocket manifests ATPase activity and facilitates client protein maturation, while the C-terminus appears to exhibit allosteric properties.4 In addition to its potential as an anti-cancer target, regulation of Hsp90 also exhibits promising activity that may be utilized for the treatment of numerous neurodegenerative disorders, including Alzheimer’s and Parkinson’s Disease, in which protein aggregation is a common etiology.5,6
Natural product Hsp90 inhibitors currently being evaluated in anti-cancer models include geldanamycin (GDA) and radicicol (RDC) (Figure 1). Both of which bind to the N-terminal ATP binding site and exhibit potent in vitro activity.7 However, various side effects have slowed their clinical development. In addition, the natural products novobiocin and coumermycin A1 have been identified as C-terminal inhibitors of Hsp90 and structure–activity relationship (SAR) studies for these compounds have provided insight into key structural features necessary for optimal Hsp90 inhibitory activity.8-10 The promise that Hsp90 holds as a therapeutic target is illustrated by the number of research groups in both academia and industry that developed high-throughput screens in an effort to identify new inhibitory scaffolds that are amenable to rapid syntheses.11-15
Figure 1.
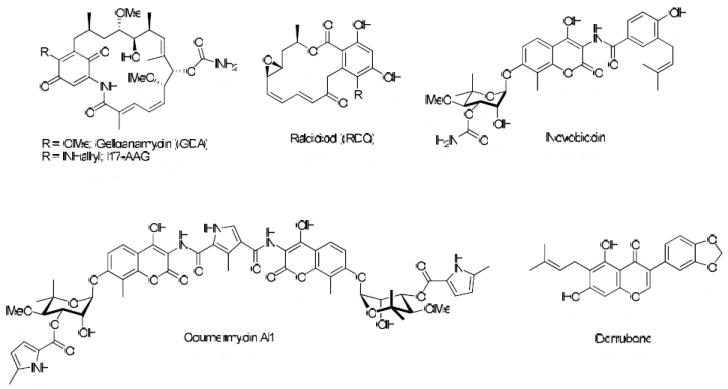
Natural Product inhibitors of Hsp90.
Utilizing a high-throughput screen recently developed with rabbit reticulocyte lysate, a library of small molecules was screened for their ability to inhibit Hsp90 via the renaturation of thermally denatured firefly luciferase, which is dependent upon Hsp90.16 In the presence of an Hsp90 inhibitor, the natural bioluminescence of luciferin is significantly reduced as a consequence of the inability to renature luciferase. Both GDA and novobiocin significantly reduce the renaturation of luciferase in this assay, indicating that it is a suitable assay for identification of both N- and C-terminal inhibitors of Hsp90. As an example of the utility of this assay, the natural product derrubone was recently identified and characterized as an Hsp90 inhibitor (Figure 1).17
During this screen, several compounds that contain a naphthoquinone core were identified as “hits” based on their ability to inhibit the renaturation of luciferase at 20 μM (HTS1-3, Table 1). The similar core structures present in these inhibitory scaffolds suggested these compounds bind to the same region of Hsp90 and provided preliminary SAR directly from the high throughput screen. Similar anti-proliferative and Her2 degradation activity in in vitro assays further supported optimization of the naphthoquinone scaffold. Interestingly, HTS1 demonstrated 4-fold greater anti-proliferative activity against MCF-7 cells (ER + human breast cancer cell line) compared to SKBr3 cells (ER -, Her2 over-expressing human breast cancer cell line), indicating this scaffold may provide a useful probe to study estrogen-dependent cancers. This manuscript details the synthesis, optimization, and in vitro activity of the naphthoquinone scaffold as a new class of Hsp90 inhibitors.
Table 1.
In Vitro results of HTS hits with the naphthoquinone scaffold.
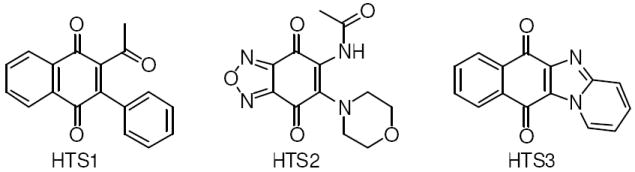 | ||||
|---|---|---|---|---|
| Compound | Luciferase Assaya | Her2 ELISA IC50c | Anti-Proliferation IC50c | |
| MCF-7 | SKBr3 | |||
| HTS1 | 0.25b | 3.3 ± 1 | 0.21 ± 0.02 | 0.82 ± 0.02 |
| HTS2 | 0.38 | 1.2 ± 0.2 | 19 ± 4 | 2.9 ± 0.5 |
| HTS3 | 0.02 | 1.2 ± 0.3 | 0.83 ± 0.13 | 0.87 ± 0.23 |
All values are reported in μM
Values represent the IC50 of a representative luciferase refolding assay.
Values are mean ± SEM of three separate experiments performed in triplicate.
