

Abstract
Recent advances indicate that maintaining a balanced level of autophagy is critically important for neuronal health and function. Pathologic dysregulation of macroautophagy has been implicated in synaptic dysfunction, cellular stress, and neuronal cell death. Autophagosomes and autolysosomes are induced in acute and chronic neurological disorders including stroke, brain trauma, neurotoxin injury, Parkinson’s, Alzheimer’s, Huntington’s, motor neuron, prion, lysosomal storage, and other neurodegenerative diseases. Compared to other cell types, neuronal autophagy research presents particular challenges that may be addressed through still evolving techniques. Neuronal function depends upon maintenance of axons and dendrites (collectively known as neurites) that extend for great distances from the cell body. Both autophagy and mitochondrial content have been implicated in regulation of neurite length and function in physiological (plasticity) and pathological remodeling. Here, we highlight several molecular cell biological and imaging methods to study autophagy and mitophagy in neuritic and somatic compartments of differentiated neuronal cell lines and primary neuron cultures, using protocols developed in toxic and genetic models of parkinsonian neurodegeneration. In addition, mature neurons can be studied using in vivo protocols for modeling ischemic and traumatic injuries. Future challenges include application of automated computer-assisted image analysis to the axodendritic tree of individual neurons and improving methods for measuring neuronal autophagic flux.
1. Introduction
The neuron is the most highly polarized postmitotic cell, consisting of a soma and specialized axonal and dendritic projections (collectively called neurites) that form networks of arborizing intercellular synaptic connections. Maintenance of these long neuritic structures is required for the propagation of electrochemical signals across vast cellular distances. Basal physiological levels of autophagy play a critical role in maintaining neuronal health, presumably by removing effete, oxidized, or aggregated proteins and organelles [Reviewed in (Boland and Nixon, 2006; Cherra and Chu, 2008; Ventruti and Cuervo, 2007)], whereas dysregulated autophagy contributes to neurite degeneration and neuronal cell death, as shown in several in vitro and in vivo systems (Koike et al., 2008; Plowey et al., 2008; Yang et al., 2007; Yue et al., 2008; Zhu et al., 2007).
Synaptic/neuritic pathology is a prevalent theme in neurodegenerative diseases [Reviewed in (Wishart et al., 2006)] and neurite degeneration and remodeling are elicited in acute brain injuries (Ito et al., 2006). These neuritic pathologies include protein and organelle accumulation, beading/fragmentation of neurites, and disruption of microtubule networks involved in axonal transport (Fiala et al., 2007). Neuritic pathologies highlighted by silver stains or immunohistochemistry of disease-related proteins include Lewy neurites in Parkinson/Lewy body diseases (Spillantini et al., 1997), dystrophic neurites in Alzheimer disease (Nixon, 2007), huntingtin protein in cortical neurites (DiFiglia et al., 1997), amyloid precursor protein in axonopathy of head trauma (Gentleman et al., 1993), and spongiform change in prion diseases (Ironside, 1998). Dystrophic neurites, which likely exhibit abnormal retrograde and anterograde transport, are associated with dysfunctional synapses. Alzheimer disease brains exhibit decreased synaptic density (Masliah et al., 2001), even in the earliest stages of clinical disease (Scheff et al., 2006), and inhibition of hippocampal long-term potentiation has been demonstrated in animal models (Chapman et al. 1999; LaFerla & Oddo 2005; Walsh et al. 2002). Alterations in synaptic morphology are also observed in the basal ganglia of Parkinson’s disease patients (Lach et al., 1992; Machado-Salas et al., 1990), with synaptic dysfunction as a prominent feature in models based on mutations in α-synuclein [Reviewed in (Cookson and van der Brug, 2008)] or PINK1 (Kitada et al., 2007). In models of Huntington disease, the medium spiny striatal neurons demonstrate abnormal dendritic spine morphology and electrophysiology (Di Filippo et al., 2007). Synaptic pathology has also been implicated in models of prion diseases (Chiesa et al., 2005; Clinton et al., 1993; Fournier, 2008; Jeffrey et al., 2000; Kitamoto et al., 1992). As synaptic contacts form the morphological substrate for neuronal function, neuritic indices of injury should be considered as carefully as historically emphasized cell death endpoints in studies of neurological diseases.
Unlike other eukaryotic cells, neurons are almost devoid of autophagic vacuoles (AVs) and lysosomes under basal conditions (Boland and Nixon, 2006). Nevertheless, autophagy has been implicated in normal physiological neuritic/synaptic function. In the nematode, autophagy regulates levels of GABAA receptors at inhibitory synapses (Rowland et al., 2006). Autophagy-deficient knockout mice show severe axonal dystrophy with relative sparing of dendrites (Hara et al., 2006; Komatsu et al., 2007; Yue et al., 2008). Neuritic differentiation is impaired with either too much or too little mTOR activity (Zeng and Zhou, 2008), suggesting a possible role for autophagy in synaptogenesis. There is also evidence that mTOR modulates activity-dependent synaptic plasticity in the dendrites of hippocampal neurons (Gong et al., 2006), although the specific role of autophagy was not investigated.
Pathological increases in AVs and lysosomes are observed in Parkinson’s, Alzheimer’s, and Huntington’s diseases, transmissible spongiform encephalopathies, and in toxicity due to MPP+, methamphetamine, dopamine and mutations in the familial parkinsonian gene LRRK2 which encodes leucine rich repeat kinase 2 (Chu et al., 2007; Gomez-Santos et al., 2003; Larsen et al., 2002; Liberski et al., 2002; Nixon et al., 2005; Orr, 2002; Plowey et al., 2008; Rudnicki et al., 2008; Zhu et al., 2007). Brain tissue from Parkinson and Lewy body disease patients display increased mitochondrial autophagy (Zhu et al., 2003). The accumulation of autophagosomes, often in neurites, is observed following stroke and brain trauma (Adhami et al., 2006; Lai et al., 2008; Liu et al., 2008). Increased autophagosomes observed in disease states could also reflect impaired autophagosome clearance, as suggested for Alzheimer and lysosomal storage diseases (Koike et al., 2005; Nixon et al., 2005). These findings implicate disruption of the balance between degradative and biosynthetic mechanisms as a key regulator of neurite remodeling and dysfunction elicited by acute and chronic insults (Cherra and Chu, 2008). In the following sections, we will discuss protocols developed to study neuritic and somatic autophagy in neuronal cell lines, primary neuron cultures and in vivo models of brain injury.
2. Studying Neuronal Autophagy In Vitro
2.1. Differentiation of SH-SY5Y neuroblastoma cells
SH-SY5Y is a third-generation cell line cloned from a metastatic neuroblastoma removed from a 4-year-old girl (Ross et al., 1983). Dividing SH-SY5Y cells have an undifferentiated appearance and tend to aggregate in culture. We induce neuron-like differentiation by seeding and incubating the cells in 10 μM retinoic acid (RA; Sigma, St. Louis, MO, USA). RA induces a fusiform to triangular soma accompanied by elongation of slender neuritic processes (Pahlman et al., 1984). Advantages of RA-differentiation include markedly reduced cell division, and spreading out of differentiated cells uniformly across the culture surface. We store 1000X aliquots of RA in dimethyl sulfoxide (DMSO; Sigma) at 4 °C. Under these conditions, RA is stable for about 3 months; with longer storage times, it loses efficacy resulting in decreased neurite lengths in treated cells. We have also employed a differentiation protocol involving sequential exposure of cells to RA followed by brain-derived neurotrophic factor (BDNF) (Encinas et al., 2000). BDNF-differentiated SH-SY5Y cells demonstrate further elongation of neurites accompanied by increased neuritic branching compared to RA alone. However, these cultures are more cumbersome to study due to increased neurite overlap between cells, extension of neurites in and out of the focal plane, and greater variability in neurite lengths in control differentiated cultures (sometimes requiring piecing together multiple high-power fields). Consequently, we conduct the majority of our experiments in RA-differentiated SH-SY5Y cells.
2.1.1. Plating and Differentiation Protocol (Plowey et al., 2008)
SH-SY5Y cells are maintained in a humidified incubator with 5% CO2 at 37 °C in antibiotic-free Dulbecco’s modified Eagle’s medium (DMEM; Lonza, Walkersville, MD, USA) supplemented with 10% heat-inactivated fetal calf serum, 10–15 mM HEPES and 2 mM glutamine (Biowhitaker, Walkersville, MD), referred to henceforth as DMEM/FBS.
Culture chambers are seeded on Day 1 at a relatively low density of 5.0 × 104/cm2 in DMEM/FBS supplemented with 10 μM RA. Uncoated 10-cm2 plastic culture chambers are used for biochemical analyses and uncoated Lab-Tek II chambered cover glasses (#1.5 German borosilicate; Nalge Nunc International, Naperville, IL, USA) for imaging experiments.
After 3 days differentiation (Day 4), the media is refreshed and experiments are initiated. For BDNF-differentiation, the media is changed to serum-free DMEM with 50 ng/ml BDNF and experiments are initiated following an additional two days of incubation.
Because of reduced proliferation, RA cultures can be maintained for about 10 days and BDNF cultures for about 12 days at this seeding density. Maximal neurite lengths are reached by 4–5 days in RA and 2–3 days in BDNF. Because neurite length is maintained in the weeks following differentiation, these cultures can be used for chronic studies of 2–4 weeks if seeded at a lower density, although eventually an undifferentiated flat population may overgrow the plate.
2.2. NIH ImageJ assisted analysis of neurite length and arborization
The length and complexity of the axodendritic arbor in neurons can be markedly altered by pathological stresses and physiologic activity-dependent remodeling. To analyze the impact of autophagy on neurites, rapid, reproducible, and unbiased methods for measuring neurite lengths and branching complexity are desirable. A variety of approaches have been used in cell culture studies of outgrowth/differentiation (Charych et al., 2006; Dominguez et al., 2004; Hynds et al., 2002; Price et al., 2006), degeneration/injury (Bilsland et al., 2002), or dystrophy (Pigino et al., 2001). Imaging methods developed to study neurite outgrowth from neuronal explants involve counting the number of neurites that extend beyond the circumference of a circle centered at the explant (Lagreze et al., 2005). However, this method is not readily translated to studying injury-induced neurite retraction in dispersed neuronal cultures, as neuritic processes associated with an individual neuron are not separately delineable.
