

Abstract
The immunity-related GTPases (IRG), also known as p47 GTPases, are a family of proteins that are tightly regulated by IFNs at the transcriptional level and serve as key mediators of IFN-regulated resistance to intracellular bacteria and protozoa. Among the IRG proteins, loss of Irgm1 has the most profound impact on IFN-γ-induced host resistance at the physiological level. Surprisingly, the losses of host resistance seen in the absence of Irgm1 are sometimes more striking than those seen in the absence of IFN-γ. In the current work, we address the underlying mechanism. We find that in several contexts, another protein in the IRG family, Irgm3, functions to counter the effects of Irgm1. By creating mice that lack Irgm1 and Irgm3, we show that several phenotypes important to host resistance that are caused by Irgm1 deficiency are reversed by coincident Irgm3 deficiency; these include resistance to Salmonella typhimurium in vivo, the ability to affect IFN-γ-induced Salmonella killing in isolated macrophages, and the ability to regulate macrophage adhesion and motility in vitro. Other phenotypes that are caused by Irgm1 deficiency, including susceptibility to Toxoplasma gondii and the regulation of GKS IRG protein expression and localization, are not reversed but exacerbated when Irgm3 is also absent. These data suggest that members of the Irgm subfamily within the larger IRG family possess activities that can be opposing or cooperative depending on the context, and it is the balance of these activities that is pivotal in mediating IFN-γ-regulated host resistance.
Keywords: interferon, resistance, p47 GTPases
INTRODUCTION
The immunity-related GTPases (IRG; also known as p47 GTPases) are a family of vertebrate GTP-binding proteins that play important roles in host resistance to intracellular pathogens [1,2,3,4,5]. The IRG family is particularly extensive in the mouse, which has 23 different IRG genes that are divided into Irga, Irgb, Irgc, Irgd, and Irgm subfamilies [6]. The expression of most of the mouse IRG genes is strongly induced by type I and type II IFNs, resulting in high expression following bacterial, protozoal, and viral infection [1, 2, 7]. Gene targeting studies inactivating individual IRG proteins indicate that they are differentially required for IFN-induced resistance to intracellular bacteria and protozoa. This is particularly true of the Irgm subfamily, so named, as they possess a noncanonical GMS sequence in the G1 GTP-binding motif [2]. Mice lacking Irgm1 (originally named LRG-47) are markedly susceptible to all intracellular bacteria and protozoa that have been examined (e.g., Toxoplasma gondii and Salmonella typhimurium) [1, 8, 9]. Mice lacking Irgm3 [IFN-γ-inducible GTP-binding protein (IGTP)] are also susceptible to many protozoa and bacteria, although to a more limited group (e.g., T. gondii but not S. typhimurium) [9, 10]. In contrast, the proteins in the other IRG protein subfamilies, all of which possess the canonical GKS sequence in the G1 motif, seem to have more limited roles in host resistance at the physiological level as judged by gene-targeting studies. For instance, mice lacking Irgd (IRG-47) have nearly normal resistance to all of the pathogens that have been examined, with only slightly increased susceptibility to T. gondii [8]. Similarly, mice lacking Irga6 have essentially normal resistance to several protozoa and bacteria (J. C. Howard, unpublished data).
The roles of the IRG proteins in host resistance are thought to reflect, to some extent, their abilities to mediate cell autonomous control of bacteria and protozoa in host cells, including macrophages, astrocytes, and fibroblasts [11,12,13]. IRG proteins bind lipid membranes to varying extents, and importantly, they concentrate on pathogen-containing vacuoles/phagosomes in host cells, leading to the prevailing theory that they drive processing of the vacuole and thus, undermine survival of the pathogen [7, 14, 15]. The exact effect of the IRG protein on vacuole processing seems to vary with the particular IRG protein and the pathogen. Irgm1, for instance, is required for IFN-γ-induced lysosomal fusion and/or autophagy of Mycobacterium tuberculosis containing vacuoles in macrophages [16,17,18]. Irgm3 and Irga6 are required for IFN-γ-stimulated vesiculation or break-down of the T. gondii vacuole in macrophages and astrocytes [12, 19, 20]. It is not yet clear whether the IRG proteins all promote a similar pathway of vacuole processing or whether they promote several distinct pathways. It should be noted that localization of IRG proteins to the vacuole is not a strict requirement for altering pathogen survival; for instance, Irgm1 does not localize detectably to the T. gondii vacuole in astrocytes and macrophages, yet it has a profound effect on survival of T. gondii in those cells [13, 15].
Beyond the effects on vacuole/phagosome-bound pathogens, additional functions have been described for Irgm1 that also may be important for its role in host resistance. These include the regulation of adhesion and motility of IFN-γ-activated macrophages in vitro [9], the production of proinflammatory cytokines in response to infection [21], and the expansion of hematopoietic stem cells following infection [22].
The basis for the dominant role of the Irgm proteins, relative to the GKS IRG proteins in instructing host resistance at the physiological level, is not fully understood. Clues have come from recent studies that suggest that Irgm proteins may regulate the function of GKS IRG proteins in some respects. For instance, yeast two-hybrid studies have shown that IRG proteins are able to form homotypic and heterotypic complexes in a guanine nucleotide-dependent manner, the interacting partners including GMS (Irgm) complexes with GKS proteins [23]. Within the same studies, it was also shown that when the Irgm proteins are depleted by small interfering RNA targeting, GKS IRG proteins become mislocalized and fail to load appropriately on T. gondii vacuoles. Thus, based on these and other data, Irgm proteins, of which there are three, seem to regulate a large number of GKS IRG proteins, of which there are at least 15, perhaps explaining in part the marked phenotype of Irgm-deficient mice and cells relative to GKS IRG protein-deficient mice/cells.
Among the three Irgm proteins, the important role of Irgm1 in host resistance is particularly striking. In fact, in some contexts, Irgm1-deficient mice or cells exhibit phenotypes that are even more marked than those seen in the absence of IFN signaling. This is surprising given that Irgm1 and the other IRG proteins are perceived as downstream IFN effectors that are tightly regulated at the transcriptional level by IFN [1, 2]. However, IFN-γ-stimulated, Irgm1-deficient macrophages manifest marked decreases in adhesive and motile behavior, which are not seen in the similar macrophage populations in the absence of IFN-γ [9]. Additionally, Irgm1-deficient mice exhibit impaired lymphocyte expansion during chronic infection processes, which is not seen in mice lacking IFN-γ [22]. Irgm1-deficient mice also display profound susceptibility to intracellular bacteria, which in some cases, is more striking than that seen in IFN-γ-deficient mice (G. A. Taylor and C. G. Feng, unpublished studies). To date, these important aspects of Irgm1 function have yet to be explained. In the current work, we address the underlying mechanism, specifically, the hypothesis that IFN-γ may stimulate the expression of a factor or factors that function to counter the activity of Irgm1. We show that this is in fact the case, while showing surprisingly that an important opposing factor is another Irgm protein, Irgm3. By creating Irgm1/Irgm3 doubly deficient mice and characterizing important IFN-γ-stimulated functions, we show that Irgm3 functions to counter the effects of Irgm1 in a number of settings related to intracellular bacterial resistance. Our results emphasize the pivotal roles of Irgm proteins in coordinating IFN-γ-mediated host defense, while establishing a striking diversity among them.