Results and Discussion
Chemistry
Optimization of the naphthoquinone scaffold was based upon the structural features present in compounds HTS1–HTS3 that were combined to develop a library of analogues to further probe SAR for Hsp90 inhibition. Library synthesis began with commercially available 2-amino-3-chloro-1,4-naphthoquinone, 1. The original synthetic route for these compounds included Boc protection of the amine, conversion of the chloride to the corresponding iodide and subsequent Suzuki coupling with various aryl boronic acids. Numerous attempts to protect the amine were unsuccessful, highlighting its reduced nucleophilicity as a consequence of its participation in the electron deficient naphthoquinone ring system. This reduced activity combined with previous reports of successful Suzuki couplings of vinyl chlorides conjugated to electron-withdrawing groups18,19 altered the original route and led to direct coupling of the starting naphthoquinone to a series of arylboronic acids instead (Figure 2). Reflux of 1 in the presence of 4 mol% tetrakis(triphenylphosphine)palladium(0), an arylboronic acid, and sodium carbonate resulted in the phenylnaphthalene diones, 2a-i, in moderate yield (25-46%) (Scheme 1).
Figure 2.
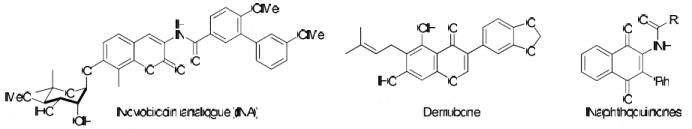
Structural similarity of novobiocin analogue (NA), derrubone, and naphthoquinone scaffolds.
Scheme 1.
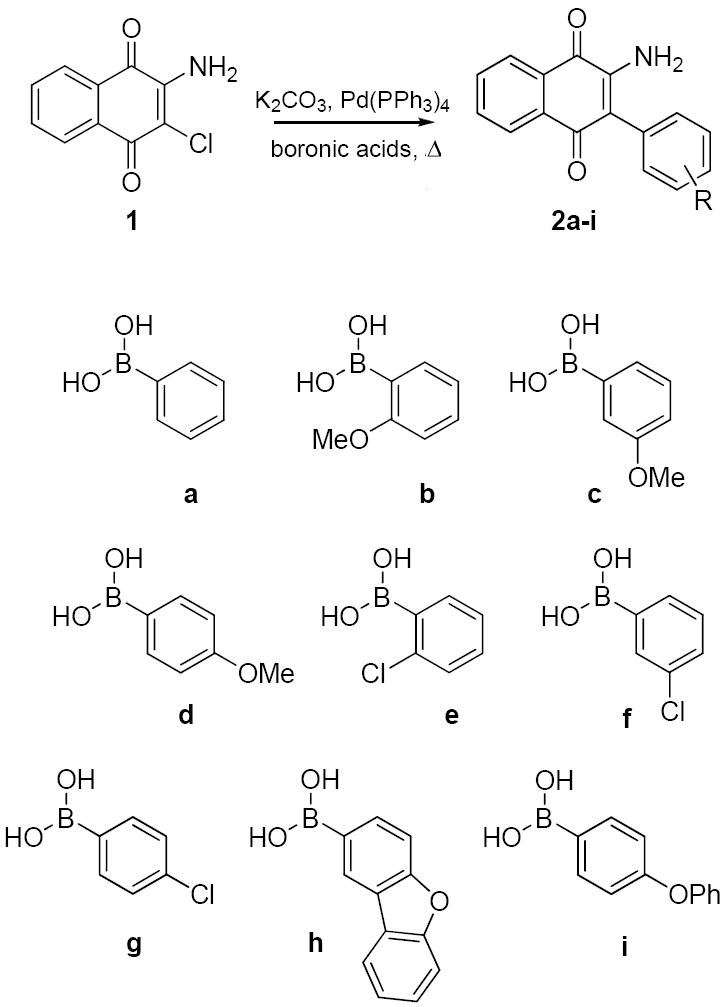
Suzuki coupling of naphthoquinone analogues.
To probe the size of the binding region for the amide side chain of HTS2, we prepared both acetamide and benzamide derivatives. The unreactive nature of the amine prevented use of standard peptide coupling conditions and thus, more reactive conditions were employed to provide the requisite acetamides (3a-i) and benzamides (4a-i) (Scheme 2). Reflux of the arylated amines, 2a-i, in acetyl chloride with catalytic sulfuric acid afforded the acetamides in quantitative yield.20 In contrast, treatment of the amine with sodium hydride and benzoyl chloride produced the benzamide compounds, 4a-i, in moderate yield (35-62%). In addition, 1 was acetylated (5) or benzylated (6) to provide compounds that contain chlorine in the 3-position.
Scheme 2.
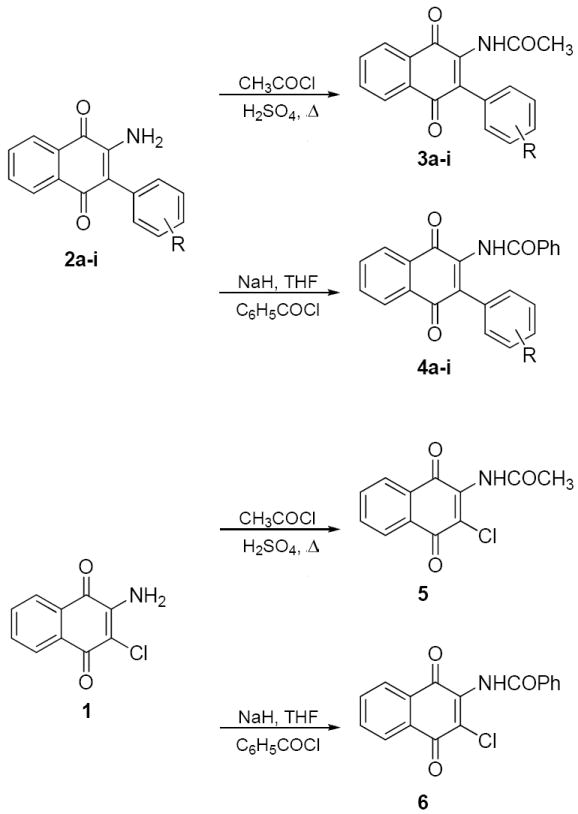
Preparation of acetamide and benzamide naphthoquinone analogues.