Commercial software such as Metamorph (Molecular Devices, Sunny-Vale, CA) and Image Pro Plus/Express (Media Cybernetics, Silver Springs, MD) have been employed to measure neurites and branching complexity in primary neurons, which involve manually drawing polygons around the neurite(s) and soma of neurons to be measured (Gerecke et al., 2004). In addition, many researchers use NIH ImageJ software for data quantification. Advantages of ImageJ include the open access nature of its source code, availability of an online manual in macro programming and maintenance of a large repository of macros available in the public domain (http://rsb.info.nih.gov/ij/macros/).
In the subsections here, we discuss two protocols that we have developed using the NeuroJ plug-in for the public access software ImageJ (NIH, Bethesda, MD, USA), along with their advantages and limitations. Both protocols involve transient transfection of a random subset of neuronal cells in culture to delineate the neuritic arbor of individual cells. The first involves intensity-assisted tracing of neurites of individual neurons showing little overlap (Plowey et al., 2008), and the second reflects our early efforts towards automated measurements that can be used to analyze more densely transfected cells in a higher throughput manner.
2.2.1. GFP transfection to delineate neuronal cells for quantitative analysis
Overlap of neuritic processes from multiple cells hampers confident documentation of the neuritic arbors of individual neurons and differentiated cell lines (Fig. 11.1A). While one approach may involve plating cells at very low densities, we find that neuronal cells are not well maintained in the absence of sufficient cellular contacts. In our studies, quantitative analysis of neuritic extensions of individual cells is made possible by transfection with plasmids encoding soluble, monomeric GFP to highlight the cell bodies and neuritic arbors. This approach capitalizes on relatively low transfection efficiencies that neuronal cells exhibit with cationic lipid-based methods (Lipofectamine 2000; Invitrogen, Carlsbad, CA, USA), allowing us to achieve adequate spatial resolution between transfected cells for confident image analysis at plating densities supportive of neuronal health in culture. We routinely use the pEGFP-C1 (Invitrogen) vector (Fig. 11.1B–C). Although pMAX GFP (Amaxa Biosystems) exhibits more intense fluorescence amenable to low-power analysis, transient expression result in cytosolic GFP granules in the soma of differentiated cells. These may reflect either aggregation or accumulation in lysosomes, both of which appear similar by fluorescence microscopy (Katayama et al., 2008). Thus, we prefer using the pEGFP-C1 plasmid, which remains diffusely distributed (Katayama et al., 2008; Plowey et al., 2008).
Figure 11.1.
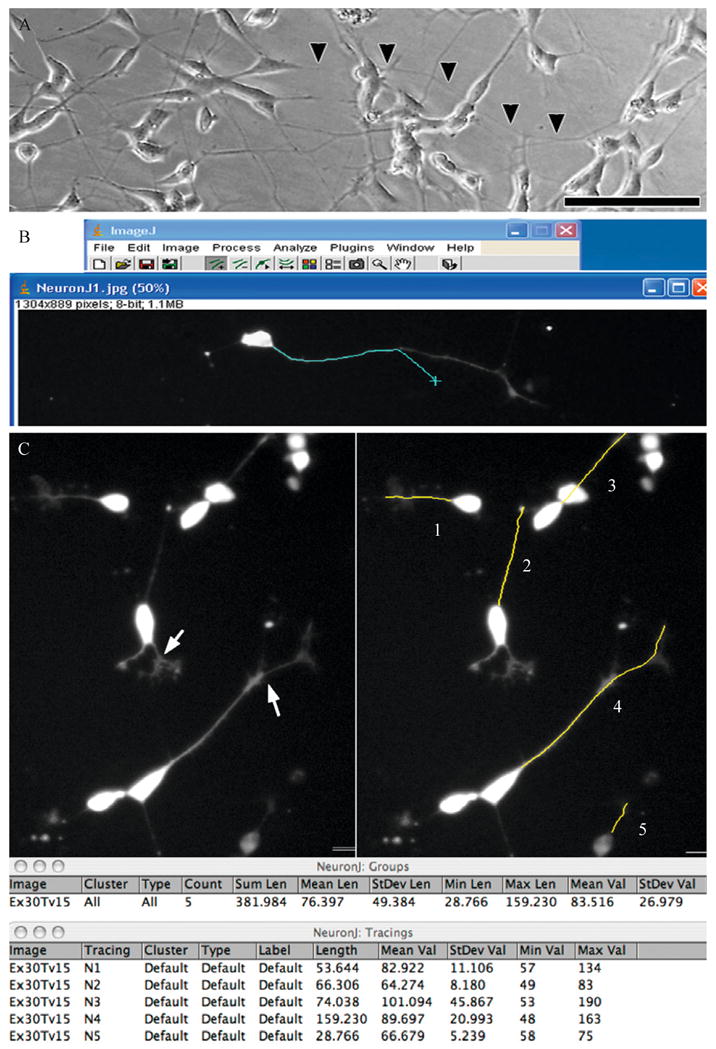
Using the intensity-tracing algorithm in NIH ImageJ. (A) Phase image of differentiated SH-SY5Y cells showing extensive overlap of processes making it difficult to determine the end of a neuritic process (arrowheads) that merges with processes of other cells. Scale: 100 μm. Transfection of RA/BDNF (B) or RA-differentiated (C) cultures with EGFP-C1 allows visualization of the neuritic arbors associated with individual neurons. (B) Using the NeuronJ plug-in for NIH ImageJ, the user clicks at the base of the neurite and as the mouse (cyan X) is moved toward the distal end, the software automatically follows the subtle curves of the structure allowing the user to simply move the mouse to the end of the structure without having to manually trace it. (C) The arrows in the left panel illustrate examples of branch points, and as seen on the right, the user has input in deciding whether to end the measurement at the branch point for primary segment determinations, or select the longer branch to determine the maximal extent of neurite elongation from the soma. As shown in the data table, individual NeuronJ tracings can be grouped to obtain summated and average lengths of all traced neurites in a group. Multiple neurites extending from a single multipolar neuron may be defined as a group to obtain summated neurite lengths per cell (not shown). The software is compatible with both Macintosh and PC operating systems (Panel B: Windows XP running ImageJ 1.36b and NeuronJ 1.1.0; Panel C: MacBook Pro OSX 10.4.11 running ImageJ 1.38X with Java 1.5.0_13 and NeuronJ 1.2.0).
2.2.2. Lipofectamine Transfection Protocol (Plowey et al., 2008; Dagda et al., 2008)
The following protocol is effective for transient transfection of differentiated or undifferentiated SH-SY5Y cells with a variety of DNA plasmids (pEGFP-C1, pMAX GFP, GFP-LC3, GFP-ERK2). See section 2.5.1 for copy ratio considerations when cotransfection of more than one plasmid is desired, and Section 2.6 for combining with siRNA protocols.
Cells should be seeded on 2-well Lab-Tek II chambered cover glasses as described previously. They can be differentiated prior to or after transfection, and we often design experiments so that the final days of differentiation occur during the 2–3 day posttransfection period necessary to allow time for protein expression.
-
We have found that delivery of 1–1.6 μg of DNA/well in a final lipofectamine concentration of 0.1% is effective at transfecting both SH-SY5Y and primary neurons without causing measurable injury to the cells. For 4-well chambered slides, 0.5–0.8 μg is used per well.
For example, 1 μg of high quality DNA (260 nm:280 nm absorbance ratio > 1.5) in Tris-EDTA is mixed in 100 μl of OptiMEM (Invitrogen), and 1% Lipofectamine is prepared separately in the same volume of OptiMEM.
The Lipofectamine in OptiMEM is incubated for 5 min prior to the addition of the DNA/OptiMEM. The transfection mixture is then allowed to sit for 20 min at room temperature prior to being added to the cells.
Because the cells are grown in 1-ml volumes, 200 μl of DMEM/FBS is removed, and replaced with the entire contents of the transfection mixture, distributed in a dropwise fashion around the well in a spiral pattern. If concurrent RA differentiation is desired, we reduce the OptiMEM volumes in step 2 to 50 μl each (doubling the initial DNA and Lipofectamine concentrations) such that 100 μl of total DNA/Lipofectamine/OptiMEM is added to 900 μl of DMEM/FBS/RA in order to minimize dilution of the RA.
The cells are incubated in the DNA/Lipofectamine/OptiMEM-containing medium for at least 48 h prior to imaging to allow time for protein expression.
This protocol yields transfection efficiencies of 25%–35% in undifferentiated SH-SY5Y cells, 10% in RA-differentiated cells, and 2%–5% in primary mouse neurons. Forty to fifty random low-power fields are captured blind to the experimental conditions using an inverted epifluorescence microscope and images are quantified as described subsequently.
2.2.3. NeuroJ tracing of neurites for quantification of length and branchpoints
The NeuroJ/ImageJ system provides the ability to measure neurite lengths and branching with accuracy by manually defining neurites for a small sample size of neurons. The JAVA source code of NeuroJ allows the user to interactively analyze running averages and statistics of branch point numbers, neurite lengths and fluorescence intensities during the analysis. We define maximal neurite length as the length from the hillock to the tip of the most distant terminal branch, beginning the measurement at the hillock of each primary neurite and selecting the branch at each branchpoint that would yield the most distant terminal end. For RA-differentiated SH-SY5Y cells, the majority are unipolar or bipolar. In contrast, BDNF-differentiated cells are occasionally multipolar and show more elaborate neurite branching.
We employ the NeuroJ extension (Meijering et al., 2004) of NIH ImageJ to rapidly quantify neurite lengths in our images. This shareware plug-in utilizes an intensity-tracing algorithm that facilitates measurements of neuritic processes in gray-scale images (Meijering et al., 2004). This is an important feature that significantly reduces variability between users and fatigue-related error, as the user needs only to identify the beginning and end of the neurite rather than attempting to trace the whole structure (Fig. 11.1B). Occasionally, a few extra mouse clicks may be required to assist the program in spanning regions that are out of focus or exhibit ambiguous signal intensity.
Neurite branching is another parameter of neurite morphology (Dominguez et al., 2004). We calculate neurite branchpoint number by summing up the number of times a secondary or tertiary neurite branch diverges from the longest primary path. Treatment induced modulation of neurite branchpoints is observed as an index of altered complexity in BDNF-differentiated SH-SY5Y cells. However, in RA-differentiated cells, neurite branchpoints are relatively few even under control conditions (Fig. 11.1C). For analysis of more complex primary neurons, our pEGFP-C1 transfection protocol would also yield images suitable for Sholl analysis of individual neurons (Gutierrez and Davies, 2007) following creation of a binary mask for each transfected neuron.