MATERIALS AND METHODS
Mice and cell culture
Irgm1 (LRG-47)-deficient [Irgm1 knockout (KO)] and Irgm3 (IGTP)-deficient mice (Irgm3 KO) were generated as described previously [8]. These strains were intercrossed to produce Irgm1/3 KO mice that were homozygous for disruptions at the Irgm1 and Irgm3 loci. All strains were generated and maintained on C57Bl/6 and 129SvImJ genetic backgrounds. The mice were maintained according to Institutional Animal Care and Use Committee-approved protocols at the Durham VA and Duke University Medical Centers (Durham, NC, USA).
Primary murine bone marrow macrophages (BMM) were isolated from the tibia and femurs of 2- to 4-month-old mice. BM was flushed from the bones using a 27G needle fitted to a syringe filled with DMEM (Life Technologies, Gaithersburg, MD, USA), the marrow was dispersed by drawing through the needle three to four times, and red cells were lysed with associated tyrosine kinase lysing buffer (Life Technologies). Adherent cells were cultured for 6 days in BMM medium [DMEM supplemented with 10% (v/v) FBS, Hyclone, Logan, UT, USA] and 30% (v/v) L929 cell-conditioned medium. The cells were cultured on Petri dishes that were not cell culture-treated, which resulted in cultures that were loosely adherent and easily removed from the plates with cell dissociation buffer (#13150-016, Invitrogen/Gibco, Carlsbad, CA, USA). Twenty-four hours prior to all experiments, the cells were placed in medium lacking L929-conditioned media [DMEM supplemented with 10% (v/v) FBS]. Wild-type (WT) and Irgm1-deficient BMM were found to be at least 95% positive for the F4/80 macrophage marker.
In vivo S. typhimurium infection
S. typhimurium were cultured overnight in Luria-Bertani (LB) broth without shaking. For in vivo studies, bacteria were injected i.v. at 6 × 105 cells per mouse in a volume of 0.1 ml PBS. The aroA– SL7731 strain was used [24, 25]; resistance of this strain is dependent on IFN-γ-induced mechanisms [25], and it is more easily handled than the fully virulent strain. Bacterial counts were determined by excising spleens and livers under sterile conditions, homogenizing portions of the organs in PBS, and plating serial dilutions of the homogenate on LB agar plates. Colony counts were assessed the following day, from which the total bacterial load per organ was calculated.
In vitro bacterial survival assays
The virulent strain SL1344 of S. typhimurium [26] was cultured overnight in LB broth without shaking. BMM were plated in 24-well plates at 0.35 × 106 cells per well. They were exposed to varying concentrations of IFN-γ (EMD Biosciences/Calbiochem, San Diego, CA, USA) for 24 h. The cells were infected with S. typhimurium at a multiplicity of infection (moi) of 2 bacteria per macrophage. The bacteria were added as a suspension in DMEM; the plates were centrifuged at 250 g for 5 min, incubated for an additional 10 min, and washed three times with DMEM to remove extracellular bacteria. The cells were then incubated for various times in medium supplemented with 6 μg/ml gentamycin. To quantify bacterial levels, the cells were washed with DMEM three times and then lysed with 0.2% (v/v) Triton X-100 in PBS. Dilutions of the lysates were plated on LB plates, and colony-forming units were counted. The assay was performed in triplicate for each condition.
In vivo T. gondii infection
Mice were injected i.p. with 0.5 ml PBS containing 20 cysts of the avirulent ME49 strain of T. gondii that had been prepared from the brains of infected C57BL/6 mice. The mice were monitored daily for survival.
T. gondii culture and in vitro infection
The choramphenical acetyltransferase-GFP T. gondii strain [27] was maintained by serial passage on HS27 human foreskin fibroblasts (American Type Collection, Manassas, VA, USA; CRL-1634), grown in DMEM, supplemented with 10% FBS. Tachyzoites were harvested after 3–5 days in culture when the fibroblast host cell layer reached 80–90% lysis. Suspensions of parasites were cleared of fibroblast debris by centrifugation at 50 g for 5 min, and parasites were enumerated using a hemocytometer.
Subconfluent cultures of BMM were plated on polylysine-coated coverslips. The cells were allowed to adhere for 24 h, after which, they were exposed to 100 U/mL IFN-γ for another 24 h. The cells were then infected with T. gondii by adding tachyzoites at a moi of 5:1, spinning parasites onto the coverslips at 400 g for 2 min at room temperature, followed by an additional 10-min incubation at 37ºC, and finally, washing the coverslips three times with PBS. Following incubations for the times indicated in the text, the cells were prepared for immunocytochemistry as described below.
Western blotting
Western blots were performed according to standard protocols [8, 28] using anti-Irga6 rabbit polyclonal antisera [15], anti-Irgb6 rabbit polyclonal antisera raised against the peptide KKVGPYISEPPEYWEA, or antiactin antisera (Chemicon, El Segundo, CA, USA; #MAB1501).
Histology
Liver tissues were isolated from mice that had been infected with S. typhimurium for 6 days. Tissues were preserved in buffered formalin and then processed for paraffin sectioning and H&E staining.
Adhesion assay
BMM were plated on uncoated glass coverslips in 24-well plates at 0.35 × 106 cells per coverslip. Twenty-four hours after plating, the cells were exposed to 100 U/ml IFN-γ (EMD Biosciences/Calbiochem) for an additional 24 h. The cells were then washed with PBS without Ca2+/Mg2+ four times to remove loosely adherent cells, fixed with 4% (w/v) paraformaldehyde/PBS for 10 min, stained with 0.5% (w/v) crystal violet/methanol for 10 min, and washed five times with water to remove excess stain. Finally, the cells were solubilized in methanol overnight at 4ºC, and the absorbance at 595 nm was read as a measure of adherent cells.
Transwell motility assay
Standard transwell devices in a 24-well format (Corning, Corning, NY, USA), which had an upper chamber separated from a lower chamber by a polycarbonate membrane containing 5 μm pores, were used. The lower surface of the porous membrane was coated with human fibronectin. Twenty-four hours prior to the experiment, BMM were placed in medium that lacked L929-conditioned media but contained in some cases, 100 U/ml IFN-γ. These cells were dislodged with cell dissociation buffer, enumerated with a hemocytometer using trypan blue to exclude dead cells, and then placed in the upper chamber of transwell devices in medium lacking L929-conditioned media and IFN-γ. [Note that after removing IFN-γ from activated WT BMM, the cells retained high Irgm1 expression for at least 24 h (data not shown)]. BMM medium (containing L929-conditioned media, a source of the chemoattractant M-CSF/CSF-1) was placed in the lower chamber of the transwell device. The cells were then allowed to migrate through to the lower chamber and adhere to the underside of the membrane for 4 h, at which point, the cells were fixed with 70% (v/v) ethanol and 4′,6-diamidino-2-phenylindole (DAPI)-stained. Cells on the upper surface of the polycarbonate membrane were removed with cotton swabs, and those that had migrated through the membrane and were bound to the lower surface of the membrane were enumerated by cell imaging and integrated morphometry analysis.