Structural similarity shared between the naphthoquinone core scaffold, the central coumarin ring of novobiocin, and the isoflavone of derrubone have led to our current hypothesis that these compounds may bind Hsp90 in a similar region. Previous structure–activity relationship studies for the benzamide moiety of novobiocin from our laboratory identified the biaryl functionality present in novobiocin analogue (NA) as exhibiting improved anti-proliferative activity over the novobiocin prenylated benzamide (Figure 2).10 Based on the improved inhibitory activity of the biaryl moiety when incorporated into novobiocin analogues and in an effort to identify synergistic structural modifications between these three classes of inhibitors, the biaryl acid 9 was prepared as previously described,10 converted to the corresponding acid chloride and coupled with the phenylnaphthalene dione 2d to give the tri-aryl naphthoquinones, 11 (Scheme 3). The para-methoxy dione was chosen as the core scaffold for benzamide SAR for two reasons: (1) its structural similarity to the benzamide of NA and (2) previously elucidated SAR for derrubone21 identified this functionality as important for Hsp90 inhibition. The starting dione 1 was also coupled directly to the biaryl acid to give analogue 12 for direct comparison of the necessity of the p-methoxy aryl ring in the presence of the biaryl functionality. In addition, 2d was coupled with a variety of benzoyl chlorides that contain substitutions at the ortho-, meta-, and para- positions to determine optimal substitutions at these positions of the benzamide ring (Scheme 4).
Scheme 3.
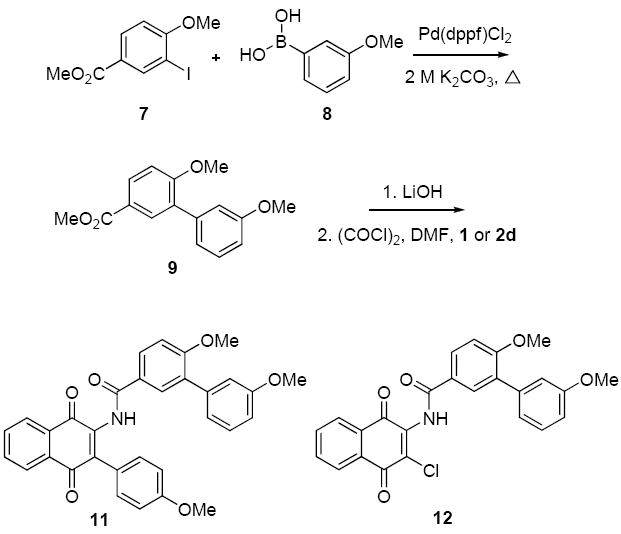
Preparation of biaryl naphthoquinone analogues.
Scheme 4.
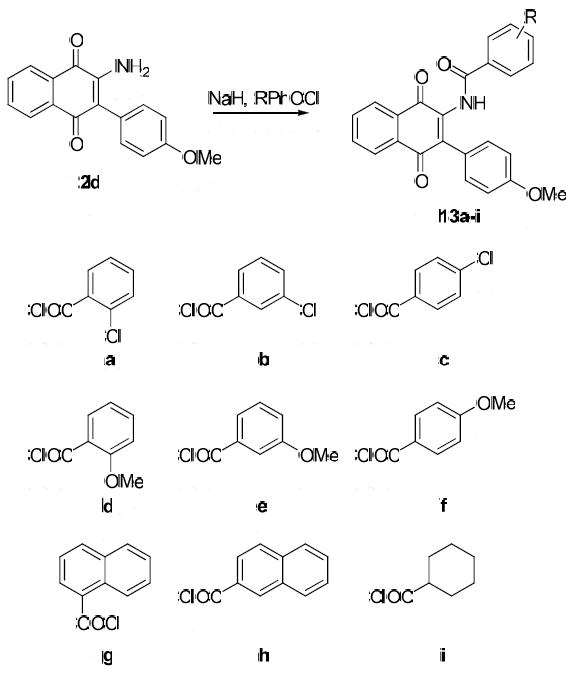
Preparation of Benzamide Analogues.