2.2.4. Semiautomated Measure Neurites macro for high-throughput analysis of neurites
Whereas NeuroJ-assisted tracing is an excellent method to assess neurite morphology, this procedure still requires manually defining neurites of individual neurons. We have been developing alternative methods for measuring the effects of autophagy on neurite abundance (incorporating both length and complexity), using the automated custom Record Macro function of ImageJ (Fig. 11.2). We created the Measure Neurites macro, a custom ImageJ macro for measuring neurite lengths in a high-throughput manner, which is available for download at the ImageJ website (http://imagejdocu.tudor.lu/author/rkd8/). The macro uses the Outlines Algorithm component of the Analyze Particles function of ImageJ, an algorithm used to outline interconnected pixels around the object to be analyzed. Once it is downloaded from the ImageJ website, the text file should be moved to the macro folder of the ImageJ program (c:/programs/ImageJ/macro).
Figure 11.2.

Using the Measure Neurites macro. The Measure Neurites macro was used to analyze an epifluorescent micrograph captured at 20X magnification of mouse cortical neurons (7 days in vitro; DIV7). Mouse primary neurons were seeded at a relatively high density of 1.5 × 105/cm2 on DIV0, and transiently transfected with GFP on DIV5. RGB images of GFP-expressing neurons were processed by the Measure Neurites macro at 48 h following transfection. The green channel was extracted from the RGB image and inverted, so the highly fluorescent pixels appear black. The image is manually thresholded such that pixels that exceed the threshold intensity are highlighted in red, while those falling below the threshold remain in greyscale (A). The macro traces the thresholded neurites using the Outlines algorithm (B) and computes the number of neurites measured, average neuritic area, and average neuritic perimeter per image, and the longest neuritic perimeter. Notice that background fluorescence (left side of panel (A) or regions of neurites that are incompletely thresholded due to low fluorescence or uneven illumination (right side of panel A) are not traced by the macro and are thus excluded from the analyses (B). The macro computes average neuritic length by substracting the average soma perimeter from the average total perimeter (soma plus neurites) divided by the number of objects being counted to produce summated neuritic perimeters, which can be divided by 2 to approximate neurite lengths. Alternatively, one can normalize neuritic perimeters or lengths by the number of transfected neurons included in the thresholded image (e.g., GFP neurons containing DAPI stain).
The Measure Neurites macro is optimized to measure neurites of GFP transfected, differentiated SH-SY5Y cells, and primary cortical neurons. However, the macro can be customized depending on the neuronal population being analyzed by changing the value of the average soma perimeter (line 34) that is subtracted from the total average cellular perimeter lengths to obtain average neurite perimeters. In brief, the macro opens up RGB images, splits them into individual channels, and performs photographic inversion of the green channel. The macro allows the user time to manually threshold the pixels (set a minimum pixel value for the structures to be analyzed in a given epifluorescent field) to trace the neurites (Fig. 11.2A–B). The macro then computes the number of neurites, summation of the areas and perimeters of soma and neurites, and the average neuritic area and perimeter per epifluorescent field. The results can be pasted onto an Excel spreadsheet. The summated neurite perimeters for each image is divided by 2, assuming negligible contributions of neurite thicknesses, to yield the summed total of neurite lengths, then normalized to the number of cells analyzed in the image (determined by counting the number of green neurons with DAPI stained nuclei), prior to statistical analysis.
Prior to running the macro, it is important to set the correct scale of the image being analyzed using the Set Scale command under the Analyze pull-down menu. The scale is to be assigned as pixels/μm. It is also important to determine if the average soma perimeter of the cells being studied is accurately reflected in line 34 of the macro as discussed previously. The quality and uniformity of the background in the image to be analyzed will determine whether the majority of neurites will be traced by thresholding. As the macro obtains summated lengths of primary, secondary, and tertiary neurites, it estimates a higher neurite number compared to manual identification; although average lengths in RA differentiated, predominantly unipolar SH-SY5Y cells show only ~1 micron differences when the same set of images is used to directly compare the two methods. In terms of time efficiency, the Measure Neurites macro is best used for images with higher neuron densities or complex neuritic arbors because more technical replicates are required to develop consistent user thresholding. For images with fewer neurons with minimal overlap, the manual identification method is still faster and more precise.
Although this is still a work in progress, a strength of the macro is that it can be used as a template for further optimization by modifying the code to fit new conditions. Updates and detailed instructions on how to use our most current version of the macro will be available on the website (http://imagejdocu.tudor.lu/author/rkd8/).
2.3. Studying neuritic and somatic autophagy in culture models of Parkinsonian injury
The conversion of cytosolic LC3-I to LC3-II, which is covalently bound to membranes of AVs,is considered to be the most specific marker of autophagy to date (Klionsky et al., 2008). As illustrated elsewhere in this volume, the repertoire of available tools used to measure autophagy in cells includes counting LC3 puncta, measuring conversion of LC3-I to LC3-II by western blot, electron microscopy, and lysosomal markers for later stages of maturation.
2.3.1. Fluorescent markers of autophagic vacuoles
Visualization of GFP-LC3 puncta has become popular due to its technical simplicity, convenience, and reliability, though potential caveats relating to GFP aggregation and/or autophagy induced by transfection-related injury need to be considered (Klionsky et al., 2008). The average number of GFP-LC3 puncta per cell is frequently used to quantify autophagic activity in the soma. In neurites, determining the number of axonal GFP-LC3 puncta per μm length requires high-resolution, high-magnification epifluorescence imaging, as autophagic puncta in neurites are generally smaller than those observed in the soma and fluoresce to a lesser degree (Fig. 11.3).
Figure 11.3.

Features of the GFP-LC3 macro. A representative image of four SH-SY5Y cells transfected with GFP-LC3 and loaded with MitoTracker dye to label mitochondria. Cells were treated with 6-hydroxydopamine for four hours to induce autophagy (A). The cell of interest is traced using the polygonal tracing tool of ImageJ for analysis. The green channel is then extracted from the RGB image and manually thresholded to trace the majority of the somatic GFP-LC3 puncta (colored red within the grey region of interest (B). The macro then traces the thresholded GFP-LC3 puncta by employing the Outlines algorithm (C) and computes the total number of GFP-LC3 puncta analyzed in the field, the average GFP-LC3 puncta size (μm), and percent area occupied by GFP-LC3 puncta. Notice that cytosolic and nuclear GFP-LC3 fluorescence, and background pixelation are not thresholded and are therefore excluded from analysis. (D) Representative neurite of a RA-differentiated SH-SY5Y cell transfected with GFP-LC3. Note that GFP-LC3 puncta are smaller and less numerous than somatic puncta. (E) The GFP-LC3 macro identifies neuritic GFP-LC3 puncta (colored red) for further analysis as described earlier.
Using the protocol described in section 2.2.1, approximately 10% of RA differentiated SH-SY5Y cells are transfected with GFP-LC3 using 0.1% lipofectamine in DMEM media containing 10% fetal bovine serum. If transfection is performed prior to RA differentiation, 25%–35% of differentiated cells will express GFP-LC3. These transfection efficiencies result in little neuritic overlap between cells, ideal for image-based counting of GFP-LC3 puncta, yet is sufficient for detection of a GFP-LC3-II shift as a biochemical measure of autophagy in transfected cells (Plowey et al., 2008) (see sections 2.3.2–2.3.3). In primary cultures, only 2%–5% of neurons are transfected with GFP-LC3 using Lipofectamine 2000. While this is sufficient for image analysis of GFP-LC3 puncta and the axodendritic arbor of individual neurons, transgenic mice or viral transduction methods may be necessary for biochemical studies of autophagy in genetically altered primary neurons.
We have also constructed stable cell lines of GFP-LC3-expressing SH-SY5Y cells (Dagda et al., 2008), which can be RA differentiated and maintain the same pattern of autophagic responses as the parent line. While these cells are useful for somatic studies or biochemical analysis, they are not as amenable to quantitative image analysis of neuritic autophagy due to extensive overlap of fluorescent structures. If stable cell lines are used, multiple clones should be studied, as the stable clones vary with regard to the degree of neuronal versus fibroblastic versus epithelioid differentiation.
In terms of studying early versus late AVs, combined studies using GFP-LC3 and LC3 N-terminally fused to monomeric red fluorescent protein (RFP-LC3) may be needed. Neurons show higher basal levels of RFP-LC3 puncta per cell than seen with GFP-LC3, probably because the RFP moiety is more stable to lysosomal conditions (Kimura et al., 2007) Other markers to analyze later stages of autophagy include monodansylcadaverine (MDC) (50 μM final concentration in media, SIGMA, St. Louis, MO) or Lyso-Tracker Red DND-99 (100 nM, Molecular Probes, Eugene, CA) (also see the chapter by Vázquez and Colombo in volume 452). Briefly, we use the following protocol:
10 mM working stocks of MDC in DMSO is added directly onto cells in medium (50 μM final MDC concentration) and incubated for 30 min at 37°C, 5% CO2.
Cells are washed with PBS containing glucose prior to imaging of MDC-labeled vacuoles.
To visualize lysosomes, a 1 mM stock of LysoTracker Red DND-99 in DMSO is prepared in medium (100 nM, or a 10,000X dilution from 1 mM stocks). The medium of cells is replaced with medium containing LysoTracker Red DND-99 and cells are incubated for 30 min at 37 °C in a 5% CO2 tissue culture incubator.
The medium is refreshed with DMEM/FBS prior to imaging.
MDC-labeled structures colocalize with lysosomes but not GFP-LC3 (Munafo and Colombo, 2001), consistent with observations that GFP-LC3 fluorescence is diminished in lysosomal environments (Kimura et al., 2007). MDC labels a subset of mCherry-LC3 puncta in vivo and does not label any structures not labeling with mCherry-LC3, indicating specificity for autolysosomes (Iwai-Kanai et al., 2008). A limitation is that the fluorescence of lysosomal markers tends to quench rapidly. The combined use of early and late AV markers such as GFP-LC3 and MDC form a useful complement in studying autophagic maturation (Chu, 2006).