Cell staining
BMM were plated onto fibronectin-coated coverslips, cultured for ∼24 h, and then cultured an additional 24 h in the presence of 100 U/ml IFN-γ. The cells were fixed with 4% paraformaldehyde (w/v) in PBS for 15 min and permeabilized with 0.2% (w/v) saponin in PBS for 10 min. As indicated in the text, the cells were then stained with phalloidin-conjugated Alexafluor-594 to stain actin (Molecular Probes, Eugene, OR, USA), anti-Irga6 rabbit polyclonal antisera, anti-Irgb6 rabbit polyclonal antisera, or anti-T. gondii antisera (Biodesign, Saco, ME, USA; #C65203M), along with Alexafluor-conjugated secondary antibodies (Molecular Probes). Cell imaging and analysis were performed using an Olympus IX70 microscope linked to a computer outfitted with AutoQuant 9.3 and MetaMorph 6.2.3.5 software.
Statistical analysis
Student’s paired t-test was used to assess statistical significance.
RESULTS
Generation of Irgm1/3-deficient mice
We generated mice that possessed targeted disruptions in the Irgm1 (LRG-47) and Irgm3 (IGTP) genes by crossing Irgm1 KO mice with Irgm3 KO mice [8, 10]. This strategy was repeated to independently generate the doubly deficient strain on a mixed C57Bl/6-129SvImJ genetic background, on a pure C57Bl/6 background, and on a pure 129SvImJ background. On all backgrounds, Irgm1/3 KO mice bred normally and displayed no overt phenotype. In the experiments described below, we found no differences in the phenotypes of the Irgm1/3 KO mice on the different genetic backgrounds, with the exception of experiments involving S. typhimurium, where only minor differences were noted, likely related to the well-documented difference in Salmonella susceptibility between the C57Bl/6 and 129SvIm/J strains [29, 30].
Response of Irgm1/3-deficient mice to S. typhimurium
We assessed the response of the Irgm1/3 KO mice to S. typhimurium. We had found previously that mice lacking Irgm1 or Irgm3 alone display different responses to S. typhimurium: Irgm1 KO mice are acutely susceptible as reflected in increased mortality, increased hepatic and splenic bacterial loads, impaired granuloma maturation, and impaired IFN-γ-induced killing of the bacteria in macrophages; in contrast, Irgm3 KO mice display nearly normal susceptibility, as well as normal, IFN-γ-regulated intracellular killing in macrophages [9]. In the current studies, we found again that on the 129SvImJ background, Irgm1 KO mice had significantly increased splenic and hepatic bacterial loads at 6 days following S. typhimurium infection, in contrast to Irgm3 KO mice, in which hepatic bacterial titers were statistically the same as those in WT mice (Fig. 1A). Surprisingly, bacterial loads in the tissues of Irgm1/3 KO mice were not elevated but were comparable with those in WT mice (Fig. 1A). We repeated these studies using mice derived on the C57 Bl/6 background and obtained similar results (Fig. 1B): Irgm1 KO mice showed significantly elevated hepatic and splenic bacterial loads; bacterial levels in Irgm3 KO mice were again not elevated but slightly lower than those in WT mice; and bacterial levels in Irgm1/3 KO mice were not as elevated as those in Irgm1 KO mice but were intermediate between the levels in Irgm1 and Irgm3 KO mice. Thus, when mice lack Irgm1 and Irgm3, the marked susceptibility displayed by mice lacking Irgm1 alone is completely or partially reversed.
Fig. 1.

Response to S. typhimurium of mice lacking Irgm1, Irgm3, and Irgm1/3. (A) Groups of seven mice of the indicated genotypes on the 129SvIm/J genetic background were inoculated i.v. with 106 CFU S. typhimurium. Six days later, spleen and liver were isolated and used for CFU determination. Error bars indicate sd; **, statistically significant differences relative to WT levels (P<0.05). The differences between the bacterial levels in the spleen and liver in the WT and Irgm1 KO mice were statistically significant (P=0.003 and 0.0163, respectively) but not between WT and Irgm3 KO mice (P=0.59 and 0.17) or WT and Irgm1/3 KO mice (P=0.79 and 0.41). (B) Groups of six WT, five Irgm1 KO, seven Irgm3 KO, and seven Irgm1/3 KO mice on the C57Bl/6 genetic background were inoculated i.v. with 106 CFU S. typhimurium. Six days later, spleen and liver were isolated and used for CFU determination. Error bars indicate sd; **, statistically significant differences relative to WT levels (P<0.05). The differences between the bacterial levels in the spleen and liver in the WT and the other genotypes were statistically significant, with the exception of WT versus Irgm1/3 KO in the liver. For spleen and liver, WT versus Irgm1 KO, P = 0.0003 and 0.0004, respectively; for WT versus Irgm3 KO, P = 0.023 and 0.025; and for WT versus Irgm1/3 KO, P = 0.004 and 0.095. (C) The mice inoculated in A were also used for spleen weight determination at 6 days postinfection. The weights from control, uninfected mice were also determined for each genotype (four to five mice per group). Error bars indicate sd; **, statistically significant differences relative to WT levels (P<0.05). Among the infected mice, the difference in the weights of spleens between WT and Irgm1 KO mice was statistically significant (P=0.013), as was the difference between WT and Irgm3 KO (P=0.015), and the difference between WT and Irgm1/3 KO was not (P=0.52). (D) The mice inoculated in B were also used for spleen weight determination at 6 days postinfection. The weights from control, uninfected mice were also determined for each genotype (four to eight mice per group). Error bars indicate sd; **, statistically significant differences relative to WT levels (P<0.05). Among the infected mice, the difference in the weights of spleens between WT and Irgm1 KO mice was statistically significant (P=0.009); the difference between WT and Irgm3 KO (P=0.12) and WT and Irgm1/3 KO (P=0.70) was not. The data presented in A–D are representative data from individual experiments selected from a total of six experiments using mice of the different genetic backgrounds. (E) Groups of three mice of the indicated genotypes on the C57Bl/6 background were inoculated i.v. with 106 CFU S. typhimurium. At 6 days postinfection, liver tissue was processed for H&E staining. Inflammatory foci (100 per sample) were enumerated in a blinded manner and scored in one of three categories, as indicated in the figure: immature foci containing >50% polymorphonuclear neutrophils (PMNs), intermediate foci containing 10–50% PMNs, or mature granulomas composed of <10% PMNs/>90% epithelioid cells. Error bars indicate sd; **, statistically significant differences relative to WT levels (P<0.05). Differences in the numbers of foci for each category between WT and Irgm1 KO livers were statistically significant (P=0.021, 0.017, and 0.008) but not between WT and Irgm3 KO or WT and Irgm1/3 KO livers (P>0.05 in all cases).