Biological Activity
The individual boronic acids (Scheme 1) were chosen to (1) probe the hydrogen bond donor and acceptor properties for this region of the molecules as well as (2) to determine the potential for hydrophobic and -stacking attributes of the aromatic ring. Each compound was tested for its anti-proliferative activity against two distinct human breast cancer cell lines, an estrogen receptor positive cell line, MCF-7, and an estrogen receptor negative, Her2 over-expressing cell line, SKBr3. In addition, the ability to downregulate the Hsp90 client protein, Her2, was measured via ELISA assay in the SKBr3 cell line.22 There were two general trends observed for the anti-proliferative activity manifested by both the acetamide 3a-i, 5 (Table 2) and benzamide 4a-i, 6 (Table 3) series of compounds: (1) the analogues were more active against the MCF-7 cell line, particularly for the benzamide analogues and (2) the analogues containing biaryl ethers (3h-i and 4h-i) were significantly less active than the naphthoquinones incorporating a single phenyl group. In addition, both series of compounds demonstrated reduced anti-proliferative activity as compared to the most active HTS hits (HTS1 and HTS3), however, their comparable activity in the Her2 ELISA assay (low micromolar IC50 values) suggests they are equipotent at Hsp90 inhibition. Nonspecific protein binding in vitro or increased redox activity of the HTS hits may account for the observed anti-proliferation discrepancies. Both the acetamide and the benzamide appear to be well tolerated, suggesting that the binding region for this portion of the scaffold may be amenable to additional functionalities of varying size. In addition, the anti-proliferative activity of 5 against the SKBr3 cell line (0.6 ± 0.003 μM) was significantly increased compared to MCF-7 cells and Her2 degradation, suggesting the potential for an additional target for this compound within the SKBr3 cell line.
Table 2.
In vitro activity of acetamide naphthoquinones.
| Compound | Her2 ELISA IC50a | Anti-Proliferation IC50 | |
|---|---|---|---|
| MCF-7 | SKBr3 | ||
| HTS1 | 3.3 ± 1 | 0.21 ± 0.02 | 0.82 ± 0.02 |
| 3a | 3.2 ± 2.0 | 1.6 ± 0.5 | 1.5 ± 0.4 |
| 3b | 2.5 ± 1.4 | 2.2 ± 0.7 | 1.7 ± 0.3 |
| 3c | 4.8 ± 1.4 | 2.7 ± 0.7 | 3.9 ± 0.7 |
| 3d | 2.2 ± 0.1 | 2.2 ± 0.3 | 2.7 ± 0.3 |
| 3e | 2.3 ± 0.2 | 3.0 ± 0.3 | 3.6 ± 0.2 |
| 3f | 2.0 ± 0.2 | 1.3 ± 0.5 | 2.5 ± 0.1 |
| 3g | 5.3 ± 2.0 | 1.4 ± 0.2 | 1.8 ± 0.4 |
| 3h | 13.3 ± 2.6 | 2.4 ± 0.3 | 6.8 ± 0.5 |
| 3i | 24.6 ± 1.5 | 2.5 ± 0.2 | 12.1 ± 0.4 |
| 5 | 6.6 ± 0.9 | 1.7 ± 0.1 | 0.6 ± 0.003 |
Values are mean ± SEM of three separate experiments performed in triplicate and reported in μM.
Table 3.
In vitro activity of benzamide naphthoquinones.
| Compound | Her2 ELISA IC50a | Anti-Proliferation IC50a | |
|---|---|---|---|
| MCF-7 | SKBr3 | ||
| HTS1 | 3.3 ± 1 | 0.21 ± 0.02 | 0.82 ± 0.02 |
| 4a | 3.8 ± 0.6 | 2.1 ± 0.4 | 6.8 ± 0.1 |
| 4b | 3.0 ± 1.4 | 2.2 ± 0.1 | 1.9 ± 0.7 |
| 4c | 2.7 ± 0.7 | 1.4 ± 0.1 | 2.6 ± 0.02 |
| 4d | 2.8 ± 0.9 | 1.4 ± 0.02 | 4.0 ± 1.8 |
| 4e | 1.8 ± 0.4 | 1.8 ± 0.1 | 4.0 ± 0.4 |
| 4f | 6.3 ± 1.5 | 4.8 ± 2.0 | 12.7 ± 0.6 |
| 4g | 1.8 ± 0.5 | 2.6 ± 0.1 | 8.5 ± 0.6 |
| 4h | >100 | 6.0 ± 0.1 | >100 |
| 4i | >100 | 23 ± 1.9 | >100 |
| 6 | 30.1 ± 1.1 | 1.8 ± 0.3 | 2.1 ± 0.1 |
Values are mean ± SEM of three separate experiments performed in triplicate and reported in μM.
Naphthoquinone analogues containing the 3-position p-methoxy aryl functionality and varying benzamides were prepared and evaluated for two specific reasons: (1) to probe the structural requirements for improving activity at each position of the benzamide ring and (2) to apply SAR developed for other Hsp90 inhibitory scaffolds to the naphthoquinone series. The biaryl benzamide that exhibits increased anti-proliferative activity when incorporated in novobiocin analogues10 was completely inactive when combined with the p-methoxy aryl ring (11, IC50>100 μM, see Table 4). The increased steric bulk of the tri-aryl ring system appears too large to be accomodated in the proposed binding site and is thus unable to maintain favorable binding interactions. The improved anti-proliferative activity of compound 12 (IC50= 1.7 – 3.0 μM) compared to 11 may suggest that the flexibility of the biaryl benzamide moiety allows it to adopt an orientation in which the m-methoxy phenyl ring of the benzamide can occupy the binding site normally occupied by the 3-position aryl functionalities. In general, these analogues 13a-i also exhibit increased anti-proliferative activity against MCF-7 cells over SKBr3 cells. Hydrogen bond acceptors were well tolerated at both the ortho and para positions, however, meta substituents were detrimental to Her2 degradative activity. A 6- and 15-fold decrease in activity was demonstrated for the meta chloro (13b) or methoxy (13e) substituents, respectively. The 3-fold decrease in Her2 activity exhibited by 13g compared to the corresponding benzamide (4d) suggests that the increased steric bulk of the naphthalene ring system prevents optimal binding site interactions. In addition, orientation of the naphthalene ring is important as evidenced by complete loss of Her2 degradative activity for the 2-napthyl analogue (13h). Finally, 13i exhibits Hsp90 inhibitory activity comparable to the corresponding benzamide (4d) suggesting the aryl ring is not essential for inhibitory activity.