2.3.2. Semiautomated GFP-LC3 macro for measuring autophagy in fluorescent images (Dagda et al., 2008)
Taking a similar approach that we used previously to create a MetaMorph journal to analyze the size distribution of MDC-labeled autolysosomes (Zhu et al., 2007), we have made an NIH ImageJ macro called GFP-LC3 available for download (http://imagejdocu.tudor.lu/author/rkd8/), which performs automated quantification of the number and size of GFP-LC3 puncta (Dagda et al., 2008) (Fig. 11.3). It is important to analyze GFP-LC3 puncta only from high-resolution, high-quality (low autofluorescence) TIFF images captured at 60X–90X magnification when using this macro. Images captured at 40X magnification are inadequate, as the decreased resolution of GFP-LC3 puncta in these images will result in identification of overlapping or clustered puncta as a single large particle by the macro. This is particularly evident in somatic measures of puncta, where clustering is more frequent.
Prior to using this macro, it is critical to assign the correct scale (pixels/μm) of the epifluorescence micrograph images being analyzed, using the Set Scale command found in the Analyze pull-down menu. The macro has been optimized to measure specific GFP-LC3 puncta larger than 0.2 but less than 10 μm, a size range that is consistent with phagophores and autophagosomes while excluding background pixels and potential nuclear GFP fluorescence from the analysis. The macro performs the following commands:
First, the macro allows the user time to select the region of interest or the cell to be analyzed using the polygon selection tool. The macro then splits the RGB image into individual channels (blue, green, and red), closes the blue and red channels, and extracts the green channel to grayscale followed by photographic inversion (GFP fluorescence converted to black pixels over a white background).
-
The macro will allow the user time to assign the regions of interest to be measured by manually thresholding for minimal and maximal pixel values using the Threshold function under the Image pull-down menu, Adjust submenu (Fig. 11.3A–C). Consistency in the application of thresholding across cells is important. A threshold value of two standard deviations from the background fluorescence is enough to trace the majority of GFP-LC3 puncta.
Please note that GFP-LC3 puncta in axons and dendrites must be thresholded and measured independently of somatic GFP-LC3 puncta for each neuron to be analyzed. This is due to the fact that GFP-LC3 puncta in neurites are smaller and less bright, and cannot be detected using thresholds set according to somatic puncta.
Once pixels are thresholded, the macro employs the Measure Particles algorithm to record GFP-LC3 puncta number, area, size (expressed as radii length in μm, calculated using the formula r = square root (area/π)), and fraction of cellular area occupied by GFP-LC3 puncta. Results are displayed in the Results window and can be transferred to an Excel spreadsheet using the copy and paste functions of ImageJ. The macro is conveniently looped to restart as many times as necessary to allow the user to analyze multiple neurons in the same field without closing the image being analyzed. This macro can also be adjusted to quantify number and area occupied by other organelles such as mitochondria (Dagda et al., 2008).
2.3.3. LC3 shift Western blot
As the analysis of LC3 gel shift has been extensively reviewed (Mizushima and Yoshimori, 2007) (see also the chapter by Kimura et al., in volume 452), this section will focus on highlighting considerations for transfected cells or neurons. The lipidated form of LC3 (LC3-II) exhibits faster electrophoretic mobility by SDS-PAGE compared to LC3-I, and the LC3-II/LC3-I ratio is a measure of steady state autophagosome content in cells. Some suggest that the LC3-II/β-actin ratio may be more reliable, as LC3-I expression can be transcriptionally regulated and there is variability in how well LC3-I is recognized by different antibodies (reviewed in (Klionsky et al., 2008)).
Because neurons typically show reduced transfection efficiency compared to other cell types, analysis of genetic models of neurodegeneration by western blot may be limited to virally transduced or stable cell lines. In transiently transfected differentiated cell lines, however, a disease associated mutant protein can be cotransfected with GFP-LC3 (see section 2.5). The GFP tagged LC3-I and GFP-LC3-II bands can be detected by immunoblotting with a polyclonal rabbit anti-GFP IgG (1:5000; Invitrogen, Carlsbad, CA) (Plowey et al., 2008). This GFP-LC3-II shift is a biochemical measure of autophagy in the transfected subpopulation, and is inhibited by RNAi knockdown of Atg7 (Plowey et al., 2008).
2.3.4. Measurements of autophagic flux
To measure autophagic flux, as compared to steady-state levels of AVs, LC3-II turnover or GFP-LC3 puncta are measured at different time points in the presence and absence of bafilomycin A1 (10 nM for SH-SY5Y cells), which inhibits lysosomal acidification and fusion of autophagosomes with the lysosome, or with cell permeable lysosomal protease inhibitors E64-D (40 μM for SH-SY5Y cells) and pepstatin (25 μM for SH-SY5Y cells). For in vivo studies of autophagic flux, chloroquine has been employed to block degradation (Iwai-Kanai et al., 2008). The fraction of AVs that is elevated in response to the presence of fusion/degradation inhibitors represents an estimate of flux, but the length of the bafilomycin treatment must be titrated to ensure that the assay is not approaching saturation under basal control conditions. Autophagic flux in axons and neurites is more difficult but may be possible using the image-based methods of sections 2.3.1–2.3.2 for quantifying GFP-LC3 puncta in the presence or absence of degradation inhibitors. The additional factor of AV trafficking into and out of the region of quantification would also need to be considered. Clearly, additional methods for studying autophagic flux in neuronal cells are needed both in vitro and in vivo.
2.4. Methods for measuring mitochondrial autophagy
Mitochondria undergo selective autophagic degradation in a process called mitophagy (Kissova et al., 2004; Rodriguez-Enriquez et al., 2004). In neurons, autophagy is critical for the turnover of aged or subtly impaired mitochondria (Kiselyov et al., 2007), whereas pathological mechanisms may cause excess degradation of neuronal mitochondria (Xue et al., 2001; Zhu et al., 2007). To measure the effects of environmental or genetic factors on neuronal mitophagy, one can employ both image-based and biochemical techniques.
2.4.1. Image-based methods for analyzing mitophagy (Dagda et al., 2008)
To analyze for mitophagy using GFP-LC3, RA differentiated SH-SY5Y cells are prepared on uncoated chambered Lab-tek II cover glasses. Cover glasses coated with 100 μg/ml per well of poly-D-lysine (SIGMA, St. Louis, MO) are used to culture primary neurons.
The cultures are transiently transfected with GFP-LC3 (1 μg of DNA in 0.10% Lipofectamine) in neurobasal media as described in sections 2.2.1 and 2.3.
To visualize mitochondria, GFP-LC3 expressing cultures are labeled with 100 nM MitoTracker Red dye 580 (diluted from 1 mM stocks in DMSO directly into medium overlying cells) (Molecular Probes, Eugene, CA) or 200 nM tetramethylrhodamine methyl ester (TMRM) (mixed into media to create a working stock that is used to replace the growth media) (Sigma, St. Louis, MO) for at least 30 min in 37 °C, 5% CO2.
The cultures are washed with warm medium prior to imaging mitochondria using an epifluorescence or laser confocal microscope.
Alternatively, cells can be cotransfected with a mitochondrially targeted monomeric RFP (mito-RFP) to visualize mitochondria. A word of caution is that transient overexpression of inner mitochondrial membrane targeted RFP may result in cell injury/increased basal autophagy/mitophagy in neurons (Dagda & Chu, unpublished observations). Statistical significance for measuring effects of an environmental or genetic factor on mitophagy can be achieved by analyzing high quality images captured at a high magnification (60X), taking images from at least 25 cells per condition (Dagda & Chu, unpublished observations). Mitophagy in axons is expressed as the percent of GFP-LC3 or RFP-LC3 puncta that colocalize with mitochondria within a given unit length.
Late stages of mitophagy (mitochondria undergoing lysosomal degradation) are followed by delivery of mitochondria to lysosomes. To this end, neurons can be colabeled with 250 nM MitoTracker Green FM dye (Molecular Probes, Eugene, CA) or transiently transfected with mitochondrially targeted GFP (mito-GFP) to visualize mitochondria and loaded with LysoTracker Red DND-99 (Molecular Probes, Eugene, CA) to label lysosomes followed by a wash with warm media. To determine whether lysosomal degradation of mitochondria depends on autophagy, mitophagy is analyzed in neurons treated with bafilomycin A (cannot be used with LysoTracker Red) or RNAi targeting the autophagic machinery (Zhu et al., 2007) (See section 2.6, below). The use of 3-methyladenine (3-MA) to inhibit autophagy is not optimal for this kind of analysis as not all forms of mitophagy are dependent on Beclin 1/class 3 phosphoinositide 3-kinase signaling (Chu et al., 2007).
2.4.2. Western blot of mitochondrial proteins
Mitophagy can be analyzed by quantifying cellular levels of mitochondrial proteins by immunoblotting in the presence or absence of autophagy inhibitors or autophagy-related gene (Atg) RNAi (Dagda et al., 2008). Depending on the time frame and context of the experiment, mitochondrial biogenesis may or may not play a confounding role to be considered. For example, a study of the Parkinsonian toxin MPP+ used immunofluorescence and Western blot analysis of proteins residing in different compartments of the mitochondria, and the effects of MAP kinase inhibitors were confirmed using electron microscopy (Zhu et al., 2007). The 60-kDa mitochondrial complex IV protein and p110 mitochondrial membrane antigen antibodies can also be used to stain mitochondria in paraffin embedded human brain tissues (Zhu et al., 2003). Intermembrane space proteins such as cytochrome c or apoptosis inducing factor are less reliable as they are lost from mitochondria that have undergone permeability transition during apoptosis (Chu et al., 2005).
2.5. Autophagy in genetic models of Parkinson’s disease
In addition to toxin models of parkinsonian injury (Dagda et al., 2008; Zhu et al., 2007), we found that autophagy actively contributes to the neurodegenerative phenotype in a genetic model of Parkinson’s disease based on mutation in the LRRK2 gene (Plowey et al., 2008). Mutations in LRRK2 have been detected in familial and apparently sporadic cases of parkinsonism (Kay et al., 2006). The protocols described herein would be generally applicable to other genetic models of neurological diseases that use transient transfection to achieve expression of a disease-associated mutant protein.
2.5.1. Optimizing cotransfection conditions (copy ratio)
To measure autophagy in mutant-LRRK2 transfected cells, we cotransfect the pEGFP-LC3 plasmid with either the wild type full-length HA-tagged LRRK2 cDNA subcloned into the pcDNA 3.1 vector backbone or one of several mutant LRRK2 constructs. Following media refreshment, we modify the basic transfection protocol in Section 2.2.1 to cotransfect the pEGFP-LC3 and pcDNA3-LRRK2 plasmids in a 1:15 mass ratio (MacLeod et al., 2006) (equivalent to a 1:4 molar ratio) in OptiMEM medium (Invitrogen) (5% of culture volume) mixed 1:1 with 2% Lipofectamine prepared separately in the same volume (final lipofectamine concentration of 0.1%). These conditions yield an overall transfection rate of 10%–15% and a 75%–90% coexpression rate as determined by immunolabeling for HA and GFP.
