

Abstract
Laminins have dramatic and varied actions on neurons in vitro. However, their in vivo function in brain development is not clear. Here we show that knockout of laminin γ1 in the cerebral cortex leads to defects in neuritogenesis and neuronal migration. In the mutant mice, cortical layer structures were disrupted, and axonal pathfinding was impaired. During development, loss of laminin expression impaired phosphorylation of FAK and paxillin, indicating defects in integrin signaling pathways. Moreover, both phosphorylation and protein levels of GSK-3β were significantly decreased, but only phosphorylation of AKT was affected in the mutant cortex. Knockout of laminin γ1 expression in vitro, dramatically inhibited neurite growth. These results indicate that laminin regulates neurite growth and neuronal migration via integrin signaling through the AKT/GSK-3β pathway, and thus reveal a novel mechanism of laminin function in brain development.
Keywords: Neuronal migration, cortical development, laminin, neurite growth Paxillin, FAK, AKT, GSK-3β
Introduction
The mammalian adult cerebral cortex exhibits an organized laminar structure with six neuronal layers. During development, waves of neuroblasts generated from the ventricular neuroepithelium migrate towards the pia to form successive cortical layers (Rakic, 1990). Earlier-born neurons travel short distances possibly by somal translocation (Nadarajah et al., 2001; Nadarajah and Parnavelas, 2002) whereas later-born neurons migrate further, through the older neuronal layers by migrating along radial glial fibers (Angevine and Sidman, 1961; Sidman and Rakic, 1973). The radial glial network is formed by glial processes extending from the ventricle toward the surface of the brain with end-feet attaching to the pia basement membrane (Marin and Rubenstein, 2003).
Neuronal migration is composed of two phases: leading process extension and forward replacement of the soma and nucleus into the leading process (Tsai and Gleeson, 2005). Many factors such as cell adhesion molecules and extracellular matrix (ECM) proteins can regulate the direction and extension of neuronal processes during development (Hopker et al., 1999; Kiryushko et al., 2004) . Laminins are major components of ECM and are heterotrimeric molecules composed of an α, β and γ chain (Timpl, 1996). They are expressed in the cerebral cortex and play important roles in neuronal plasticity, degeneration and regeneration (Chen et al., 2003; Chen and Strickland, 1997; Grimpe et al., 2002; Indyk et al., 2003; Nakagami et al., 2000; Yin et al., 2003). Laminins also participate in neurite outgrowth and axon pathfinding in vitro (Bonner and O'Connor, 2001; Gomez and Letourneau, 1994; Kuhn et al., 1995; Letourneau et al., 1988; Luckenbill-Edds, 1997; McLoon et al., 1988; Rogers et al., 1986; Timpl and Brown, 1994). For example, hippocampal neurons will extend several neurites in vitro, but the first neurite to reach a laminin substrate develops preferentially and becomes the axon (Esch et al., 1999; Menager et al., 2004). Laminins are also involved in neuronal migration and brain development (Colognato and Yurchenco, 2000; Liesi et al., 2001; Luckenbill-Edds, 1997; Miner et al., 1998). During cortical development, laminin is deposited along the radial glial fibers (Liesi, 1985; Liesi, 1990), and antibodies raised against a neurite outgrowth-promoting site of the laminin γ1 chain inhibit neuronal migration (Liesi et al., 1995; Liesi et al., 1992). In vitro, when neurons migrate on laminin, they first extend processes and then move the nucleus inside the process that has formed (Liesi, 1992). This process is very similar to neuronal migration along radial glial fibers in vivo (Tsai and Gleeson, 2005).
Even though the in vitro effect of laminin on neurons is well studied, their in vivo function in the CNS is not clear. Mice with a targeted deletion of the nidogen-binding site of the laminin γ1 chain show instability of the pial basement membrane in the brain and abnormal neuronal migration (Halfter et al., 2002). This study revealed a nidogen-binding dependent function of laminin γ1 in brain development. Since global knockout of laminin γ1 expression leads to early embryonic lethality (embryos die at E5.5) (Mitchell et al., 2001; Skarnes et al., 1995; Smyth et al., 1999), but mice with deletion of the nidogen-binding site of laminin γ1 survive until birth (Willem et al., 2002), some essential functions of laminin γ1 must be nidogen-binding independent.
To study the role of laminin in cerebral cortex development, we disrupted laminin expression in the brain using the Cre-loxP system (Chen and Strickland, 2003; Yu et al., 2005). The brains of these mice have a disrupted cerebral cortical layer structure, and cortical neurons show shorter neurites. Knockout of laminin γ1 perturbs neuronal migration and impairs integrin and AKT/GSK-3β signaling, but cell proliferation and neuronal cell death are similar between mutant and control embryos. Our results indicate that laminin plays a critical role in neuronal morphogenesis and migration in vivo.
Materials and Methods
Mouse lines and analysis of Cre activity
The mutant mice used were homozygous for a floxed laminin γ1 allele (Chen and Strickland, 2003) and carried the Cre recombinase transgene under the Calcium-Calmodulin-dependent protein Kinase II α promoter (Dragatsis and Zeitlin, 2000) (CaMKII/Cre:fLAMγ1 mice). Cre recombinase activity was monitored by using the LacZ/EGFP double reporter mouse line (Novak et al., 2000), and visualization of EGFP (described below). The floxed allele, Cre and EGFP were detected by genotyping mouse tail genomic DNA (Chen and Strickland, 2003).
Histological analysis, TUNEL staining, BrdU incorporation, and Golgi staining
All animals were maintained according to Animal Welfare guidelines at the Rockefeller University. Histology (Chen and Strickland, 2003; Chen and Strickland, 1997), TUNEL staining (Yu et al., 2005), BrdU incorporation assay (Yu et al., 2005), and Golgi staining using the FD Rapid GolgiStain kit (FD NeuroTechnologies Inc., Baltimore, MD) were performed as described.