Table 4.
In vitro activity of p-OMe/Benzamide derivatives.
| Compound | Her2 ELISA IC50a | Anti-Proliferation IC50 | |
|---|---|---|---|
| MCF-7 | SKBr3 | ||
| HTS1 | 3.3 ± 1 | 0.21 ± .02 | 0.82 ± .02 |
| 11 | >100 | >100 | >100 |
| 12 | 1.7 ± 0.04 | 3.0 ± 0.4 | 1.7 ± 0.5 |
| 13a | 2.0 ± 0.06 | 2.2 ± 0.1 | 5.5 ± 0.9 |
| 13b | 12.9 ± 0.4 | 3.4 ± 1.5 | 9.5 ± 2.3 |
| 13c | 3.0 ± 0.2 | 3.3 ± 1.2 | 3.0 ± 1.7 |
| 13d | 5.8 ± 0.8 | 2.1 ± 0.1 | 7.9 ± 0.8 |
| 13e | 44.2 ± 4.0 | 1.8 ± 0.1 | 6.4 ± 0.2 |
| 13f | 2.4 ± 0.08 | 1.5 ± 0.3 | 6.4 ± 0.1 |
| 13g | 9.9 ± 0.9 | 2.2 ± 0.3 | 5.8 ± 1.1 |
| 13h | >100 | 6.8 ± 0.6 | >100 |
| 13i | 2.5 ± 1.2 | 1.9 ± 0.6 | 4.8 ± 1.3 |
Values are mean ± SEM of three separate experiments performed in triplicate and reported in μM.
Compounds representing each series of naphthoquinone analogues were tested for their ability to inhibit Hsp90-dependent refolding of firefly luciferase in the HTS screening assay (Table 5). As a general trend, Hsp90 inhibition with these compounds compared favorably with their anti-proliferative and Her2 degradation activities. The acetamide derivatives evaluated were more active than the corresponding benzamide derivatives, suggesting that the smaller functionality provides improved inhibitory activity. The inhibition of luciferase renaturation elicited by the most active compound, 3g (IC50 = 0.2 ± 0.03 μM), compared favorably to that observed from the original high throughput screen. As expected, 11 was unable to inhibit luciferase refolding, while 12 exhibited low micromolar activity, further supporting the hypothesis that the presence of three bulky aryl moieties prevents optimal binding/inhibition of Hsp90. In addition, the low micromolar activity exhibited by 13i corresponds well to its in vitro activity suggesting that the aryl ring may not be essential for activity.
Table 5.
Firefly luciferase renaturation.
| Compound | IC50a |
|---|---|
| HTS1 | 0.25b |
| 3b | 1.6 ± 0.5 |
| 3g | 0.2 ± 0.03 |
| 4b | 39 ± 16 |
| 4g | 1.4 ± 0.3 |
| 5 | 5.0 ± 1.5 |
| 11 | 86 ± 22 |
| 12 | 1.6 ± 0.4 |
| 13e | 2.5 ± 1.6 |
| 13f | 24.0 ± 15 |
| 13i | 1.4 ± 0.3 |
Values are mean ± SEM of three separate experiments performed in triplicate and reported in μM.
Value represents the IC50 value from a luciferase refolding assay
The naphthoquinones 3g, 12, and 13a were further evaluated for their ability to downregulate numerous oncogenic Hsp90 client proteins, including Her2, Raf-1, and Akt in MCF-7 cells via western blot analysis as shown in Figure 3. All three compounds induced the degradation of these client proteins in a concentration-dependent manner while producing no similar effects on actin, a non Hsp90-dependent client protein used as a positive control. 13a induced Hsp90 client protein degradation at concentrations comparable to its anti-proliferative activity (IC50s ~5–10 μM). By contrast, both 3g and 12 were slightly less active than 13a at client protein degradation (IC50s ~10–20 μM). The concentrations at which 3g decreased levels of Hsp90-dependent client proteins correlates directly with its activity in cell-based assays rather than its ability to inhibit luciferase refolding.
Figure 3.