2.5.1.1. Visualization of GFP-LC3 and LRRK2 co-transfected cells (Plowey et al., 2008)
Following 2 days of expression after transfection, the cells are briefly and gently rinsed with warmed Dulbecco’s Phosphate Buffered Saline (DPBS) (Invitrogen).
After aspiration of washing medium, 3.9% paraformaldehyde in phosphate buffered saline [137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4·7H2O, and 1.4 mM KH2PO4] (PFA) is added to the wells, and the cells are incubated for 20 min at room temperature.
The fixed cells are permeabilized by washing in PBST (PBS containing 0.1% Triton X-100) for 15 min on a rocker at room temperature.
LRRK2-transfected cells are incubated overnight at 4 °C on a rocker table with a mouse monoclonal HA-tag primary IgG (1:1000 dilution; Covance, Emeryville, CA, USA). The cells are then brought to room temperature for 30 min, washed 4 times with PBST for 5 min each, followed by incubation for 1 h at room temperature on a rocker with a polyclonal Cy3-labeled donkey antimouse IgG (1:1000 dilution; Jackson Immunolabs).
The cells are then washed twice in PBST and twice in PBS. Nuclei are counterstained in a 1:500 dilution of DAPI in PBS (2.5 mg/ml stock solution in DMSO stored at −20 °C) for 5 min. The cells are then washed 3 times for 5 min each with PBS prior to imaging. Fifty random 60X objective oil-immersion images of LRRK2-positive cells, or in the case of vector transfected cells, 50 random images of GFP-LC3 transfected cells are digitally captured for data analysis.
2.5.2. Quantitative analysis of somatic and neuritic autophagy
The basic methods of identification and quantification of GFP-LC3 puncta in mutant-LRRK2 cotransfected cells are similar to the threshold-based ImageJ techniques described for toxin models in section 2.3.2. Because of the diffuse nature of GFP-LC3-I distribution in cells, neurites are well highlighted in these cells and the brighter puncta are well visualized against this background (Plowey et al., 2008). Cotransfection of mutant LRRK2 plasmid with pEGFP-C1 is used as a control to ensure that the mutant plasmid does not induce puncta through aggregation of GFP, and no protein aggregates are observed by electron microscopy. We find no significant differences in mean neurite lengths as measured in pEGFP-C1 versus GFP-LC3 transfected cells.
With mutant LRRK2 expression, we see significant increases in the numbers of AVs per neurite, the number of AVs per unit length of neurite, the number of AVs per cell body and the percent cytoplasmic area of the cell body occupied by GFP-LC3-labeled AVs (Plowey et al., 2008). The latter parameter reflects the observation that somatic AVs are increased in number as well as size, whereas neuritic AVs are small and increased in number alone. Because only 10% of cells express LRRK2 constructs, the standard LC3 mobility shift assay is not sensitive enough to detect increased autophagy in the transfected cell population among a background of untransfected cells. However, we use GFP immunoblotting to detect a GFP-LC3 mobility shift indicative of increased autophagy in LRRK2-co-expressing cells as described in Section 2.3.3.
2.5.3. Special considerations for protein aggregation disease
A recent study demonstrated that cytosolic protein aggregates can be labeled with GFP-LC3 even in the absence of progression to autophagy (Kuma et al., 2007). This phenomenon has the potential to be misinterpreted. Although the authors note differences between transient and stable transfectants, the phenomenon is most likely related to levels of overexpression or using high levels of lipofectamine rather than representing an intrinsic advantage of stable lines, which can be confounded by clonal selection bias for cells equipped to survive chronic expression of a toxic gene product. As lipofectamine-related GFP-LC3 puncta are typically transient (Klionsky et al., 2008), including appropriate time-matched transfection controls or verification of findings using other methods are recommended. Use of lipidation-deficient mutants of GFP-LC3 (Tanida et al., 2008), or demonstration that the observed increases in GFP-LC3 puncta are inhibited by siRNA knockdown of core autophagy machinery proteins, or confirmatory transmission electron microscopy (TEM) studies may be used to verify that the observed increases in GFP-LC3 puncta correspond to increases in AVs (Plowey et al., 2008).
2.6. Autophagy modulation using RNA interference against Atg proteins
Because short double-stranded siRNAs are effectively transfected into >90% of differentiated neuronal cells (Plowey et al., 2008), standard Western blot analysis can be used to assess efficacy of RNAi knockdown at the time that toxin is administered, or the disease-related genes are expressed, and for the duration of the injury (Zhu et al., 2007). Note that increased plating density, higher lipofectamine concentrations and/or use of antibiotics in the maintenance of cells may all serve to reduce siRNA transfection efficiency, potentially due to enhanced cytotoxicity. We typically maintain lines under antibiotic-free conditions, as antibiotics can impair mitochondrial metabolism or mask suboptimal sterile technique, increasing the potential for mycoplasma contamination. Even when the population of interest reflects a smaller percentage of randomly distributed cells transfected to initiate injury a few days after RNAi, as in our study of LRRK2 (G2019S)-induced neurite remodeling, standard western blot analysis can still be used to assess efficacy of the siRNA. Control experiments using fluorescently labeled dsRNA confirm that siRNA remains uniformly distributed in >90% of the smaller population of cells expressing HA-tagged LRRK2 (G2019S) (Plowey et al., 2008). Thus, there is no evidence of preferential transfection of cells depending upon RNAi status, nor that subsequent DNA plasmid transfection might affect the stability of the siRNA. Alternatively, the effects of Atg7 siRNA on autophagy can be monitored specifically in transfected cells by co-transfecting mutant LRRK2 with GFP-LC3 and following the inhibition of the GFP-LC3-II shift as described above (Plowey et al., 2008).
Interpretation of 3-MA in the context of neurite remodeling is complex as 3-MA (5 mM) effectively inhibits Akt phosphorylation, an important neuronal prosurvival factor, as well as several other death regulatory protein kinases (Xue et al., 1999; Zhu & Chu, unpublished observation). The DMF or DMSO concentrations necessary to achieve 5–10 mM doses of this drug that are typically used in autophagy studies must also be considered. Moreover, phosphoinositide 3-kinase signaling plays an important role in regulating neurite outgrowth (Kimura et al., 1994), potentially by preventing the inhibition of anterograde axonal transport by glycogen synthase kinase 3 (Pigino et al., 2003). These effects would confound the analysis of experiments seeking to inhibit autophagy using pharmacological methods.
We highlight herein a general timeline for using siRNA to study the role of autophagy during neurite remodeling induced by transient expression of a disease-causing mutant protein. During the entire timeline, cells are maintained in 10% fetal bovine serum DMEM containing retinoic acid (10 μM). Western blot is used to study efficacy of protein knockdown, and the techniques described previously to confirm reduction of injury-induced neuritic GFP-LC3 puncta, prior to analysis of neurite length and complexity.
Cells are plated on day 1 in DMEM/FBS/RA, and transfected with siRNA targeting either Atg7 or LC3 using 0.1% lipofectamine on day 2. On day 3, the DMEM/FBS/RA media is refreshed. On day 4, the insult being studied is applied in DMEM/FBS/RA (e.g., neurotoxin or transfection with the mutant gene of interest; typically cotransfection with a fluorescent marker is used to delineate neuritic processes of the transfected subpopulation). On day 6, the cells are fixed with paraformaldehyde for GFP-LC3 imaging and neurites are measured as described in sections 2.5.1–2.5.2. The effects of Atg7 knockdown on autophagy are confirmed by monitoring GFP-LC3 puncta or GFP-LC3 gel shift, while MDC is used to confirm effects of reduced LC3 expression on autophagy.
2.7. Use of GFP-LC3 transgenic mice for primary neuron culture studies of autophagy
To develop sensitive and specific tools for the detection and quantification of autophagy in primary cortical neurons, our initial studies employed the classic stimulus of nutrient deprivation. Primary neurons from rats or mice are cultured using standard methods (Du et al., 2004). Nutrient deprivation is induced by replacing standard culture media with custom media lacking D-glucose, sodium pyruvate, L-glutamine, L-glutamate, and L-aspartate, and without the fatty acid–containing supplement B27 (Gibco). Neurons can be cultured in 96-well plates for rapid-throughput analysis of the effect of pharmacological or molecular interventions using the 3-[4,5-dimethyl thiazol]-2,5-diphenyltetrazolium bromide (MTT) assay, on larger plates or dishes for analysis of proteins by Western blot (Du et al., 2004), or on glass chamber slide coverslips for immunofluorescence analysis (Zhu et al., 2007). We have used primary cortical neurons from GFP-LC3+/− transgenic reporter mice, identified at birth using a headset equipped for epifluorescent illumination, to track dynamic formation of autophagosomes under conditions of nutrient deprivation and following in vitro stretch-induced traumatic injury (Fig. 11.4). The fluorescent signal can be enhanced using immunolabeling with anti-GFP antibody, or visualized directly for live cell assays.
Figure 11.4.

Detecting autophagy in neurons during nutrient deprivation and after dynamic stretch-induced injury in vitro. Primary cortical neurons from GFP-LC3+/−transgenic mice were subjected to nutrient deprivation (media without glucose, pyruvate, glutamate, glycine, aspartate, or fatty acids) or dynamic stretch (4.7 psi × 100 ms, ~50% strain). Autophagosomes are identified by discrete, high fluorescent intensity, punctuate labeling (arrows).
3. Studying Brain Autophagy In Vivo
3.1. Models of traumatic brain injury
Although the role for autophagy has yet to be established, it is clear that autophagy is increased in brains after acute traumatic injury. Upregulation of beclin 1 was reported within injured hippocampal and cortical neurons after closed head injury in mice (Diskin et al., 2005). We recently reported increased LC3-II and formation of autophagosomes and secondary lysosomes after focal traumatic brain injury (TBI) in mice (Lai et al., 2008). In addition, increased formation of Atg12-5 conjugates is observed after fluid percussion injury in rats (Liu et al., 2008).