In the same studies, we also examined the spleen size in Irgm1, Irgm3, and Irgm1/3 KO mice at 6 days following S. typhimurium infection, as spleen size normally increases substantially following infection with this bacterium, reflecting a robust, inflammatory response. On the 129SvIm/J and C57Bl/6 backgrounds, there was a failure of the spleens of Irgm1 KO mice to increase to the same extent as those in WT mice, likely reflecting an insufficient, inflammatory response in the absence of Irgm1 (Fig. 1, C and D). In contrast, in Irgm3 KO mice, there was a robust increase in spleen size, which on the 129SvImJ background, was significantly greater than that in WT mice and on the C57Bl/6 background was statistically equivalent to that in WT mice. In the Irgm1/3 KO mice on both genetic backgrounds, the spleen sizes were larger than those in Irgm1 KO mice and intermediate between those of Irgm1 KO and Irgm3 KO mice. Therefore, the more robust increase in spleen size noted in Irgm1/3 KO mice, relative to the spleen size in Irgm1 KO mice, paralleled the normal control of bacterial growth/survival in the spleen and liver (Fig. 1, A and B).
We also assessed the histological appearance of granulomas in the livers of WT, Irgm1 KO, Irgm3 KO, and Irgm1/3 KO mice (on the C57Bl/6 background) at 6 days postinfection. Overall, the number of inflammatory lesions/granulomas correlated with the hepatic bacterial titers (Fig. 1B) with abundant areas of inflammation in Irgm1 KO mice and far fewer areas in WT, Irgm3 KO, and Irgm1/3 KO mice. The lesions in WT mice were primarily well-organized granulomas composed mainly of epithelioid and multinucleated giant cells of macrophage origin (Fig. 1E, and data not shown). In contrast, in the Irgm1 KO mice, the foci were predominantly immature, displaying less organization, while having a much higher PMN content and a rudimentary epithelioid cell response (Fig. 1E, and data not shown). Again, in keeping with overall control of bacterial growth, Irgm3 KO and Irgm1/3 KO mice displayed mature granulomas rich in epithelioid cells (Fig. 1E, and data not shown).
Normal IFN-γ-induced control of S. typhimurium in Irgm1/3 KO macrophages
We have demonstrated previously that IFN-γ-induced control of S. typhimurium is impaired in macrophages (BMM) that lack expression of Irgm1, and IFN-γ-induced control is normal in Irgm3 KO macrophages [9]. As in vivo control of this bacterium by Irgm1 may be related in part to its ability to regulate survival of the bacteria in macrophages, it was important in the current studies to examine the ability of Irgm1/3 KO macrophages to restrict bacterial growth. The experiments were performed using BMM, which were derived from mice on the C57Bl/6 background, as the effect of Irgm1 deficiency is more robust in cells from this background (G. A. Taylor, unpublished). In keeping with the data published previously [9], IFN-γ-induced control of S. typhimurium survival was greatly muted in Irgm1 KO BMM (Fig. 2). In contrast, IFN-γ-induced control was intact in Irgm3 KO and Irgm1/3 KO BMM. Thus, although absence of Irgm1 resulted in loss of IFN-γ-stimulated bacterial control, absence of Irgm3 and Irgm1 restored normal cell-autonomous control of this bacterium.
Fig. 2.

Normal IFN-γ-induced suppression of S. typhimurium in Irgm1/3 KO macrophages. BMM of the indicated genotypes were cultured for 24 h under control conditions or in the presence of 100 U/ml IFN-γ, as indicated. The cells were subsequently infected with S. typhimurium SL1344 at a moi of 2:1. At 2, 6, and 8 h postinfection in medium containing gentamycin to inhibit extracellular bacterial growth, CFUs were determined from triplicate samples. Shown are the composite results from five experiments. To illustrate the effect on intracellular bacterial survival, the data are displayed as the ratio of the CFU remaining at 6 and 8 h to the CFU present at the initial 2-h time-point. The error bars indicate sd; ***, statistically significant differences relative to WT levels at that time-point (P<0.05). In IFN-γ-activated cells, the differences between values in WT and Irgm1 KO BMM were statistically significant at 6 and 8 h (P=0.012 and 0.0048, respectively), and none of the differences between WT and Irgm3 KO BMM or WT and Irgm1/3 KO BMM were significant. In BMM cultured under control conditions, none of the data for the Irgm1 KO, Irgm3 KO, or Irgm1/3 KO BMM was statistically different from those in the WT BMM at 6 or 8 h.
Normal adhesion, motility, and morphology of Irgm1/3 KO macrophages
We have noted previously that in the absence of Irgm1, the adhesion and motility of IFN-γ-activated macrophages are strikingly altered, as is their morphology [9]. In the current work, we examined Irgm1/3 KO macrophages to determine whether they also displayed these phenotypes. Irgm1/3 KO macrophages were first examined for their ability to adhere to glass coverslips (Fig. 3A). As opposed to IFN-γ-activated Irgm1 KO BMM that displayed impaired adherence to uncoated glass, as shown previously [9], activated Irgm1/3 KO BMM showed normal adherence comparable with that seen in activated WT and Irgm3 KO BMM (Fig. 3A). Next, Irgm1/3 KO macrophages were examined for their ability to exhibit motility using standard transwell assays. Although motility was markedly impaired in IFN-γ-activated Irgm1 KO BMM, it was normal and comparable with that of WT cells in activated Irgm1/3 KO BMM (Fig. 3B). Finally, the morphology of adherent, IFN-γ-activated BMM, which lacked Irgm1 and/or Irgm3, was examined on fibronectin, a substrate on which we have noted a marked change in morphology of Irgm1 KO BMM previously [9]. Regarding the overall shape of the cells, a variety of morphologies was noted among the cells, as is typical of macrophage populations (Table 1). The distributions of these morphologies were similar among the WT, Irgm3 KO, and Irgm1/3 KO BMM, and they were markedly different among the Irgm1 KO BMM (Table 1). In general, the WT, Irgm3 KO, and Irgm1/3 KO cells showed a higher degree of polar and stellate cells. In comparison, the Irgm1 KO cells assumed a flattened, apolar shape more frequently; further, among the Irgm1 KO cells that could be characterized as polar or stellate, the cellular protrusions/lammelipodia tended to be broader and flatter. At a finer cell level, the WT, Irgm3 KO, and Irgm1/3 KO cells displayed areas of actin-dense membrane ruffles on the perimeters of the cells (Fig. 4). In contrast and in keeping with our previous observations [9], the Irgm1 KO BMM displayed a marked decrease in membrane ruffling (Fig. 4). In summary, the substantial changes in cell adhesion, motility, and morphology, which were readily apparent in activated macrophages lacking Irgm1, were reversed when the cells also lacked Irgm3.