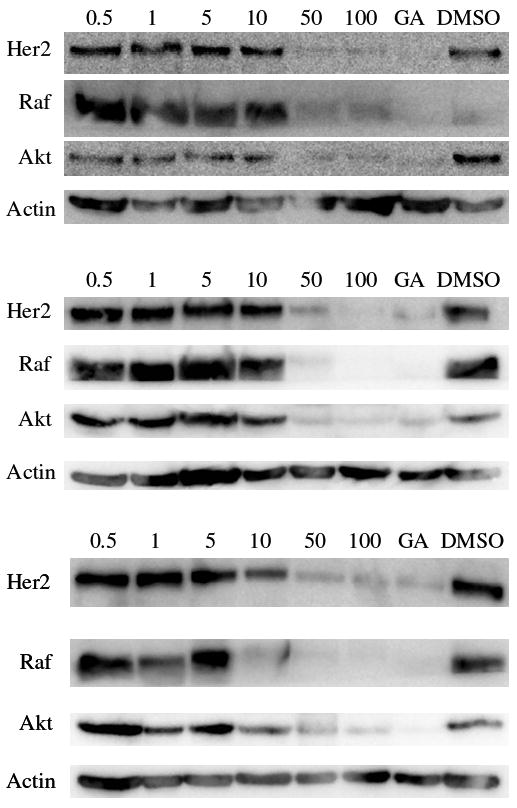
Induced degradation of Hsp90 client proteins via 3g (top), 12 (middle), or 13a (bottom) inhibition of Hsp90. Compound, at varying concentrations, (μM) was evaluated for its ability to downregulate several client proteins as described in the Experimental Section. GA (500 nM) and DMSO were used as positive and negative controls, respectively.
Conclusion
Identification of the 1,4-naphthoquinone scaffold as a new class of Hsp90 inhibitors led to the synthesis and evaluation of a rationally–designed series of analogues that contain the naphthoquinone core. Structure–activity relationship studies for these compounds have provided insight into the functionalities that produce optimal in vitro activity. While both the benzamide and acetamide derivatives were active in cell-based assays, the acetamides were more potent in the luciferase refolding assay, a direct measure of Hsp90 inhibition. Compounds that contained three aryl rings branched from the central naphthoquinone core were less active, suggesting that increased steric bulk prevents these analogues from binding similarly to the proposed site. Based on these analogues, the rational design and evaluation of naphthoquinone mimics lacking the redox-active quinone moiety are currently underway in an effort to further develop this class of inhibitors.
Experimental Section
2-amino-3-phenylnaphthalene-1,4-dione (2a)
A solution of 2-amino-3-chloro-naphthoquinone (300 mg, 1.4 mmol) and tetrakis(triphenylphosphine)palladium(0) (4 mol%, 64.7 mg, 56 μmol) in 11 mL THF:2M K2CO3 (10:1) was stirred at rt for 30 min. Phenyl boronic acid (341 mg, 2.8 mmol) in THF (3 mL) was added and the solution stirred for 30 min at rt. The solution was heated to reflux and stirred for 12 hr. The solution was cooled, filtered through celite, and diluted with EtOAc (50 mL). The EtOAc was washed with H2O (50 mL) and saturated aqueous sodium chloride (50mL), dried (Na2SO4), filtered, and concentrated. Chromatography (SiO2, Hex:EtOAc, 3:1) afforded 2a as a red solid (34%).1H NMR (CD2Cl2, 400 MHz) 8.13 (t, J = 7.3 Hz, 2H), 7.80 (t, J = 7.3 Hz, 1H), 7.61 (t, J = 7.3 Hz, 1H), 7.54 (t, J = 7.9 Hz, 2H), 7.41 (m, 3H) 5.25 (broad, NH2). 13C NMR (CD2Cl2, 500 MHz) 182.2, 181.9, 145.5, 135.0, 133.6, 133.3, 132.6, 131.0, 130.5 (2C), 129.3 (2C), 128.4, 126.7, 126.1, 117.13. νmax 3138, 3022, 1674, 1622, 1572, 1555, 1354, 1230, 1047, 733 cm-1. ESI-HRMS m/z calculated for C16H12NO2 [M+H]+ 250.0868, found 250.0860.
N-(1,4-dioxo-3-phenyl-1,4-dihydronaphthalen-2-yl)acetamide (3a)
To a solution of 2a (10 mg, 0.04 mmol) in 5 mL acetyl chloride at rt was added a catalytic amount of concentrated sulfuric acid. The solution was refluxed for 15 mins (or until the solution changed from deep red to yellow). The acetyl chloride was removed and the mixture redissolved in EtOAc (5 mL) and H2O (5 mL). The aqueous layer was removed, the organic layer washed with saturated aqueous sodium chloride (5 mL), dried (Na2SO4), and concentrated. Preparative TLC (Hex:EtOAc, 2:1) afforded 3a in quantitative yield as a yellow oil. 1H NMR (CD2Cl2, 400 MHz) 8.17 (d, J = 6.8 Hz, 2H), 7.83 (m, 2H), 7.75 (s, 1H), 7.46 (m, 2H), 7.38 (m, 3H), 2.01 (s, 3H). 13C NMR (CD2Cl2, 500 MHz) 183.9, 182.7, 166.7, 138.0, 135.0, 134.9, 134.0, 133.9, 132.7, 131.0, 129.7 (2C), 128.7, 128.2 (2C), 127.2, 126.5, 24.2. νmax 3109, 3058, 2962, 1664, 1612, 1595, 1491, 1477, 1326, 1294, 1157, 1016, 731 cm-1. ESI-HRMS m/z calculated for C18H14NO3 [M+H]+ 292.0974, found 292.0973.