For our studies, adult male C57BL/6J mice are subjected to moderate-severe TBI to the left parietal cortex using a controlled cortical impact device (Satchell et al., 2003). For ultrastructural studies, mice are perfused with 2% paraformaldehyde/0.01% glutaraldehyde in phosphate buffered saline (PBS) and prepared for electron microscopy using standard methods (Lai et al., 2008). In control mice, ultrastructural evidence for autophagy is rarely detected; however, observation of normal brain tissue raises an important caveat relevant specifically to brain tissue. Researchers should be aware that neurites cut in cross section within brain tissues often appear as double membrane, organelle and/or vesicle containing, structures (examples in Figs. 11.5A–B). Autophagosomes can therefore be difficult to discriminate from cell processes in coronal section with dissolution of intralumenal contents. Identification of true autophagosomes requires criterion based on size (generally 400–1000 nm in diameter) and lack of synaptic clefts or myelin sheaths. Location within a larger, clearly defined profile representing a dendrite or myelinated axon also helps (examples in Figs. 11.5C–D). Autophagosomes with double membranes containing cellular debris and multilamellar bodies are readily detectible in the injured cortex, hippocampus, and thalamus 2 h and 24 h after TBI. Interestingly, after TBI, AVs are most prominent in cell processes and axons, raising the possibility that autophagy may be involved in axonal damage and/or dendritic pruning (or play roles in glial processes) after TBI, consistent with autophagy-related neurite degeneration described in other injuries (Plowey et al., 2008; Yang et al., 2007).
Figure 11.5.
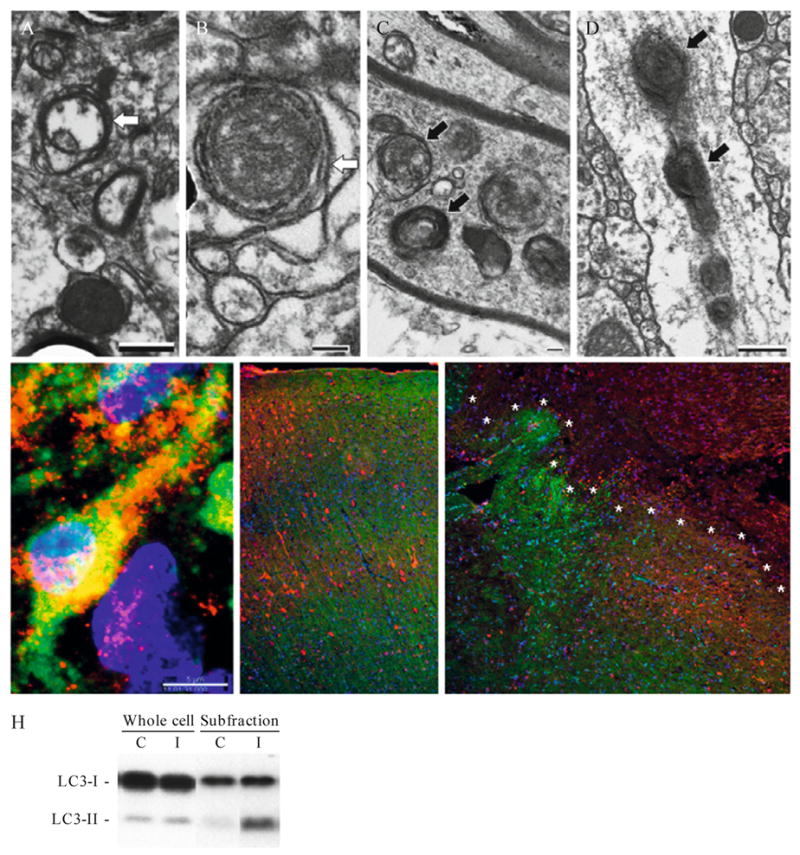
Detecting autophagy after traumatic brain injury (TBI) in vivo. (A and B) Transmission EM from naive mice showing double-membrane structures likely representing neurites in cross section (white arrows). (C and D) Transmission EM from mice 24 h after TBI showing secondary lysosomes and autophagosomes within axons and dendrites (black arrows). (E) Confocal immunohistochemical labeling for LC3 (red), Fluorojade C (green) and DAPI (blue) in a male PND 17 rat 24 h after TBI. Punctate LC3 labeling is shown in a Flurojade-C-positive neuron in the CA3 hippocampus. (F and G) Immunohistochemical labeling for GFP in GFP-LC3+/− mice. Note the loss of green fluorescence but retention of immunolabeling for GFP (red) in the injured region (demarcated by asterisks) from a mouse 24 h after TBI (G) compared to the control (F). (H) Western blot for LC3 in brain tissue. LC3-II is enriched in the P2 subfraction vs. the whole cell lysate (C = control, I = injured).
Autophagosomes in brain can be evaluated using confocal immunohistochemistry and antibodies against LC3. Because current antibodies identify both LC3-I and -II, discrimination relies on identification of discrete, punctate labeling. Again, the caveat of misidentifying LC3-I enriched neurites cut in cross-section applies. Nonetheless, using stringent criteria perhaps with the addition of other labels, such as Fluorojade C, which labels degenerating neurons, one can track autophagy by immunofluorescence in brain with a reasonable degree of confidence. The example shown in Fig. 11.5E is an ipsilateral CA3 hippocampal neuron 24 h after TBI in a postnatal day 17 rat immunostained for LC3 (red) and colabeled with Fluorojade C (green) and DAPI (blue). Punctate LC3 labeling within degenerating neurons in vulnerable regions after TBI is suggestive of autophagosome formation, and supports a role for autophagic stress in acute neurodegenerative diseases such as TBI. Due to fluorescence overlap, biomarkers other than Fluorojade C are required for colabeling of autophagy in dying or stressed cells when examining tissues from GFP-LC3 transgenic mice, unless one has access to a microscope equipped with a spectral analyzer.
One can also use GFP-LC3+/− mice to track autophagy in brain; however, we have found this to be more complicated than use in vitro. First, the endogenous GFP signal appears less robust in brain tissue sections (10 micron thick), and typically needs to be enhanced using anti-GFP immunofluorescence. The second, and perhaps more important reason, is that after TBI the milieu within injured brain tissue appears to quench the endogenous GFP signal. Shown in Fig. 11.5F–G are coronal brain sections from normal and injured GFP-LC3+/− mice, respectively. The sections were colabeled using an antibody against GFP (red) to demonstrate that the lack of green fluorescence was not due to a lack of GFP. The quenching of GFP in injured tissue may be related either to increased oxidative stress and/or reduced pH, both of which are known to occur in damaged tissue after TBI.
An LC3 Western gel shift can be used to detect and semiquantify autophagy after TBI. For Western blot examination of LC3-II after TBI, mice are perfused with ice-cold saline. The left dorsal hippocampus and overlying cortex are dissected and processed for subcellular fractionation as described previously (Lai et al., 2008). We find in cultured neurons and after in vivo TBI that LC3-II is concentrated in the mitochondria-enriched, P2 fraction (Fig. 11.5H). TBI results in increased LC3-II at 2, 24, and 48 h, peaking at 24 h, versus control mice (Lai et al., 2008). Similar subfractionation techniques are also used by other investigators to detect changes in LC3-II and Atg12-5 conjugates in rats after TBI (Liu et al., 2008).
3.1.1. Western blot analysis for LC3-II in vivo (Lai et al., 2008)
Animals are anesthetized then transcardially perfused with ice-cold saline.
Brains are removed and regions of interest are dissected, placed in microcentrifuge tubes and homogenized in 10 volumes of lysis buffer (20 mM HEPES-KOH, pH 7.4, 10 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 250 mM sucrose, 1 μM DTT, 1 mM PMSF, 2 μg/ml aprotinin).
Lysates are centrifuged at 1025×g for 15 min at 4 °C to pellet nuclei (P1). The S1 supernatant fractions are centrifuged at 735×g at 4 °C for 10 min; the resulting supernatant fractions are further centrifuged at 10,000×g at 4 °C for 15 min to pellet autophagosomes, mitochondria, and small organelles (P2). Samples are stored at −80 °C in 10% glycerol.
Proteins are loaded into 15% acrylamide gels (30 μg/well) and separated electrophoretically, then transferred to a polyvinyl difluoride membrane overnight. The transferred membranes are incubated in a 1:1000 dilution of a monoclonal antibody that recognizes both LC3-I and -II (Clone 51-11; MBL International, Woburn, MA) at room temperature for 1 h. Membranes are washed in PBS containing 0.1% Tween 20, then incubated in the appropriate secondary antibody (1:3000) for 1 h, incubated in chemiluminescence reagents, and exposed to X-ray film.
The relative optical densities for LC3-I and LC3-II can be semiquantified using standard image analysis. Membranes are stripped and probed for cytochrome oxidase as a loading control for the P2 subfraction.
3.1.2. Is autophagy increased after TBI in humans?
To begin to address the question, we performed LC3 and beclin 1 Western blot analysis on brain tissue samples obtained from 5 adult patients resected for management of refractory intracranial hypertension after TBI (Clark et al., 2008). We also examined control brain tissue from 5 adult patients dying of causes unrelated to CNS trauma. An LC3 shift could be detected in 4/5 TBI and 3/5 control patients. Signal intensity for LC3-II was less robust than that observed in cultured neurons or experimental TBI, perhaps related to the fact that whole-cell lysates were used, since subfractionated samples were not available. Punctate LC3 labeling consistent with autophagy was observed immunohistochemically. Autophagy has also been demonstrated in brain tissue from patients with Parkinson, diffuse Lewy body, Alzheimer, and Creutzfeldt-Jakob diseases (Anglade et al., 1997; Nixon et al., 2005; Sikorska et al., 2004; Zhu et al., 2003). The data suggest that autophagy occurs in adult human brain during a variety of diseases, including TBI, warranting further study.
3.2. Models of ischemic brain injury
Ischemic brain injury is either focal or global. Focal ischemic injury results from occlusion of one or more cerebral vessels as occurs in stroke. Focal ischemia is further divided into permanent or temporary. Temporary ischemia occurs when an occluded vessel regains patency (from thrombolytic therapy or percutaneous manipulation). Global injury is typically the result of complete cessation of total cerebral circulation as occurs in cardiac arrest, caused by asphyxia, electrically induced ventricular fibrillation, compression of the great vessels leaving the heart, or chemical cardiac paralysis using potassium (animal models of cerebral ischemia are reviewed in Durukan and Tatlisumak, 2007; Ginsberg and Busto, 1989; Kim, 1997; Traystman, 2003). Total cerebral ischemia secondary to cardiac arrest is an insult shared by both the brain and other major organs. Thus, cardiac arrest models more closely resemble clinical events, but important caveats include the need to consider effects of multisystem organ dysfunction and extracerebral toxins on progression of brain injury.