Fig. 3.

Normal adhesion and motility in Irgm1/3 KO macrophages. (A) Adhesion: BMM of the indicated genotypes were cultured on uncoated glass coverslips and then exposed to 100 U/ml IFN-γ or control conditions for 24 h. Adherence was assessed by washing off loosely adherent cells, staining the remaining adherent cells with crystal violet, solubilizing them, and measuring absorbance of the lysate as an indicator of adherence. The data were expressed as the percentage of adherent, IFN-γ-treated cells relative to adherent cells cultured under control conditions. The study was repeated three to five times for each of the different genotypes; shown are average values with error bars indicating sem, and **, statistical significance (P<0.05) relative to the value for WT cells. The difference between WT and Irgm1 KO BMM was statistically significant (P=0.015), but the differences between WT and Irgm3 KO (P=0.75) and WT and Irgm1/3 KO (P=0.88) were not. (B) Motility: BMM of the indicated genotypes were maintained under control conditions or were activated with 100 U/ml IFN-γ for 24 h; they were then placed in equal numbers in the upper chambers of transwell apparatuses. The medium in lower chambers contained M-CSF as a chemoattractant. Cells that migrated through the membrane and attached to the fibronectin-coated underside over a 4-h period were stained with DAPI, and those that remained in the upper chamber were removed by swabbing. The cells were imaged and quantified by measuring the total DAPI-positive area on the filter, using software image analysis. The data are expressed as the percent of motile IFN-treated cells relative to motile control cells. The experiment was repeated three to six times; shown are average values with error bars indicating sem, and **, statistical significance (P<0.05) relative to the value for WT cells. The difference between WT and Irgm1 KO BMM was statistically significant (P=0.003) but not between WT and Irgm3 KO BMM (P=0.64) or WT and Irgm1/3 KO BMM (P=0.45).
TABLE 1.
Distribution of Cellular Morphologies among Macrophages Lacking Irgm1 and/or Irgm3
| Cell morphology | WT | Irgm1 KO | Irgm3 KO | Irgm1/3 KO |
|---|---|---|---|---|
| Polar | 52.0a | 20.4 | 54.5 | 50.9 |
| Polar, flattened | 14.0 | 30.2 | 10.0 | 16.3 |
| Stellate | 24.6 | 5.3 | 20.0 | 15.2 |
| Stellate, flattened | 2.2 | 8.5 | 0.7 | 3.5 |
| Apolar, flattened | 4.7 | 24.0 | 5.9 | 8.6 |
| Rounded, partially lifted | 2.5 | 11.5 | 8.8 | 5.4 |
Data displayed as percent-positive cells based on >400 cells counted per genotype.
Fig. 4.

Loss of Irgm1 KO macrophage morphology in Irgm1/3 KO macrophages. BMM of the indicated genotypes were plated at equal densities on fibronectin-coated coverslips and then exposed to 100 U/ml IFN-γ for 24 h. The cells were then fixed, stained with Alexafluor594-conjugated phalloidin to stain actin, and imaged using deconvolution microscopy.
Effect of Irgm1 and Irgm3 deficiency on resistance to T. gondii
We next addressed the effect of Irgm1/3 double deficiency on resistance to the protozoan pathogen, T. gondii. We had found previously that Irgm1 KO and Irgm3 KO mice were acutely susceptible to i.p. infection with the normally avirulent ME49 strain of T. gondii, to the extent that their responses were indistinguishable from those of IFN-γ-deficient mice [8, 10]. In the current study, the Irgm1/3 mice were also found to be highly susceptible to T. gondii, and these mice died rapidly following infection within the same time-frame as Irgm1 KO and Irgm3 KO mice (Fig. 5). Thus, mice lacking Irgm1 and Irgm3 maintain acute susceptibility to this protozoan parasite, which contrasts with the response of the doubly deficient mice to S. typhimurium.
Fig. 5.

Loss of resistance to T. gondii in mice lacking Irgm1, Irgm3, and Irgm1/3. Groups of six mice of the indicated genotypes on the 129SvIm/J genetic background were inoculated i.p. with 20 cysts of the ME49 strain of T. gondii. The mice were then monitored for their survival for 40 days.
Effect of Irgm1 and Irgm3 deficiency on localization and expression of other IRG proteins
Recent evidence has suggested that the absence of IRG proteins in the Irgm subfamily (Irgm1, Irgm2, and Irgm3) leads to altered localization of Irga6 (IIGP) and Irgb6 (TGTP) in fibroblasts [2, 23]. Importantly, Irgm control over these and other GKS proteins localization may allow them to load correctly on the T. gondii vacuole, where they seem to trigger vesiculation of the vacuole and the demise of the pathogen [23]. In the current studies, we addressed how complete absence of Irgm1, Irgm3, or both would affect basal localization of Irgb6 and Irga6, as well as their loading on T. gondii vacuoles.
In IFN-γ-stimulated WT BMM, Irgb6 and Irga6 localized strongly to the perinuclear region with a pattern characteristic of an endoplasmic reticulum (ER) protein (Fig. 6). In the absence of Irgm1 or Irgm3, Irgb6 and Irga6 staining was slightly weaker overall and acquired a punctate appearance that included some large, intensely stained puncta. The punctate-staining pattern was more pronounced for Irgb6. In Irgm1/3 KO cells, staining appeared more punctate for Irga6 and Irgb6, and the overall intensity of staining became even weaker (to the extent that exposure times were necessarily increased substantially in Irgm1/3 KO cells to obtain readily visible images; Fig. 6). Similar results were obtained from BMM (Fig. 6) and primary embryonic fibroblasts [mouse embryonic fibroblasts (MEF); data not shown]. The nature of the redistribution of the protein was uncertain, although significant quantities of Irgb6 and Irga6 seemed to remain in the ER. For instance, further immunostaining experiments indicated that the majority of the large Irgb6 and Irga6 puncta in Irgm1/3 KO cells did not localize to a lysosome-associated membrane protein 1-positive compartment (late endosome/lysosome) or an LC3-positive compartment (autophagic vacuole); however, they apparently retained some costaining with the ER marker protein disulfide isomerase, an observation that was consistent with their continued presence in biochemically enriched ER membranes (data not shown). Nevertheless, the key observation was that the absence of Irgm1, Irgm3, and to a greater extent, Irgm1 and Irgm3 leads to a redistribution of Irgb6 and Irga6 into punctate bodies.
Fig. 6.

Altered Irga6 and Irgb6 staining in cells lacking Irgm1 and/or Irgm3. BMM of the indicated genotypes were plated at equal densities on fibronectin-coated coverslips and then exposed to 100 U/ml IFN-γ for 24 h. The cells were then fixed, stained with anti-Irga6 or anti-Irgb6 antibodies, and imaged. Note that as the immunostaining of Irgm1/3 KO BMM was significantly weaker than staining in WT, Irgm1, and Irgm3 KO BMM, the exposure times were increased in the Irgm1/3 KO BMM: For Irga6 staining, exposure times were 500 msec for WT, Irgm1 KO, and Irgm3 KO BMM and 1000 msec for Irgm1/3 KO BMM; for the Irgb6 staining, exposure times were 300 msec for WT, Irgm1 KO, and Irgm3 KO BMM and 1000 msec for Irgm1/3 KO BMM. This study was representative of three.