N-(1,4-dioxo-3-phenyl-1,4-dihydronaphthalen-2-yl)benzamide (4a)
To a solution of 2a (7.5 mg, 0.03 mmol) in anhydrous THF was added sodium hydride (4.3 mg of a 56% dispersion, 0.1 mmol) and the mixture stirred at rt for 30 min. Benzoyl chloride (5.6 mg, 4.6 μL, 0.04 mmol) was added and the mixture stirred at rt for 1 hr. The THF was removed and the mixture redissolved in EtOAc (5 mL) and H2O (5 mL). The aqueous layer was removed, the organic layer washed with saturated aqueous sodium chloride (5 mL), dried (Na2SO4), and concentrated. Preparative TLC (benzene or benzene/EtOAc, TLC plate developed 8-10 times) afforded 4a as a red oil (56%). 1H NMR (CD2Cl2, 400 MHz) 8.47 (s, 1H), 8.21 (t, J = 6.7 Hz, 2H), 7.86 (m, 2H), 7.77 (d, J = 7.8 Hz, 2H), 7.60 (t, J = 6.7 Hz, 2H), 7.40-7.52 (m, 5H), 7.34 (m, 1H). 13C NMR (CD2Cl2, 500 MHz) 183.8, 182.8, 163.9, 138.4, 135.1, 134.4, 134.1, 134.0, 133.9, 133.0, 132.9, 131.0, 129.6 (2C), 129.2 (2C), 128.7, 128.2 (2C), 128.0 (2C), 127.3, 126.6. νmax 3059, 3034, 2869, 1663, 1502, 1474, 1294, 1273, 1070, 1026, 914, 717 cm-1. ESI-HRMS m/z calculated for C23H16NO3 [M+H]+ 354.1130, found 354.1155.
3’,6-dimethoxy-N-(3-(4-methoxyphenyl)-1,4-dioxo-1,4-dihydronaphthalen-2-yl)biphenyl-3-carboxamide (11)
Acid 1010 (43 mg, 0.17 mmol) was dissolved in anhydrous CH2Cl2 (5 mL) under argon at rt. Oxalyl chloride (42.6 mg, 28.8 μL, 0.34 mmol) and catalytic DMF were added and the mixture stirred at rt for 12 hr. The CH2Cl2 was removed and the resulting yellow oil was dryed under high vacuum for 3 hr and used without further purification. To a solution of 2d (13.5 mg, 0.05 mmol) in anhydrous THF (5 mL) was added sodium hydride (12.9 mg of a 56% dispersion, 0.15 mmol) and the mixture stirred at rt for 30 min. Biaryl acid chloride (17 mg, 0.06 mmol) was added and the mixture stirred at rt for 1 hr. The THF was removed and the mixture redissolved in EtOAc (5 mL) and H2O (5 mL). The aqueous layer was removed, the organic layer washed with saturated aqueous sodium chloride (5 mL), dried (Na2SO4), and concentrated. Preparative TLC (Hex:EtOAc 2:1) afforded 11 as a red oil (46%). 1H NMR (CD2Cl2, 400 MHz) 8.39 (s, 1H), 8.16 (t, J = 7.4 Hz, 2H), 7.80 (m, 3H), 7.73 (s, 1H), 7.40 (d, J = 8.8 Hz, 1H), 7.35 (t, J = 8.8 Hz, 1H), 7.07 (t, J = 8.3 Hz, 3H), 6.92 (d, J = 8.1 Hz, 4H), 3.89 (s, 3H), 3.85 (s, 3H), 3.81 (s, 3H). 13C NMR (CD2Cl2, 500 MHz) 184.1, 182.8, 163.6, 160.4, 160.1, 159.8, 139.2, 138.3, 134.9, 134.1, 133.8, 132.9, 131.2 (2C), 131.1, 131.0, 130.7, 129.4, 129.4, 127.2, 126.5, 126.3, 126.2, 122.3, 115.7, 113.7 (2C), 113.2, 111.4, 56.2, 55.7, 55.5. νmax 2956, 2916, 2846, 1663, 1608, 1574, 1520, 1478, 1292, 1285, 1186, 667 cm-1. ESI-HRMS m/z calculated for C32H26NO [M+H]+ 520.1760, found 520.1761.
Anti-proliferative effects of naphthoquinones
MCF-7 and SKBr3 cells were maintained in a 1:1 mixture of Dulbecco’s Modified Eagle’s Medium:Ham’s F-12 (Gibco) supplemented with non-essential amino acids, L-glutamine (2 mM), streptomycin (500 μg/ml), penicillin (100 units/ml) and 10% fetal bovine serum. Cells were grown to confluence in a humidified atmosphere (37 °C, 5% CO2). Cells (2000/well, 100 μL) were seeded in 96-well plates and allowed to attach overnight (37 °C, 5% CO2). Compounds or GA at varying concentrations in DMSO (vehicle) were added (1% DMSO final concentration) and cells returned to the incubator (37 °C, 5% CO2) for 72 hr. At 72 hr, the number of viable cells was determined using an MTS/PMS cell proliferation kit (Promega) per the manufacturer’s instructions. Cells incubated in 1% DMSO were used as 100% proliferation and values were adjusted accordingly.
Her2 ELISA of naphthoquinones
SKBr3 cells were grown as described above and seeded (3000 cells/well) in 96-well plates and allowed to attach overnight (37 °C, 5% CO2). Compounds, at varying concentrations, were added and the plates returned to the incubator for 24 hr. Media was removed and the cells washed three times with ice-cold buffer (PBS with 1% Tween). Methanol (-20°C) was added and the plates placed at 4°C for 10 min to permeabilize and fix the cells. The plates were washed again with ice-cold buffer and incubated in blocking buffer (5% BSA in PBST) for 1 hr at rt. The plates were incubated with a Her2 specific antibody (rabbit IgG; 1:500 dilution in blocking buffer) at 4°C overnight. The plates were washed again and incubated at room temperature for 2 hr in the presence of an HRP-conjugated anti-rabbit IgG (1:1000 in blocking buffer). Plates were rinsed, chemiluminescent reagent was added and the plates immediately read on a luminometer (Molecular Devices).