Luxol Fast Blue staining
Luxol Fast Blue staining was performed overnight at 60 °C with 0.1% Luxol Fast Blue in 95% ethanol and 0.05% acetic acid. Color was developed by alternate rinses in 0.05% Li carbonate and 70% ethanol. The brain sections were dehydrated, and mounted in DPX (Sigma- Aldrich, St Louis, MO).
Immunostaining
Immunostaining was performed as described (Chen and Strickland, 2003; Chen and Strickland, 1997). Primary antibodies used were: calretinin (1:1000, Chemicon MAB1914, Temecula, CA), laminin I (1:2000, Sigma, St Louis, MO), laminin γ1 (1:500, Chemicon MAB1914, Temecula, CA), neurofilament (1:2000, Chemicon AB5539, Temecula, CA), TuJ1 (1:500, Covance, Berkeley, CA), BrdU (1:500, Abcam, Cambridge, UK) and SMI32 (1:2500, Sternberger Monoclonals Inc, Berkeley, CA).
Western blot analysis
Western blot analysis was performed as described (Yu et al., 2005). Antibodies against p-FAK, FAK, p-paxillin, paxillin, p-Akt, Akt, p-GSK-3β, GSK-3β, from Cell Signaling (Beverly, MA) were used at 1:1000 dilution. Seven control and mutant embryonic brains were used for each Western blot and were repeated three times. For loading controls, each membrane was re-probed with anti-β-actin antibodies (Sigma, St Louis, MO 1:8000). The Western blot films were digitized using a scanner (Microtek, Carson, California). The signal intensity of the Western Blot film was quantified by NIH Image and normalized to actin. The differences of signal intensity between control and mutant samples for each Western blot were analyzed by Student's t-Test.
Cortical cell culture
Cortical cell cultures were prepared from E18.5 embryos homozygous for the floxed laminin γ1 allele (Siao and Tsirka, 2002). After plating, adenovirus expressing LacZ (Vector Biolabs, Philadelphia, PA, for control) or Cre (Vector Biolabs, Philadelphia, PA, to knockout laminin γ1 gene expression) were added to the culture medium. The cultures were maintained in serum free medium. Three days after culture, the cover slips were fixed briefly and stained with anti-laminin and TuJ1 antibodies. For quantitative analysis, the length of the longest neurites of 150 randomly selected neurons from 3 different cultures in each group were measured using Axiovision 4 (Carl Zeiss) under a Carl Zeiss Axiovert 200 microscope, the differences between control and mutant neurons were analyzed by Student's t-test.
Results
Spatial and temporal relationship between Cre expression and laminin γ1 gene disruption in the mutant cerebral cortex during development
We have demonstrated previously that recombination of the laminin γ1 gene occurs in CaMKII/Cre:fLAMγ1 mice (hereafter termed mutant mice) in the hippocampus, spinal cord and peripheral nerves, but not in muscle or heart (Chen and Strickland, 2003). To monitor the expression of the Cre recombinase within the cerebral cortex, we used the double reporter mouse strain LacZ/EGFP (Z/EG) (Novak et al., 2000). In this mouse strain, in the absence of Cre activity, only LacZ is expressed, and after Cre excision of the LacZ gene, only EGFP is expressed. We generated mice that were homozygous for the floxed laminin γ1 allele and also carried both the CaMKII-Cre and the Z/EG transgenes. In these mice, Cre activity was indicated by EGFP expression. As shown in Figure 1, there was very little EGFP expression in the cerebral cortex on or before E12.5 (Fig. 1A); however, EGFP was broadly expressed in the cerebral cortex at E14.5 (Fig. 1B). EGFP was not expressed in the ventricular zone (arrow heads in Fig. 1B). EGFP expression pattern in the cortex remained consistent thereafter, but was broader and in more neurons (data not shown). To determine the spatial and temporal disruption of laminin γ1 gene expression and the cell types that expressed EGFP, the brain sections from E12.5 and E14.5 embryos were stained with antibodies against laminin γ1, TuJ1(Neuron) and calretinin (Cajal-Retzius cells). At E12.5, there was no significant change in laminin γ1 expression in the mutant cortex (data not shown); however, at E14.5, in areas expressing EGFP, laminin γ1 expression was decreased (arrows in Fig.1C and D, images are from regions equivalent to boxed area 1 in panel B of Fig. 1). However, in the adjacent areas that did not show EGFP expression, laminin γ1 expression was high (arrowheads in Fig. 1C and D). Since laminin is an extracellular matrix protein, disruption of its expression in some but not all cells could result in a decreased expression in the surrounding areas of these cells. A comparison of the intensity of laminin γ1 immunoreactivity between the areas that show EGFP expression in the cortex of the mutant embryos and the corresponding regions of the control embryos (E14.5) revealed a significant decrease in the cortex of mutant embryos (Fig. S1). These results showed a general correlation between Cre expression (indicated by EGFP expression) and laminin γ1 gene disruption (laminin γ1 immunoreactivity decrease).
Figure 1. Spatial and temporal relationship between Cre expression and laminin γ1 disruption in developing cerebral cortex of mutant mice.

Embryos homozygous for the floxed laminin γ1 allele and also carrying both CaMKII-Cre and Z/EG transgenes at E12.5 (A) and E14.5 (B-M) were used. Cre expression is indicated by EGFP reporter gene (green). E14.5 embryonic brain sections were stained with antibodies against TuJ1 (E, F, G and H), calretinin (J) and laminin γ1 (C, D, L, M). Images from C to F are from brain regions equivalent to boxed area 1 in panel B after immunohistochemistry as indicated. Images from G to M are from brain regions equivalent to boxed area 2 in panel B. Scale bars for A and B is 100 μm; C and D is 20 μm; E and F is 10 μm and G-M is 10 μm.