As the immunostaining experiments (Fig. 6) suggested decreased levels of Irgb6 and Irga6 overall in Irgm1/3 KO cells, protein expression was also assessed by Western blotting (Fig. 7). In BMM (Fig. 7) and MEF (not shown), levels of Irgb6 and Irga6 protein were reduced modestly in Irgm1 and Irgm3 KO cells and reduced to a greater extent in Irgm1/3 KO cells, corroborating the immunostaining studies.
Fig. 7.
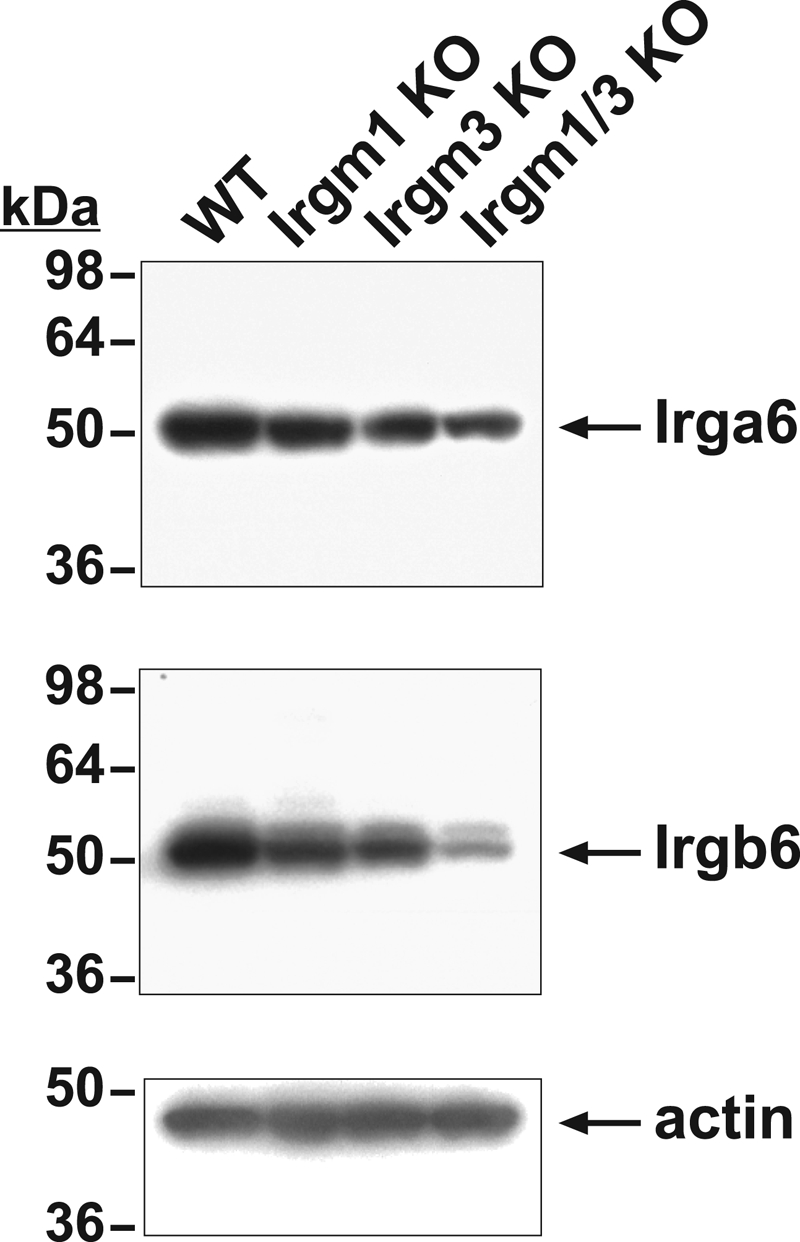
Decreased amounts of Irga6 and Irgb6 expression in macrophage lacking Irgm1 and/or Irgm3. BMM of the indicated genotypes were exposed to 100 U/ml IFN-γ for 24 h and then processed for Western blotting with the indicated antibodies. Shown at the left are the positions of the MW markers. This study is representative of three.
Finally, WT, Irgm1 KO, and Irgm1/3 KO BMM were infected with T. gondii, and the ability of Irga6 and Irgb6 to localize to the T. gondii vacuole was assessed by immunostaining. In WT cells, there was strong staining of Irga6 and Irgb6 on most T. gondii vacuoles (Fig. 8). The accumulation of Irga6 and Irgb6 on T. gondii vacuoles in WT cells was dramatic, with a major portion of total cellular Irga6 and Irgb6 redistributing from the ER to the vacuole, leaving a weakly stained ER. In Irgm1 KO cells, the fraction of strongly staining Irga6- and Irgb6-positive vacuoles decreased, although some strongly staining vacuoles remained (Fig. 8). In addition, in Irgm1 KO cells, there was redistribution of both GKS proteins into punctate bodies and a slight reduction in overall staining levels (as shown above in Fig. 6). In the Irgm1/3 KO cells, vacuoles that were strongly stained with Irga6 and Irgb6 were even less frequent. Additionally, in these cells, although weak Irga6 and Irgb6 staining of the vacuoles remained, this staining appeared clumped and spread unevenly across the vacuole (Fig. 8, and not shown). Thus, in summary, there was an additive impact of Irgm1 and Irgm3 deficiency on loading of T. gondii vacuoles with IRG GKS proteins.
Fig. 8.

Altered Irga6 and Irgb6 accumulation on the T. gondii vacuole in cells lacking Irgm1 and/or Irgm3. WT, Irgm1 KO, or Irgm1/3 KO BMM were plated at equal densities on polylysine-coated coverslips and then exposed to 100 U/ml IFN-γ for 24 h. The cells were infected with T. gondii and incubated for 1 h. The cells were fixed and stained with anti-T. gondii, anti-Irga6, and/or anti-Irgb6 antibodies as indicated and then imaged. Shown are the T. gondii staining (green), Irga6 or Irgb6 staining (red), and an overlay. Note that T. gondii vacuoles that were strongly stained with Irga6 and Irgb6 were reduced in the Irgm1 KO and Irgm1/3 KO cells. (Over 225 vacuoles were scored for each genotype and each IRG marker. For Irga6, there were 14.7% strongly stained T. gondii vacuoles in WT cells, 8.1% in Irgm1 KO cells, and 6.5% in Irgm1/3 KO cells. For Irgb6, there were 14% strongly stained T. gondii vacuoles in WT cells, 8.0% in Irgm1 KO cells, and 6.0% in Irgm1/3 KO cells.) This is representative of two studies.