3.2.1. Autophagy in cerebral hypoxic-ischemic injury
In 1995, Nitatori et al., found evidence of autophagy using electron microscopy in gerbils treated with temporary bilateral occlusion of the common carotid arteries. The next contribution to this field did not occur until 10 years later when increased LC3-II was reported following transient middle cerebral artery occlusion in mice (Degterev et al., 2005) and in a mouse model of neonatal cerebral hypoxic ischemia (Zhu et al., 2005, 2006). GFP-LC3 transgenic mice subjected to unilateral common carotid artery occlusion plus hypoxia (Levine/Vanucci procedure) (Adhami et al., 2006) exhibit increased immunofluorescence with punctuate redistribution consistent with autophagy, autophagy-like vacuoles by EM and decreased cytoplasmic LC3-I by Western blot. Rats exposed to middle cerebral artery occlusion for 90 min (Rami et al., 2008) exhibit increases in beclin 1 expression at 6–48 h following ischemia by Western blot and immunohistochemistry, with partial co-localization of caspase-3 and Beclin 1 and LC3 redistribution and colocalization with beclin 1 in ischemic neurons. Our unpublished data confirm that detection of LC3-II in models of global cerebral ischemia is enhanced by examination of the P2 fraction as described in section 3.1.1.
3.2.2. Causality, the next frontier of in vivo autophagy studies
The preceding studies clearly demonstrate that autophagy is invoked following cerebral ischemia. However, whether autophagy is playing a protective or harmful role cannot be inferred from these studies. Studies demonstrating that chronic inhibition of constitutive autophagy (e.g., in Atg knockout mice) creates a neurodegenerative phenotype suggest that inhibition of autophagy might exacerbate hypoxic ischemic injury. In contrast, other studies show that inhibition of autophagy may be protective following hypoxic ischemic injury, implicating excessive autophagy as a mechanism promoting cell death. Neonatal hypoxia-ischemia in mice elicits an increase in LC3-II (Western blot and immunohistochemistry) and formation of autophagosomes (electron microscopy) (Koike et al., 2008). As expected, the same insult in Atg7 knockout mice does not show features of autophagy. More important, the Atg7 knockout mice show dramatically less neuronal death. Atg7-deficient mice also show decreased caspase-dependent neuronal death further supporting a link between autophagy and regulation of apoptosis. Thus, at least in immature rodents, autophagy appears to exacerbate hypoxic-ischemic neuronal cell death.
4. Future Perspectives and Challenges
Evidence supporting a role for autophagy in traumatic, hypoxic-ischemic, and neurodegenerative brain disorders is just emerging and necessarily incomplete. Additional tools, such as specific inhibitors of autophagy and better methods to measure autophagic flux are needed to advance this field. The mechanism of autophagy-dependent regulation of apoptosis and the relationship of autophagic processes with neurite remodeling, synaptic (dys)function and regenerative biosynthetic processes will also be important to characterize (Cherra and Chu, 2008). This is highlighted by the prominence of autophagosomes and dysfunction in neurites after TBI—an acute neurodegenerative disease—as well as in Alzheimer’s disease and in neurotoxin and mutant LRRK2-mediated models of Parkinson’s disease. Finally, improved nonbiased, automated algorithms for image-based quantification of neuritic health and complexity are needed. The development of new brainbow mice expressing different fluorophores in adjacent neurons (Livet et al., 2007) coupled with advances in two photon imaging (St. Croix et al., 2006) may pave the way toward visualizing the axodendritic arbor of individual neurons for studies of autophagy and neurodegeneration in vivo.
Acknowledgments
We thank Jason Callio for technical assistance and maintenance of transgenic mouse colonies. The authors are supported by funding from the National Institutes of Health (R01 AG026389, R21 NS053777, K18 DC009120 to CTC; F32 AG030821 to RKD; K08 HD040848 to RWH; R01 NS038620 to RSBC).
References
- Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, Davis RJ, Mizushima N, et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- Bilsland J, Roy S, Xanthoudakis S, Nicholson DW, Han Y, Grimm E, Hefti F, Harper SJ. Caspase inhibitors attenuate 1-methyl-4-phenylpyridinium toxicity in primary cultures of mesencephalic dopaminergic neurons. J Neurosci. 2002;22:2637–2649. doi: 10.1523/JNEUROSCI.22-07-02637.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Nixon RA. Neuronal macroautophagy: From development to degeneration. Mol Aspects Med. 2006;27:503–519. doi: 10.1016/j.mam.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Charych EI, Akum BF, Goldberg JS, Jornsten RJ, Rongo C, Zheng JQ, Firestein BL. Activity-independent regulation of dendrite patterning by post-synaptic density protein PSD-95. J Neurosci. 2006;26:10164–10176. doi: 10.1523/JNEUROSCI.2379-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherra SJ, III, Chu CT. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008;3:309–323. doi: 10.2217/14796708.3.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Dossena S, Nowoslawski L, Roth KA, Ghetti B, Harris DA. Bax deletion prevents neuronal loss but not neurological symptoms in a transgenic model of inherited prion disease. Proc Natl Acad Sci USA. 2005;102:238–243. doi: 10.1073/pnas.0406173102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol. 2006;65:423–432. doi: 10.1097/01.jnen.0000229233.75253.be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: Implications for neurodegeneration and cell death. Autophagy. 2007;3:663–666. doi: 10.4161/auto.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Zhu JH, Cao G, Signore A, Wang S, Chen J. Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J Neurochem. 2005;94:1685–1695. doi: 10.1111/j.1471-4159.2005.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Bayir H, Chu CT, Alber SM, Kochanek PM, Watkins SC. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy. 2008;4:88–90. doi: 10.4161/auto.5173. [DOI] [PubMed] [Google Scholar]
- Clinton J, Forsyth C, Royston MC, Roberts GW. Synaptic degeneration is the primary neuropathological feature in prion disease: A preliminary study. Neuroreport. 1993;4:65–68. doi: 10.1097/00001756-199301000-00017. [DOI] [PubMed] [Google Scholar]
- Cookson MR, van der Brug M. Cell systems and the toxic mechanism(s) of α-synuclein. Exp Neurol. 2008;209:5–11. doi: 10.1016/j.expneurol.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress. Autophagy. 2008;4:770–782. doi: 10.4161/auto.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of non-apoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Di Filippo M, Tozzi A, Picconi B, Ghiglieri V, Calabresi P. Plastic abnormalities in experimental Huntington’s disease. Curr Opin Pharmacol. 2007;7:106–111. doi: 10.1016/j.coph.2006.08.010. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Diskin T, Tal-Or P, Erlich S, Mizrachy L, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Closed head injury induces upregulation of Beclin 1 at the cortical site of injury. J Neurotrauma. 2005;22:750–762. doi: 10.1089/neu.2005.22.750. [DOI] [PubMed] [Google Scholar]
- Dominguez R, Jalali C, de Lacalle S. Morphological effects of estrogen on cholinergic neurons in vitro involves activation of extracellular signal-regulated kinases. J Neurosci. 2004;24:982–990. doi: 10.1523/JNEUROSCI.2586-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, Graham SH, Clark RS. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem. 2004;279:38563–38570. doi: 10.1074/jbc.M405461200. [DOI] [PubMed] [Google Scholar]
- Durukan A, Tatlisumak T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav. 2007;87:179–197. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Cena V, Gallego C, Comella JX. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J Neurochem. 2000;75:991–1003. doi: 10.1046/j.1471-4159.2000.0750991.x. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Peters A, Barbas H. Mitochondrial degeneration in dystrophic neurites of senile plaques may lead to extracellular deposition of fine filaments. Brain Struct Funct. 2007;212:195–207. doi: 10.1007/s00429-007-0153-1. [DOI] [PubMed] [Google Scholar]
- Fournier JG. Cellular prion protein electron microscopy: Attempts/limits and clues to a synaptic trait. Implications in neurodegeneration process. Cell Tissue Res. 2008;332:1–11. doi: 10.1007/s00441-007-0565-5. [DOI] [PubMed] [Google Scholar]
- Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. β-amyloid precursor protein (βAPP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–144. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- Gerecke KM, Wyss JM, Carroll SL. Neuregulin-1β induces neurite extension and arborization in cultured hippocampal neurons. Mol Cell Neurosci. 2004;27:379–393. doi: 10.1016/j.mcn.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD, Busto R. Rodent models of cerebral ischemia. Stroke. 1989;20:1627–1642. doi: 10.1161/01.str.20.12.1627. [DOI] [PubMed] [Google Scholar]
- Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S. Dopamine induces autophagic cell death and α-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res. 2003;73:341–350. doi: 10.1002/jnr.10663. [DOI] [PubMed] [Google Scholar]
- Gong R, Park CS, Abbassi NR, Tang SJ. Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J Biol Chem. 2006;281:18802–18815. doi: 10.1074/jbc.M512524200. [DOI] [PubMed] [Google Scholar]
- Gutierrez H, Davies AM. A fast and accurate procedure for deriving the Sholl profile in quantitative studies of neuronal morphology. J Neurosci Methods. 2007;163:24–30. doi: 10.1016/j.jneumeth.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hynds DL, Takehana A, Inokuchi J, Snow DM. L- and D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) inhibit neurite outgrowth from SH-SY5Y cells. Neuroscience. 2002;114:731–744. doi: 10.1016/s0306-4522(02)00302-0. [DOI] [PubMed] [Google Scholar]