The cells that expressed EGFP in the cortical plate (region equivalent to boxed area 1 in panel B of Fig. 1) were TuJ1-positive cells (arrows in Fig. 1E and F) indicating neuronal identity. At E16.5 and E18.5 there were more cells expressing EGFP (Cre) and laminin γ1 was further decreased in the mutant cortices relative to the control cortices at the same developmental stages (Fig. S2). Wherever Cre was expressed, laminin γ1 immunoreactivity was decreased. Since most of the EGFP expressing cells were neurons and laminin γ1 expression was decreased wherever EGFP was expressed, neurons are probably a major source of laminin expressed in the cortex. In the marginal zone near the pia (equivalent to areas in box 2 in panel B of Fig.1), the EGFP expressing cells were TuJ1-positive (arrowheads in Fig.1G and H), and also calretinin positive (arrowheads in Fig.1I, J and K) indicating that these cells are probably Cajal-Retzius cells (Graus-Porta et al., 2001; Weisenhorn et al., 1994). EGFP positive cells in these regions (arrowheads in Fig.1L and M) did not affect laminin γ1 expression in pial basement membrane (arrows in Fig.1L and M) suggesting that these cells were not meningeal cells. In areas directly beneath the pia basement membrane laminin was not expressed either in the mutant (Fig. 1L and M) or in control embryos (data not shown), which was different than areas shown in Fig. 1 C and D.
The above results show that laminin γ1 synthesis was disrupted in cortical neurons where Cre recombinase was expressed. However Cre was expressed in large groups of neurons in localized areas, but not all neurons in the cerebral cortex, which may explain why the defects occurred in localized areas (see below).
Disruption of laminin γ1 expression leads to abnormalities in the cerebral cortex
The majority of mutant mice (81%) showed severe defects in cortical cytoarchitecture (Fig. 2B compare to normal mice, Fig. 2A, mice at P0 stage). The thickness of the mutant cortex was similar to control mice. The laminar structure of the cortex was affected with cells mostly under-migrated (Fig. 2B). Occasionally, some neurons migrated to the marginal zone and formed ectopias (data not shown). The neurons that did not migrate properly gathered together forming crescent-shaped disruptions in cortical organization (Fig 2 B and D). The reason for this phenomenon is not clear. One possibility is that Cre expression in localized large groups of neurons results in all these cells not migrating properly while the adjacent wild-type cells still migrate normally forming a crescent-shaped disruption (Fig S3).
Figure 2. Disruption of laminin γ1 gene leads to cerebral cortical defects.
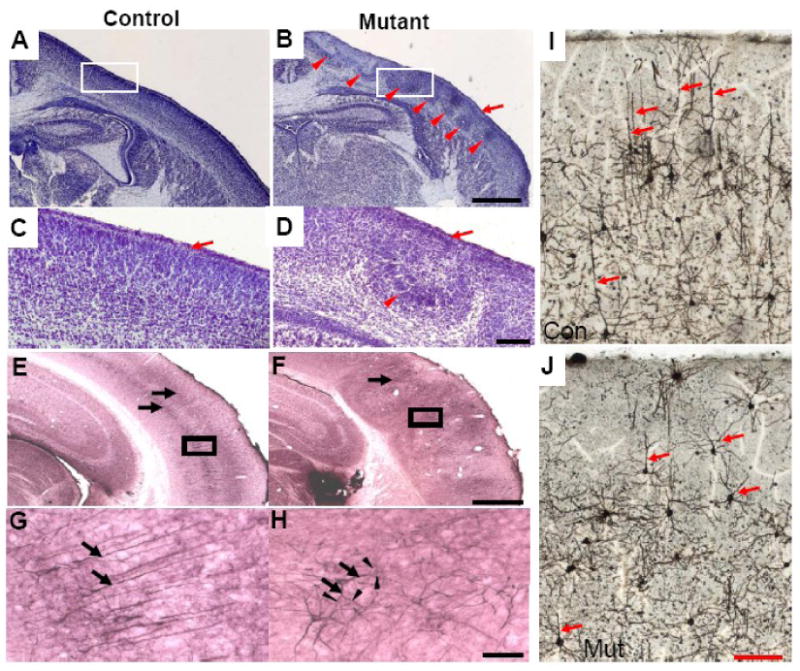
A-D, Coronal brain sections from newborn pups were analyzed by Cresyl Violet staining. Higher magnification of boxed areas in A and B are shown in C and D respectively. Arrows indicate normal marginal zone, arrowheads indicate abnormal cortical structures. E-H, adult brain sections were stained with antibodies against non-phosphorylated neurofilament (SMI 32). Arrows indicate cortical layers formed by SMI 32 positive neurons. G and H are higher magnification of the boxed areas in E and F respectively. Arrows in G indicate apical dendrites of pyramidal neurons in layer V. Arrowheads in H indicate branches from multiple dendrites (indicated by arrows) in the mutant cerebral cortex. I and J are Golgi stainings of adult control and mutant brain sections. Arrows in I and J indicate apical dendrites of pyramidal neurons. Scale bars for A and B is 0.5mm; C and D is 50 μm; G and H is 50 μm; I and J is 100 μm.
The phenotype was first observed at E14.5 and worsened until birth. After birth there was little if any further development of the defects in the cortex, since in adult mutant mice, the phenotype was similar to new born pups (data not shown).