In conclusion, these studies establish a clear dichotomy between the roles of Irgm1 and Irgm3 in different settings important for IFN-γ-stimulated host resistance. For T. gondii defense, as well as for the regulation of GKS protein expression and localization, Irgm3 deficiency exacerbated the phenotypes seen with Irgm1 deficiency. In contrast, when considering S. typhimurium defense or the adhesive and motile capacity of macrophages, Irgm3 deficiency dramatically reversed the impairments seen in the absence of Irgm1.
DISCUSSION
IFNs tightly regulate the expression of IRG proteins, which in turn, serve as downstream mediators of IFN-induced host resistance. Although several proteins in the IRG family have demonstrated roles in promoting IFN-mediated host resistance, it is the absence of members of the Irgm subfamily that has the most extensive impact on host resistance at the physiological level. Irgm1 is particularly critical, as mice lacking Irgm1 display marked reductions in host defense to every intracellular bacterium and protozoan parasite that has been examined to date [1, 31]. Surprisingly in some cases, the lack of host defense functions in the absence of Irgm1 is even more profound than that seen in the absence of IFN-γ. In the work presented here, we demonstrate that another IFN-induced factor, Irgm3, functions in several contexts to counter the function of Irgm1. This indicates that IFN-γ triggers expression of a variety of opposing and cooperative regulatory activities that are critical for instructing IFN-γ-mediated host resistance, and the Irgm proteins are pivotal among these regulators.
To address the interplay between Irgm1 and Irgm3, we created mice that were deficient in both proteins. We found for several functions central to host resistance that absence of Irgm3 in addition to Irgm1 partially or completely reversed the phenotypes seen in the absence of Irgm1 alone. This was true when resistance to S. typhimurium was examined, as absence of Irgm1 and Irgm3 reversed or ameliorated the decreased resistance caused by Irgm1 deficiency. This occurred in vivo and in IFN-γ-activated macrophages in vitro. This effect was not limited to S. typhimurium. For M. tuberculosis, the marked decrease in resistance seen in Irgm1 KO mice [16] is also reversed in mice that also lack Irgm3 (John MacMicking, personal communication). Interestingly, for Legionella pneumophila, the situation is inverted but reflects a similar interaction between Irgm1 and Irgm3: IFN-γ induced Irgm3 KO macrophages fail to restrict bacterial growth completely compared with Irgm1 KO and WT macrophages, and Irgm1/3 KO macrophages largely revert back to the WT phenotype (J. Coers, unpublished data). Additionally, in work presented here, the altered adhesion, motility, and morphology of macrophages caused by Irgm1 deficiency were reversed when Irgm1 and Irgm3 were absent. Thus, in the context of defense against these intracellular bacteria, the balance of Irgm1 and Irgm3 function seems to be key in determining immune function.
Nevertheless, there were also settings in which Irgm1 and Irgm3 seemed to have similar or additive functions. We found that mice lacking Irgm1 and Irgm3 displayed a striking decrease in resistance to T. gondii, similar to that seen when Irgm1 or Irgm3 alone was absent [8, 10]. We have also found that the situation is similar for Chlamydia trachomatis, in that embryonic fibroblasts that lack Irgm1, Irgm3, or both are impaired in their ability to control the bacterium (J. Coers, unpublished data). Further, we found in the current work that deficiency of Irgm1 and Irgm3 had an additive impact on the altered localization and decreased expression of the IRG GKS proteins, Irga6 and Irgb6. Thus, for each of these phenotypes, the functional result of Irgm1 deficiency was maintained or exacerbated by Irgm1/3 double deficiency.
It remains to be determined how Irgm1 and Irgm3 can seemingly have similar, additive functions in some aspects of host resistance and have contrasting and perhaps negating functions in others. In general, it is possible that Irgm1 and Irgm3, as well as other IRG proteins, act entirely as separate entities with variable functions that are additive in some contexts (resistance to T. gondii) but are opposing in others (resistance to S. typhimurium). However, recent data suggest that the IRG proteins interact with other IRG proteins as heterodimers/multimers with functional consequences. Yeast two-hybrid data clearly indicate that these complexes form in a guanine-nucleotide-dependent manner and importantly, in a differential manner depending on the particular IRG proteins [32]. For instance, Irgm3 appears to complex strongly with Irga6, and Irgm1 does not [32]. It is clear from the data presented here and elsewhere [2, 23] that the Irgm proteins regulate the expression and localization of GKS IRG proteins, likely as a consequence of these direct interactions. Thus, with this model, the removal of Irgm1, Irgm3, or both would likely skew the balance of heteromeric IRG protein complexes that exist in the cell and lead to altered immune function. For instance, a set of Irgm1-IRG protein complexes may drive resistance to S. typhimurium; when these complexes are absent, Irgm3-IRG complexes that inhibit resistance become dominant; and further, when these are also absent, a remaining set of IRG protein complexes (possibly involving Irgm2), which again promote resistance, becomes dominant. Clearly, a major challenge in future IRG research will be to identify the biochemical basis for the cell biological and immunological behavior of the IRG protein deficiencies described here and elsewhere.
Acknowledgments
This work was supported by a National Institutes of Health grant (AI57831; to G. A. T.), a VA Merit Review grant (to G. A. T.), and a Charles A. King Trust Postdoctoral Research fellowship (to J. C). We thank Vojo Deretic, Sudha Singh, and Brice Weinberg for helpful discussions.