- Ironside JW. Prion diseases in man. J Pathol. 1998;186:227–234. doi: 10.1002/(SICI)1096-9896(1998110)186:3<227::AID-PATH174>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Ito U, Kuroiwa T, Nagasao J, Kawakami E, Oyanagi K. Temporal profiles of axon terminals, synapses and spines in the ischemic penumbra of the cerebral cortex: Ultrastructure of neuronal remodeling. Stroke. 2006;37:2134–2139. doi: 10.1161/01.STR.0000231875.96714.b1. [DOI] [PubMed] [Google Scholar]
- Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L, Gottlieb RA. A method to measure cardiac autophagic flux in vivo. Autophagy. 2008;4:322–329. doi: 10.4161/auto.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey M, Halliday WG, Bell J, Johnston AR, MacLeod NK, Ingham C, Sayers AR, Brown DA, Fraser JR. Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathol Appl Neurobiol. 2000;26:41–54. doi: 10.1046/j.1365-2990.2000.00216.x. [DOI] [PubMed] [Google Scholar]
- Katayama H, Yamamoto A, Mizushima N, Yoshimori T, Miyawaki A. GFP-like proteins stably accumulate in lysosomes. Cell Struct Funct. 2008;33:1–12. doi: 10.1247/csf.07011. [DOI] [PubMed] [Google Scholar]
- Kay DM, Zabetian CP, Factor SA, Nutt JG, Samii A, Griffith A, Bird TD, Kramer P, Higgins DS, Payami H. Parkinson’s disease and LRRK2: Frequency of a common mutation in U.S. movement disorder clinics. Mov Disord. 2006;21:519–523. doi: 10.1002/mds.20751. [DOI] [PubMed] [Google Scholar]
- Kim HK. Experimental models of cerebral ischemia. Acta Anaesthesiol Scand Suppl. 1997;111:91–92. [PubMed] [Google Scholar]
- Kimura K, Hattori S, Kabuyama Y, Shizawa Y, Takayanagi J, Nakamura S, Toki S, Matsuda Y, Onodera K, Fukui Y. Neurite outgrowth of PC12 cells is suppressed by wortmannin, a specific inhibitor of phosphatidylinositol 3-kinase. J Biol Chem. 1994;269:18961–18967. [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Kiselyov K, Jennigs JJ, Jr, Rbaibi Y, Chu CT. Autophagy, mitochondria and cell death in lysosomal storage diseases. Autophagy. 2007;3:259–262. doi: 10.4161/auto.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279:39068–39074. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci USA. 2007;104:11441–11446. doi: 10.1073/pnas.0702717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto T, Shin RW, Doh-ura K, Tomokane N, Miyazono M, Muramoto T, Tateishi J. Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol. 1992;140:1285–1294. [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Shibata M, Waguri S, Yoshimura K, Tanida I, Kominami E, Gotow T, Peters C, Figura KV, Mizushima N, Saftig P, Uchiyama Y. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am J Pathol. 2005;167:1713–1728. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: Caution in the interpretation of LC3 localization. Autophagy. 2007:3. doi: 10.4161/auto.4012. [DOI] [PubMed] [Google Scholar]
- Lach B, Grimes D, Benoit B, Minkiewicz-Janda A. Caudate nucleus pathology in Parkinson’s disease: Ultrastructural and biochemical findings in biopsy material. Acta Neuropathol. 1992;83:352–360. doi: 10.1007/BF00713525. [DOI] [PubMed] [Google Scholar]
- Lagreze WA, Pielen A, Steingart R, Schlunck G, Hofmann HD, Gozes I, Kirsch M. The peptides ADNF-9 and NAP increase survival and neurite outgrowth of rat retinal ganglion cells in vitro. Invest Ophthalmol Vis Sci. 2005;46:933–938. doi: 10.1167/iovs.04-0766. [DOI] [PubMed] [Google Scholar]
- Lai Y, Hickey RW, Chen Y, Bayir H, Sullivan ML, Chu CT, Kochanek PM, Dixon CE, Jenkins LW, Graham SH, Watkins SC, Clark RS. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab. 2008;28:540–550. doi: 10.1038/sj.jcbfm.9600551. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Fon EA, Hastings TG, Edwards RH, Sulzer D. Methamphetamine-induced degeneration of dopaminergic neurons involves autophagy and upregulation of dopamine synthesis. J Neurosci. 2002;22:8951–8960. doi: 10.1523/JNEUROSCI.22-20-08951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberski PP, Gajdusek DC, Brown P. How do neurons degenerate in prion diseases or transmissible spongiform encephalopathies (TSEs): Neuronal autophagy revisited. Acta Neurobiol Exp (Wars) 2002;62:141–147. doi: 10.55782/ane-2002-1432. [DOI] [PubMed] [Google Scholar]
- Liu CL, Chen S, Dietrich D, Hu BR. Changes in autophagy after traumatic brain injury. J Cereb Blood Flow Metab. 2008;28:674–683. doi: 10.1038/sj.jcbfm.9600587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Salas J, Ibarra O, Martinez Fong D, Cornejo A, Aceves J, Kuri J. Degenerative ultrastructural changes observed in the neuropil of caudate nuclei from Parkinson’s disease patients. Stereotact Funct Neurosurg. 1990:54–55. 297–305. doi: 10.1159/000100226. [DOI] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- Meijering E, Jacob M, Sarria JC, Steiner P, Hirling H, Unser M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A. 2004;58:167–176. doi: 10.1002/cyto.a.20022. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to Interpret LC3 Immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Munafo DB, Colombo MI. A novel assay to study autophagy: Regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114:3619–3629. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Orr HT. Lurcher, nPIST, and autophagy. Neuron. 2002;35:813–814. doi: 10.1016/s0896-6273(02)00871-1. [DOI] [PubMed] [Google Scholar]
- Pahlman S, Ruusala AI, Abrahamsson L, Mattsson ME, Esscher T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: A comparison with phorbolester-induced differentiation. Cell Differ. 1984;14:135–144. doi: 10.1016/0045-6039(84)90038-1. [DOI] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer’s presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Pelsman A, Mori H, Busciglio J. Presenilin-1 mutations reduce cytoskeletal association, deregulate neurite growth, and potentiate neuronal dystrophy and tau phosphorylation. J Neurosci. 2001;21:834–842. doi: 10.1523/JNEUROSCI.21-03-00834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105:1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RD, Oe T, Yamaji T, Matsuoka N. A simple, flexible, nonfluorescent system for the automated screening of neurite outgrowth. J Biomol Screen. 2006;11:155–164. doi: 10.1177/1087057105283344. [DOI] [PubMed] [Google Scholar]
- Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis. 2008;29:132–141. doi: 10.1016/j.nbd.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Enriquez S, He L, Lemasters JJ. Role of mitochondrial permeability transition pores in mitochondrial autophagy. Int J Biochem Cell Biol. 2004;36:2463–2472. doi: 10.1016/j.biocel.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Ross RA, Spengler BA, Biedler JL. Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J Natl Cancer Inst. 1983;71:741–747. [PubMed] [Google Scholar]
- Rowland AM, Richmond JE, Olsen JG, Hall DH, Bamber BA. Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J Neurosci. 2006;26:1711–1720. doi: 10.1523/JNEUROSCI.2279-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki DD, Pletnikova O, Vonsattel JP, Ross CA, Margolis RL. A comparison of huntington disease and huntington disease-like 2 neuropathology. J Neuropathol Exp Neurol. 2008;67:366–374. doi: 10.1097/NEN.0b013e31816b4aee. [DOI] [PubMed] [Google Scholar]
- Satchell MA, Zhang X, Kochanek PM, Dixon CE, Jenkins LW, Melick J, Szabo C, Clark RS. A dual role for poly-ADP-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving NAD+ depletion and ribosylation of 14-3-3gamma. J Neurochem. 2003;85:697–708. doi: 10.1046/j.1471-4159.2003.01707.x. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Sikorska B, Liberski PP, Giraud P, Kopp N, Brown P. Autophagy is a part of ultrastructural synaptic pathology in Creutzfeldt-Jakob disease: A brain biopsy study. Int J Biochem Cell Biol. 2004;36:2563–2573. doi: 10.1016/j.biocel.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- St Croix CM, Leelavanichkul K, Watkins SC. Intravital fluorescence microscopy in pulmonary research. Adv Drug Deliv Rev. 2006;58:834–840. doi: 10.1016/j.addr.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Tanida I, Yamaji T, Ueno T, Ishiura S, Kominami E, Hanada K. Consideration about negative controls for LC3 and expression vectors for four colored fluorescent protein-LC3 negative controls. Autophagy. 2008;4:131–134. doi: 10.4161/auto.5233. [DOI] [PubMed] [Google Scholar]
- Traystman RJ. Animal models of focal and global cerebral ischemia. Ilar J. 2003;44:85–95. doi: 10.1093/ilar.44.2.85. [DOI] [PubMed] [Google Scholar]
- Ventruti A, Cuervo AM. Autophagy and neurodegeneration. Curr Neurol Neurosci Rep. 2007;7:443–451. doi: 10.1007/s11910-007-0068-5. [DOI] [PubMed] [Google Scholar]
- Wishart TM, Parson SH, Gillingwater TH. Synaptic vulnerability in neurodegenerative disease. J Neuropathol Exp Neurol. 2006;65:733–739. doi: 10.1097/01.jnen.0000228202.35163.c4. [DOI] [PubMed] [Google Scholar]
- Xue L, Fletcher GC, Tolkovsky AM. Autophagy is activated by apoptotic signalling in sympathetic neurons: An alternative mechanism of death execution. Mol Cell Neurosci. 1999;14:180–198. doi: 10.1006/mcne.1999.0780. [DOI] [PubMed] [Google Scholar]
- Xue L, Fletcher GC, Tolkovsky AM. Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr Biol. 2001;11:361–365. doi: 10.1016/s0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- Yang Y, Fukui K, Koike T, Zheng X. Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur J Neurosci. 2007;26:2979–2988. doi: 10.1111/j.1460-9568.2007.05914.x. [DOI] [PubMed] [Google Scholar]
- Yue Z, Wang QJ, Komatsu M. Neuronal autophagy: Going the distance to the axon. Autophagy. 2008;4:94–96. doi: 10.4161/auto.5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, Zhou JN. Roles of autophagy and mTOR signaling in neuronal differentiation of mouse neuroblastoma cells. Cell Signal. 2008;20:659–665. doi: 10.1016/j.cellsig.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Zhu C, Wang X, Xu F, Bahr BA, Shibata M, Uchiyama Y, Hagberg H, Blomgren K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005;12:162–176. doi: 10.1038/sj.cdd.4401545. [DOI] [PubMed] [Google Scholar]
- Zhu C, Xu F, Wang X, Shibata M, Uchiyama Y, Blomgren K, Hagberg H. Different apoptotic mechanisms are activated in male and female brains after neonatal hypoxia-ischaemia. J Neurochem. 2006;96:1016–1027. doi: 10.1111/j.1471-4159.2005.03639.x. [DOI] [PubMed] [Google Scholar]
- Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003;13:473–481. doi: 10.1111/j.1750-3639.2003.tb00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]