This phenotype of neuronal under-migration was different than that seen in the cerebral cortices of mice with deletion of nidogen-binding site of the laminin γ1 chain (Halfter et al., 2002). In the cerebral cortex of the mutant mice used in this study, migration of neurons into the marginal zone was rare, but most of the defects were associated with retarded neuronal migration.
To analyze the formation of specific cortical layers in adult mice, we performed immunohistochemistry using a monoclonal antibody against a non-phosphorylated epitope of neurofilament heavy chain, SMI-32 (Fig 2 E-H). This antibody labels pyramidal neurons in layers III and V (Campbell and Morrison, 1989). In the control cerebral cortex, the SMI-32 positive neurons formed two layers that were parallel to the surface of the brain (Fig. 2E). In contrast, in the mutant adult brains, layers III and V were disorganized and had a ruffled appearance (Fig 2F). To determine if the neuronal morphology was changed, we examined the SMI 32 NF-positive neurons under higher magnification. Our results showed that the neuronal morphology in either layer III or V of the mutant mouse brains was significantly different than in the controls. The neurons in layer V of the control cortex formed long straight apical dendrites radial to the surface of the brain (arrows in Fig.2G). In contrast, neurons in the mutant cortex did not form long apical dendrites (arrows in Fig.2H). Dendrites of mutant neurons formed multiple branches (arrowheads in Fig.2H). These results indicate that laminin is important for normal brain laminar formation and neuronal morphogenesis.
We further compared neuronal morphology between adult control and mutant mice by Golgi staining, as shown in Figure 2I and J. In the cerebral cortex of the control mice, pyramid neurons formed long apical dendrites toward to the surface of the brain. In contrast, in the mutant cerebral cortex, the apical dendrites of the pyramid neurons were much shorter and branched more. This result further confirmed the morphological changes of the mutant neurons as revealed by SMI-32 immunohistochemistry.
Axonal pathfinding defects in the cortex of the laminin γ1 mutant mice
Since laminin has been implicated in axonal pathfinding and axonal guidance (Garcia-Alonso et al., 1996; Hopker et al., 1999; Kafitz and Greer, 1997), we evaluated the cortex of mutant mice for evidence of defective axon pathfinding. We stained adult control and mutant mouse brains with antibodies against neurofilament. As shown in Figure 3, the antibody labeled the corpus callosum (arrowheads in Fig. 3A and B). In the mutant mouse brain there were abnormal branches from the corpus callosum extending toward the surface of the brain (arrows in Fig. 3B). To further confirm the abnormal axonal guidance, we stained the brain sections with Luxol Fast Blue which stains the myelin sheath of the myelinated axons. As shown in Figure. 3C and D, it stained the myelin sheath of the myelinated axons in the corpus callosum. However, in the mutant mouse brain, the myelinated axonal bundles from the corpus callosum extended to the cerebral cortex toward the surface of the brain (Arrows in Fig.3D). This result was consistent with the neurofilament staining and further demonstrated the abnormal axonal pathfinding in the mutant mice. The reason for the axonal pathfinding defect in laminin γ1 deficient mice is not clear. One possible explanation could be that axons release laminin which guides their pathfinding. Another possibility is that laminin is expressed by the neurons to which the axons are targeted and serves to promote axon growth. If these neurons lose laminin expression, the axons will not be able to correctly find their target.
Figure 3. Abnormal axonal pathfinding in the cerebral cortex of mutant mice.

Fixed adult control (A and C) and mutant (B and D) mouse brain sections were stained with anti-neurofilament antibody (A and B) or Luxol Fast Blue (C and D). In mutant mouse brains, axons in the corpus callosum (arrowhead) branch into the cortex toward the surface of the brain (arrows in B and D). Scale bars for A and B is 0.5 mm and C and D is 200 μm.
The above results show that disruption of laminin γ1 gene leads to abnormal brain development. The defects were only observed in embryos or mice that were homozygous for the floxed laminin γ1 allele and also carry the Cre transgene, but not in mice that were only homozygous for the floxed allele and in mice that were heterozygous for the floxed allele and also carry the Cre transgene.
There were no obvious malformations in the cerebellum of the mutant mice, where Cre-mediated recombination is minor.
Proliferation is normal and apoptosis is not induced in the cerebral cortex of the mutant mouse during development
Since laminin is involved in cell proliferation in the peripheral nervous system (PNS) (Yu et al., 2005), one possible cause for the cortical abnormalities could be defects in cell proliferation. In the developing cerebral cortex, cell proliferation occurs in the ventricular zone. As shown in Figure 4A, at E16.5, EGFP was not expressed in the ventricular zone (Fig 4Aa and Ab). At E18.5 the EGFP expression pattern was similar to E16.5. Nevertheless, we used BrdU incorporation to compare cell proliferation in the ventricular zone of control and mutant cerebral cortex. BrdU was injected into pregnant mice at gestational day 16.5, and embryos were collected fifteen minutes later for BrdU incorporation analysis. The number of BrdU positive cells was similar between control and mutant embryos in the ventricular zone (Fig. 4A c-g). Cell proliferation rates in the ventricular zone between control and mutant embryos at E12.5, E14.5 and E18.5 were also similar (data not shown). These results demonstrate that in this mutant mouse line, Cre was not expressed in the ventricular zone of the cerebral cortex during development and cell proliferation was not affected.
Figure 4. Cell proliferation is normal and apoptosis is not induced, but neuronal migration is affected in the cerebral cortex of the mutant mouse during development.