References
- Taylor G A. IRG proteins: key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol. 2007;9:1099–1107. doi: 10.1111/j.1462-5822.2007.00916.x. [DOI] [PubMed] [Google Scholar]
- Martens S, Howard J. The interferon-inducible GTPases. Annu Rev Cell Dev Biol. 2006;22:559–589. doi: 10.1146/annurev.cellbio.22.010305.104619. [DOI] [PubMed] [Google Scholar]
- MacMicking J D. Immune control of phagosomal bacteria by p47 GTPases. Curr Opin Microbiol. 2005;8:74–82. doi: 10.1016/j.mib.2004.12.012. [DOI] [PubMed] [Google Scholar]
- MacMicking J D. IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol. 2004;25:601–609. doi: 10.1016/j.it.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Taylor G A, Feng C G, Sher A. p47 GTPases: regulators of immunity to intracellular pathogens. Nat Rev Immunol. 2004;4:100–109. doi: 10.1038/nri1270. [DOI] [PubMed] [Google Scholar]
- Bekpen C, Hunn J P, Rohde C, Parvanova I, Guethlein L, Dunn D M, Glowalla E, Leptin M, Howard J C. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol. 2005;6:R92. doi: 10.1186/gb-2005-6-11-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerrahn J, Schaible U E, Brinkmann V, Guhlich U, Kaufmann S H. The IFN-inducible Golgi- and endoplasmic reticulum-associated 47-kDa GTPase IIGP is transiently expressed during listeriosis. J Immunol. 2002;168:3428–3436. doi: 10.4049/jimmunol.168.7.3428. [DOI] [PubMed] [Google Scholar]
- Collazo C M, Yap G S, Sempowski G D, Lusby K C, Tessarollo L, Woude G F, Sher A, Taylor G A. Inactivation of LRG-47 and IRG-47 reveals a family of interferon γ-inducible genes with essential, pathogen-specific roles in resistance to infection. J Exp Med. 2001;194:181–188. doi: 10.1084/jem.194.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry S C, Daniell X, Indaram M, Whitesides J F, Sempowski G D, Howell D, Oliver T, Taylor G A. Impaired macrophage function underscores susceptibility to Salmonella in mice lacking Irgm1 (LRG-47) J Immunol. 2007;179:6963–6972. doi: 10.4049/jimmunol.179.10.6963. [DOI] [PubMed] [Google Scholar]
- Taylor G A, Collazo C M, Yap G S, Nguyen K, Gregorio T A, Taylor L S, Eagleson B, Secrest L, Southon E A, Reid S W, Tessarollo L, Bray M, McVicar D W, Komschlies K L, Young H A, Biron C A, Sher A, Vande Woude G F. Pathogen-specific loss of host resistance in mice lacking the IFN-γ-inducible gene IGTP. Proc Natl Acad Sci USA. 2000;97:751–755. doi: 10.1073/pnas.97.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halonen S K, Taylor G A, Weiss L M. γ Interferon-induced inhibition of Toxoplasma gondii in astrocytes is mediated by IGTP. Infect Immun. 2001;69:5573–5576. doi: 10.1128/IAI.69.9.5573-5576.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, Howard J C. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. 2005;1:e24. doi: 10.1371/journal.ppat.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher B A, Greene R I, Henry S C, Annecharico K L, Weinberg J B, Denkers E Y, Sher A, Taylor G A. p47 GTPases regulate Toxoplasma gondii survival in activated macrophages. Infect Immun. 2005;73:3278–3286. doi: 10.1128/IAI.73.6.3278-3286.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor G A, Stauber R, Rulong S, Hudson E, Pei V, Pavlakis G N, Resau J H, Vande Woude G F. The inducibly expressed GTPase localizes to the endoplasmic reticulum, independently of GTP binding. J Biol Chem. 1997;272:10639–10645. doi: 10.1074/jbc.272.16.10639. [DOI] [PubMed] [Google Scholar]
- Martens S, Sabel K, Lange R, Uthaiah R, Wolf E, Howard J C. Mechanisms regulating the positioning of mouse p47 resistance GTPases LRG-47 and IIGP1 on cellular membranes: retargeting to plasma membrane induced by phagocytosis. J Immunol. 2004;173:2594–2606. doi: 10.4049/jimmunol.173.4.2594. [DOI] [PubMed] [Google Scholar]
- MacMicking J D, Taylor G A, McKinney J D. Immune control of tuberculosis by IFN-γ-inducible LRG-47. Science. 2003;302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- Gutierrez M G, Master S S, Singh S B, Taylor G A, Colombo M I, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Singh S B, Davis A S, Taylor G A, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- Ling Y M, Shaw M H, Ayala C, Coppens I, Taylor G A, Ferguson D J, Yap G S. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med. 2006;203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer T, Duffy A, Weiss L M, Halonen S K. IGTP is necessary for Toxoplasma vacuolar disruption and induces parasite egression in IFN{γ} stimulated astrocytes. Infect Immun. 2008;76:4883–4894. doi: 10.1128/IAI.01288-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafica A, Feng C G, Santiago H C, Aliberti J, Cheever A, Thomas K E, Taylor G A, Vogel S N, Sher A. The IFN-inducible GTPase LRG47 (Irgm1) negatively regulates TLR4-triggered proinflammatory cytokine production and prevents endotoxemia. J Immunol. 2007;179:5514–5522. doi: 10.4049/jimmunol.179.8.5514. [DOI] [PubMed] [Google Scholar]
- Feng C G, Weksberg D C, Taylor G A, Sher A, Goodell M A. The p47 GTPase Lrg-47 (Irgm1) links host defense and hematopoietic stem cell proliferation. Cell Stem Cell. 2008;2:83–89. doi: 10.1016/j.stem.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunn J P, Koenen-Waisman S, Papic N, Schroeder N, Pawlowski N, Lange R, Kaiser F, Zerrahn J, Martens S, Howard J C. Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. EMBO J. 2008;27:2495–2509. doi: 10.1038/emboj.2008.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoiseth S K, Stocker B A. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature. 1981;291:238–239. doi: 10.1038/291238a0. [DOI] [PubMed] [Google Scholar]
- VanCott J L, Chatfield S N, Roberts M, Hone D M, Hohmann E L, Pascual D W, Yamamoto M, Kiyono H, McGhee J R. Regulation of host immune responses by modification of Salmonella virulence genes. Nat Med. 1998;4:1247–1252. doi: 10.1038/3227. [DOI] [PubMed] [Google Scholar]
- Wray C, Sojka W J. Experimental Salmonella typhimurium infection in calves. Res Vet Sci. 1978;25:139–143. [PubMed] [Google Scholar]
- Striepen B, He C Y, Matrajt M, Soldati D, Roos D S. Expression, selection, and organellar targeting of the green fluorescent protein in Toxoplasma gondii. Mol Biochem Parasitol. 1998;92:325–338. doi: 10.1016/s0166-6851(98)00011-5. [DOI] [PubMed] [Google Scholar]
- Taylor G A, Jeffers M, Largaespada D A, Jenkins N A, Copeland N G, Woude G F. Identification of a novel GTPase, the inducibly expressed GTPase, that accumulates in response to interferon γ. J Biol Chem. 1996;271:20399–20405. doi: 10.1074/jbc.271.34.20399. [DOI] [PubMed] [Google Scholar]
- Govoni G, Gros P. Macrophage NRAMP1 and its role in resistance to microbial infections. Inflamm Res. 1998;47:277–284. doi: 10.1007/s000110050330. [DOI] [PubMed] [Google Scholar]
- Govoni G, Vidal S, Gauthier S, Skamene E, Malo D, Gros P. The Bcg/Ity/Lsh locus: genetic transfer of resistance to infections in C57BL/6J mice transgenic for the Nramp1 Gly169 allele. Infect Immun. 1996;64:2923–2929. doi: 10.1128/iai.64.8.2923-2929.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor G A, Feng C G, Sher A. Control of IFN-γ-mediated host resistance to intracellular pathogens by immunity-related GTPases (p47 GTPases) Microbes Infect. 2007;9:1644–1651. doi: 10.1016/j.micinf.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Hunn J P, Koenen-Waisman S, Papic N, Schroeder N, Pawlowski N, Lange R, Kaiser F, Zerrahn J, Martens S, Howard J C. Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. EMBO J. 2008;27:2495–2509. doi: 10.1038/emboj.2008.176. [DOI] [PMC free article] [PubMed] [Google Scholar]