Luciferase Renaturation Assay
The ability of the naphthoquinones to inhibit the Hsp90-dependent renaturation of firefly luciferase was determined as previously described.16
Western blot analyses of naphthoquinones
MCF-7 cells were seeded (1×106/plate) in culture dishes and allowed to attach overnight. Compounds 3g, 12, and 13a were added at varying concentration and incubated (37 °C, 5% CO2) for 24 h. Cells were harvested and analyzed for Hsp90 client protein degradation as described previously.17
Acknowledgments
This work was supported by the National Institutes of Health (NIH-1R01CA125392 and), OAES project 1975, and the Kansas Cancer Center (Postdoctoral Fellowship, M.K.H.)
Footnotes
Supporting Information Available. General synthetic procedures, full characterization of key intermediates and all final analogues. This material is free of charge via the Internet.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Blagg BS, Kerr TD. Med Res Rev. 2006;26:310–338. doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- 2.Marcu MG, Schulte TW, Neckers L. J Natl Cancer Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 3.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers L. J Biol Chem. 2000;276:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 4.Allan RK, Mok D, Ward BK, Ratajczak T. J Biol Chem. 2006;281:7161–7171. doi: 10.1074/jbc.M512406200. [DOI] [PubMed] [Google Scholar]
- 5.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H. Proc Natl Acad Sci. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen H-Y, He J-C, Wang Y, Huang Q-Y, Chen J-F. J Biol Chem. 2005;280:39962–39969. doi: 10.1074/jbc.M505524200. [DOI] [PubMed] [Google Scholar]
- 7.Hadden MK, Lubbers DJ, Blagg BSJ. Curr Top Med Chem. 2006;6:1173–1182. doi: 10.2174/156802606777812031. [DOI] [PubMed] [Google Scholar]
- 8.Burlison JA, Blagg BSJ. Org Lett. 2006;8:4855–4858. doi: 10.1021/ol061918j. [DOI] [PubMed] [Google Scholar]
- 9.Le Bras G, Radanyi C, Peyrat J-F, Brion J-D, Alami M, Marsaud V, Stella B, Renoir J-M. J Med Chem. 2007;50:6189–6200. doi: 10.1021/jm0707774. [DOI] [PubMed] [Google Scholar]
- 10.Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BSJ. J Org Chem. 2008;73:2130–2137. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- 11.Zhou V, Han S, Brinker A, Klock H, Caldwell J, Gu X-j. Anal Biochem. 2004;331:349–357. doi: 10.1016/j.ab.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 12.Cheung K-MJ, Matthews TP, James K, Rowlands MG, Boxall KJ, Sharp SY, Maloney A, Roe SM, Prodromou C, Pearl LH, Aherne GW, McDonald E, Workman P. Bioorg Med Chem Lett. 2005;15:3338–3343. doi: 10.1016/j.bmcl.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 13.Barril X, Brough P, Drysdale M, Hubbard RE, Massey A, Surgenor A, Wright L. Bioorg Med Chem Lett. 2005;15:5187–5191. doi: 10.1016/j.bmcl.2005.08.092. [DOI] [PubMed] [Google Scholar]
- 14.Avila C, Hadden MK, Ma Z, Kornilayev BA, Ye QZ, Blagg BSJ. Bioorg Med Chem Lett. 2006;16:3005–3008. doi: 10.1016/j.bmcl.2006.02.063. [DOI] [PubMed] [Google Scholar]
- 15.Kim J, Felts S, Llauger L, He H, Huezo H, Rosen N, Chiosis G. J Biomol Screen. 2004;9:375–381. doi: 10.1177/1087057104265995. [DOI] [PubMed] [Google Scholar]
- 16.Galam L, Hadden MK, Ma Z, Ye Q-Z, Yun B-G, Blagg BSJ, Matts RL. Bioorg Med Chem. 2007;15:1939–1946. doi: 10.1016/j.bmc.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hadden MK, Galam L, Matts RL, Blagg BSJ. J Nat Prod. 2007;70:2014–2018. doi: 10.1021/np070190s. [DOI] [PubMed] [Google Scholar]
- 18.Satoh N, Ishiyama T, Miyaura N, Suzuki A. Bull Chem Soc Jpn. 1987;60:3471–3473. [Google Scholar]
- 19.Boland GM, Donnelly DMX, Finet J-P, Rea MD. J Chem Soc Perkin Trans. 1996;1:2591–2597. [Google Scholar]
- 20.Hoover JRE, Day AR. J Am Chem Soc. 1954;76:4148–4152. [Google Scholar]
- 21.Hastings JM, Hadden MK, Blagg BSJ. J Org Chem. 2008;73:369–373. doi: 10.1021/jo702366g. [DOI] [PubMed] [Google Scholar]
- 22.Huezo H, Vilenchik M, Rosen N, Chiosis G. Chem Biol. 2003;10:629–634. doi: 10.1016/s1074-5521(03)00144-3. [DOI] [PubMed] [Google Scholar]