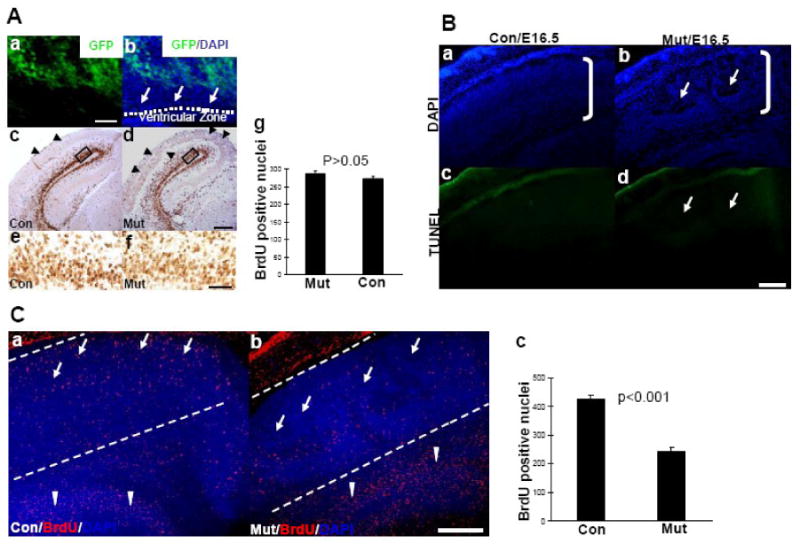
4A. Cell proliferation in the ventricular zone of mutant embryos is comparable to that of control embryos during development.
Brain sections from embryos homozygous for the floxed laminin γ1 allele and also carrying both CaMKII-Cre and Z/EG transgenes at E16.5 were analyzed for EGFP expression in the ventricular zone (Aa and b). EGFP is not expressed in this region. BrdU incorporation assay was done on E16.5 embryos. Pregnant mice were injected with BrdU at gestational day 16.5, and 15 min (A c and d) later, the embryos were collected. Brain sections were stained with anti-BrdU antibodies. The numbers of BrdU-positive nuclei in the ventricular zone of control and mutant mice were similar (A c-g). Higher magnification of the boxed areas in c and d are shown in e and f. For quantitative analysis, BrdU-positive nuclei in similar regions of the ventricular zone between control and mutant mice were counted (7 embryos for each genotype were analyzed) per microscope field. The differences between control and mutant were not significant as analyzed by Student's t test. Scale bars for panel A is: a and b 20 μm; c and d 100 μm; e and f 20 μm.
4B. Knockout of laminin γ1 expression in the cerebral cortex during development does not induce apoptosis.
E16.5 control (Ba and c) and mutant (Bb and d) embryonic brain sections were stained with TUNEL kit (Bc and d) and counterstained with DAPI (Ba and Bb). In the regions which show defects (arrows in Bb), there was no detectable cell death (arrows in Bd). Scale bars for Ba-d is 200 μm.
4C. Abnormal neuronal migration in the mutant cerebral cortex.
Pregnant mice were injected with BrdU at gestational day 15.5, and 24 hours (C a and b) later, the embryos were collected. Control (Ca) and mutant (Cb) embryonic brain sections were stained with anti-BrdU antibody and counterstained with DAPI. For quantitative analysis, BrdU-positive nuclei in the cortical plate which is between the two lines shown in C a and b were counted. One of the two lines was drawn beneath the pia (which is obvious under a microscope) and another line was approximately 500 μm away from the pia; the area between these two lines was cortical plate (roughly). The differences in BrdU-positive nuclei between control and mutant embryos were analyzed by Student's t test (7 embryos for each genotype were used). Scale bars for Ca and b is 200 μm.
Since laminin is implicated in cell death (Chen and Strickland, 1997; Yu et al., 2005) and since the regions that showed defects contained fewer cells (Fig. 2B and D, Fig. 4Bb and 4Cb), apoptotic cell death may contribute to the defects. We then examined cell death using a TUNEL assay. As shown in Figure 4B, the regions that showed defects (arrows in Fig. 4Bb) did not show signs of apoptosis (arrows in Fig. 4Bd). Examination of brain sections from E12.5, E14.5 and E18.5 embryos revealed no increased cell death in the mutant cerebral cortex (data not shown). Immunohistochemistry using antibodies against activated caspase-3 and caspase-7 showed no staining in the affected areas in the mutant mouse brain (data not shown). This result indicates that during cortical development, loss of laminin γ1 does not induce cell death.
Neuronal migration defects in the mutant cortex
Tightly controlled neuronal migration is critical for laminar formation of the cerebral cortex. Since neither cell proliferation nor apoptosis are abnormal in the mutant cerebral cortex during development, another possible cause for the cortical defects is impairment in neuronal migration. To investigate this possibility, we tracked neuronal migration in vivo, using BrdU to label neurons. We injected BrdU in pregnant mice at gestational day15.5, and 24 hours later, the position of BrdU-labeled neurons was visualized by anti-BrdU immunohistochemistry on brain sections. As shown in Figure. 4C, in the control cortex, neurons migrated throughout the cortex and reached the superficial layers (arrows in Fig.4Ca). In contrast, in the mutant cortex only few neurons migrated to the superficial layers of the cortex (arrows in Fig.4Cb). More neurons accumulated in the ventricular zone in the mutant embryos (arrowheads in Fig.4Cb) as compared to the controls (arrowheads in Fig.4Ca). Quantitative analysis revealed that there were significantly fewer neurons migrating to the cerebral cortex in the mutant than in the control mice especially in the superficial layers (Fig. 4Ca-c). These results indicated that neuronal migration in the mutant cortex is affected.
Knockout of laminin γ1 expression in the cerebral cortex impairs the phosphorylation of integrin associated molecules
The above results show that knockout of laminin γ1 expression in neurons in the cerebral cortex leads to neuronal morphology and migration abnormalities, and these phenotypes (neuronal morphological and migration abnormalities) are similar to that of integrins or FAK knockout mice (Schmid and Anton, 2003, Beggs HE, et al., 2003, 501) but with some differences (most mutant neurons under-migrate in laminin γ1 knockout mice, while in integrin or FAK knockout mice, some neurons under-migrate but most of them over-migrate into the marginal zone). It is not clear whether laminin participates in cortical development through the integrin signaling pathway and whether lack of laminin expression in the cerebral cortex affects the activation of integrin receptors. FAK is a non-receptor tyrosine kinase and is strongly activated following integrin binding to extracellular matrix proteins (Parsons, 2003) and plays important roles in integrin-mediated signaling transduction. To investigate whether the activation of FAK is affected in the mutant cerebral cortex, we compared the phosphorylation levels of FAK between control and mutant cerebral cortex. As shown in Figure. 5A, the total levels of FAK in the cortex of mutant mice were similar to that of the controls. However, the levels of phosphorylated FAK were significantly decreased, indicating that knocking out laminin γ1 expression in the cortex impaired the activation of integrin receptors and downstream molecules.
Figure 5. Signaling pathway changes in the mutant cerebral cortex.

Cerebral cortices from E16.5 control and mutant embryos were collected and proteins were extracted for western blots using antibodies as indicated. For quantitative analysis, seven mutant and control embryos were used. The intensity of the positive bands was normalized with actin and the differences between control and mutant were analyzed by Student's t-test.
Paxillin is a focal adhesion-associated adaptor protein involved in integrin-mediated signaling (Schaller, 2001). Binding of FAK to paxillin can regulate its phosphorylation (Thomas et al., 1999). Paxillin also controls cell migration (Schaller, 2001). However, its function in CNS development is not clear. To investigate whether paxillin levels changed in the mutant cerebral cortex we examined its phosphorylation level. As shown in figure 5B, the phosphorylation level of paxillin was dramatically decreased compared to that of the control. However the total protein levels were similar between these two groups. This result indicates that lack of laminin expression in the cerebral cortex during development impairs the integrin signaling pathway, including paxillin.
Impairment of AKT/GSK-3β signaling pathway in the mutant cerebral cortex
Since the AKT/GSK-3β signaling pathway is involved in neuronal morphogenesis (Jiang et al., 2005; Yoshimura et al., 2005; Zhou et al., 2004) and laminin has a role in neurite outgrowth and neuronal polarity in vitro (Arimura and Kaibuchi, 2005), we examined the phosphorylation levels of AKT in the mutant cortex. We found that the levels were significantly decreased compared to the controls at E16.5 (Fig. 5C). We also examined the expression levels of GSK-3β, as shown in Figure 5D, and found that both total protein and phosphorylated levels of GSK-3β were dramatically decreased in the mutant cerebral cortex compared to that of the control. This result indicates that lack of laminin expression in the cerebral cortex during development affects the expression and phosphorylation of GSK-3β, and the levels of phosphorylated AKT, but not the total levels of AKT.
Disruption of laminin γ1 expression affects neuronal morphogenesis in vitro
Laminin γ1 mutant mice show neuronal morphological abnormalities in their cerebral cortex (Fig. 2). However this could be a secondary effect of ectopic positioning of the neurons. To investigate whether disruption of laminin γ1 expression directly affects neuronal morphogenesis, we performed primary neuronal culture from embryos that are homozygous for the floxed laminin γ1 allele. Disruption of laminin γ1 expression was achieved by addition of adenoviruses that express Cre recombinase (Ad-CMV-Cre). Adenoviruses expressing LacZ (Ad-CMV-LacZ) were used as controls. As shown in Figure 6, the control viruses were not toxic to neurons and did not affect neuronal morphology or laminin expression (Fig.6 A-C). However, adeno-Cre viruses significantly reduced laminin expression in neurons (compare Fig. 6D to A) and caused dramatic neuronal morphological changes (compare Fig. 6E to B). Quantitative analysis revealed that disruption of laminin γ1 expression significantly blocked neurite outgrowth (Fig. 6G).
Figure 6. Knockout of laminin γ1 expression inhibits neurite outgrowth in vitro.

Cortical neurons from E18.5 embryos homozygous for the floxed laminin γ1 allele were cultured on PDL-coated cover slips. After plating, adenoviruses expressing LacZ (A-C) (for control) or Cre (D-F) (to knock out laminin γ1 gene expression) were added to the culture medium. Three days after culture, the cover slips were stained with anti-laminin (A and D) and TuJ1 (B and E) antibodies. For quantitative analysis, the longest neurites of 150 randomly selected neurons from 3 different cultures in each group were measured using Axiovision (Zeiss), the differences were analyzed by Student's t test (G). Scale bars for A-F are 20 μm.
We also performed neuronal cultures from mutant mice and found there were not obvious neuronal morphological changes. However, some cells do not undergo recombination in the mutant mice and can still secrete laminin in cultures made from these mice. This laminin can therefore rescue the defect in the cultures. In contrast, laminin produced in vivo by non-recombinant cells has limited diffusion and the mutant phenotype is manifest. Partial rescue in vivo may also explain why the phenotype is localized in specific areas. These results show that laminin γ1 plays an important role in neurite outgrowth and suggests that the morphological defects in the mutant neurons are likely due to a lack of laminin expression and not a secondary effect of abnormal migration.
Discussion
Laminins are implicated in neurite outgrowth and axonal specification in vitro (Esch et al., 1999; Luckenbill-Edds, 1997), but their in vivo function in the nervous system is not well studied. Selective disruption of the laminin γ1 gene in the cerebral cortex resulted in mutant mice which developed severe brain abnormalities. The defects included abnormal cortical anatomy, layer formation, neuronal morphogenesis and axonal pathfinding. However the prominent feature of the mutant cerebral cortex is that neurons do not migrate enough to reach the superficial layers. This phenotype is different than the over-migration of neurons into the marginal zone in mice with targeted deletion of the nidogen-binding site of laminin γ1 (Halfter et al., 2002) and laminin receptor knockout mice (Moore et al., 2002; Schmid and Anton, 2003). In these cases, cerebral cortex abnormities are due to defective pia basement membrane. This neuronal migration defect difference between laminin γ1 knockout mice and integrin or FAK knockout mice may indicate that in the absence of these molecules, some other receptors or signaling molecules could compensate for their functions. Since the Cre expression in the CaMKII-Cre transgenic mice is primarily in neurons, the phenotype in mice most likely stems from an absence of laminin in cerebral cortical neurons. The radial glial cells are in general morphologically normal in laminin γ1 knockout mice, even though occasionally there were some minor changes. The minor defects include some with irregular morphology, some radial fibers that are not straight, and a few radial fibers that do not extend to the surface of the brain. If radial glial cells contribute to the phenotype, it could be just a minor effect. Therefore the phenotypes of laminin γ1 knockout mice may reveal the function of neuronal laminin and indicate that laminins play an important role in neuronal morphogenesis and neuronal migration in vivo.
The role of neuronal and pial basement membrane laminin in cerebral cortex development
ECM proteins and their receptors are important for brain development (Costell et al., 1999; Georges-Labouesse et al., 1998; Graus-Porta et al., 2001; Moore et al., 2002) and most of them are involved in the assembly and maintenance of the integrity of the pia basement membrane. The major components of the pia basement membrane are produced by meningeal cells. Selective ablation of meningeal cells by 6-hydroxydopamine leads to cortical dysplasias (Sievers et al., 1994). Mutations in mouse genes which have a role in the formation and assembly of the pial basal lamina also result in abnormal cortical development with extrusions of neurons through breaches in the basement membrane. Mutations in perlecan lead to discontinuous expression of laminin in the pia basement membrane and cortical dysplasia (Costell et al., 1999). Deletion of the nidogen binding site of the laminin γ1 gene leads to the same phenotype and furthermore results in a disorganized radial glial scaffold (Halfter et al., 2002). Mutations in receptors for laminins, such as dystroglycan and α6 and β1 integrins have similar effects (Georges-Labouesse et al., 1998; Graus-Porta et al., 2001; Moore et al., 2002), as does lack of cortical expression of the intracellular molecule FAK (Beggs et al., 2003). All of these mutant animals have one common observed defect: laminin disruptions in the pia. Therefore, it is established that defects in the assembly or integrity of the pia basement membrane will lead to abnormal neuronal migration probably by affecting the end-feet attachment to the radial glial fibers. Since the radial glial fibers normally extend and attach to the pial basal lamina, breaches within the pia basement membrane lead to disorganization of the radial glial scaffold. The guidance of neuroblast migration by the radial glial scaffold is impaired and leads to an abnormally formed cortical plate and eventually to abnormal cortical layers.
Laminins are also expressed in cortical neurons (Grimpe et al., 2002; Indyk et al., 2003; Yin et al., 2003). However, the role of neuronal laminin is not clear. Our results show that loss of laminin in a large group (not all of them) of cortical neurons impairs neuronal morphogenesis and migration. Therefore the function of laminin in the cerebral cortex is important in two locations: at the pia basement membrane and in developing neurons. Laminin in the pia basement membrane regulates neuronal migration through the radial glia scaffold, while neuronal laminin regulates neuronal migration probably by promoting neurite extension. When neuronal laminin expression is ablated, neurons do not migrate enough to reach the superficial layers. Consistent with the hypo-migration phenotype, neuronal morphology is changed and the neurites of mutant neurons are shorter than the controls. This result suggests that when neuronal laminin is disrupted, neurite extension is affected. Since during neuronal migration, the movement of the neuronal soma follows the neuronal leading process, slowing or blocking leading process extension may impair neuronal migration.
Based on our results and others (Liesi, 1985; Liesi, 1990; Liesi, 1992; Liesi et al., 1995; Tsai and Gleeson, 2005), we hypothesize that laminins produced by migrating neurons are deposited around neurons, neuronal processes, and radial glial fibers. Laminins induce extension of the leading process of migrating neurons toward the pia, and mediate radial glia-neuron interactions. When laminin expression is abolished, the leading process extension is slowed or stopped, the glial-neuron interactions are disrupted, the cell body cannot move, which leads to a migration defect (Fig. 7A). This hypothesis is supported by the migration defects in mutant mice which suggest that neurons did not migrate far enough to reach the surface of the brain. Since this CaMKII-Cre mouse line expresses Cre in a large group of neurons in localized areas, the result is localized disruption of laminin γ1 gene expression and localized migration defects (Fig. 1 C and D).
Figure 7. Proposed model of laminin function in neuronal morphogenesis and migration.

A. Proposed model of laminin function in neuronal migration.
We hypothesize that laminin produced by migrating neurons is deposited around neuronal processes and along the radial glial fibers, and induces extension of the leading process toward the pia, and mediates glial-neuron interactions. When laminin is depleted, the leading process extension is retarded, the glial-neuron interaction is disrupted, and migration defects result. In addition, lack of laminin affects neurite growth causing abnormal neuronal morphology.
B. Signaling pathways involved in laminin function in neuronal morphogenesis.
Laminin binding to integrin receptors induces integrin clustering and activation, which then trigger phosphorylation of the downstream molecules such as FAK and paxillin. Integrin-associated molecules activate AKT by ILK. Activated AKT phosphorylates GSK-3β and inactivate its activity. Inactivated GSK-3β regulates microtubule assembly through molecules such as CRMP-2 to promote neurite elongation.
Since targeted deletion of the nidogen-binding site of the laminin γ1 appears to predominantly affect the pia basement membrane (Halfter et al., 2002), the function of neuronal laminin seems nidogen-binding independent. In the PNS, the function of laminin in axonal myelination in the spinal roots is basement membrane independent (Yang et al., 2005). In the CNS, laminin γ1 is involved in axon regeneration and its action is basement membrane independent (Grimpe et al., 2002). Therefore laminin function can be divided to two aspects: basement membrane-dependent and –independent. Neuronal laminins may exert their actions independent of basement membrane formation. Consistent with this notion, peptides from laminin chains, which would be independent of basement membrane formation, are active in various biological events including neurite growth promotion (Meiners and Mercado, 2003). Therefore, it seems likely that laminin-mediated signaling plays an important role in its function in neuronal morphogenesis and migration. It is intriguing that knockout of laminin receptors such as integrin or dystroglycan leads to cerebral cortical malformations, which seem predominantly associated with defects in the pia basement membrane. These phenotype differences between laminin γ1 and integrin or dystroglycan knockout mice may indicate that functional compensation of these receptors may occur. For example, during PNS axonal myelination, knockout of β1 integrin is functionally compensated by β4 integrin and dystroglycan (Previtali et al., 2003), while after knockout of laminin γ1, other laminin subunits or other ECM proteins are not up-regulated (data not shown).
Signaling pathways that mediate laminin function in neuronal morphogenesis and migration
The intracellular signaling pathway mediating laminin and laminin receptor functions in cortex development is not clear (Schmid and Anton, 2003). Our results show that phosphorylation of integrin-associated proteins such as FAK and paxillin are decreased in the mutant cortex. Moreover, both phosphorylation and total protein levels of GSK-3β are decreased, while only the phosphorylation levels of AKT are decreased in the cerebral cortex of mutant mice. FAK and paxillin are involved in mediating ECM/integrin functions in cytoskeleton organization and neurite extension (Bozzo et al., 1994; Huang et al., 2004; Ivankovic-Dikic et al., 2000; Mitra et al., 2005; Parsons et al., 2000; Turner, 2000; Turner et al., 2001; Yamauchi et al., 2006). Phosphorylation of FAK and paxillin are rapidly induced in fibroblasts when plated on laminin (Burridge et al., 1992; Ilic et al., 1997; Yamauchi et al., 2006). In chicken retinal neurons, 15-30 minutes after plating on laminin, phosphorylation of both paxillin and FAK were increased followed by integrin-mediated neurite outgrowth. Moreover, phosphorylation of paxillin is regulated during retinal development, indicating its in vivo function (de Curtis and Malanchini, 1997). Even though the role of FAK in cerebral cortex development is well studied (Beggs et al., 2003), the function of paxillin in cortical development is unknown. Our results show that when laminin is knocked out, phosphorylation of both FAK and paxillin are decreased, and these decreases accompany severe anatomic and neuronal morphological defects, suggesting possible functions of paxillin in cortical development.
The AKT/GSK-3β signaling pathway plays an important role in neuronal morphogenesis in vitro (Jiang et al., 2005; Shi et al., 2003; Yoshimura et al., 2005; Zhou et al., 2004) and is also involved in laminin function in neurite growth in vitro (Arimura and Kaibuchi, 2005; Menager et al., 2004; Zhou et al., 2006). Neuronal morphogenesis in vitro seems controlled by an intrinsic program, and the specification of axons seems random in the absence of particular cues. However, in the presence of substrate cues such as laminin, the neurite that first encounters laminin becomes the axon (Esch et al., 1999). The role of AKT in laminin-induced rapid neurite extension has examined by Menager (Menager et al., 2004). Using GFP-tagged AKT (AKT-PH-GFP), they showed that PI3-kinase is activated locally when neurites reach laminin. In the presence of laminin coated beads, neurons rapidly translocate AKT-PH-GFP to the site of neurite-laminin contact, which leads the neurite in contact with the laminin-bead to elongate 30-fold faster. When a second neurite from the same neuron contacts the laminin-coated beads, this neurite rapidly elongates while the first laminin-contacted neurite stops elongation (Menager et al., 2004). Addition of PI3-kinase inhibitors block laminin-induced AKT-PH-GFP accumulation and neurite elongation (Menager et al., 2004) suggesting that localized activation of PI3-kinase signaling mediates laminin-induced neurite elongation and axon specification. Since neuronal morphogenesis or polarity establishment in vivo is not random, there could be extracellular molecules that may guide neurite extension during development. Laminin may play such a role in vivo during cerebral cortex development. Even though the role of AKT/GSK-3β signaling pathway in neuronal morphogenesis and polarity establishment in vitro is well studied (Arimura and Kaibuchi, 2005; Jiang et al., 2005; Shi et al., 2003; Yoshimura et al., 2005; Zhou et al., 2006; Zhou et al., 2004), whether this signaling pathway play a role in neuronal development in vivo is unknown. Our studies show that when laminins, which induce rapid neurite extension through AKT/GSK-3β signaling pathway in vitro, are deleted from developing neurons, the phosphorylation of AKT and GSK-3β is decreased and neuronal morphogenesis is impaired. These results suggest that the AKT/GSK-3β signaling pathway plays an important role in neuronal morphogenesis in vivo during cortical development.
Based on our results, we hypothesize that the signaling pathway which mediates laminin function in neuronal morphogenesis and migration in cortical development as the following (Fig. 7): laminin binding to integrin receptors induces integrin clustering and activation, which then triggers phosphorylation of downstream molecules such as FAK and paxillin (Brakebusch and Fassler, 2003). Activation of integrin-associated molecules phosphorylates AKT through integrin-linked kinase (ILK). Active AKT phosphorylates GSK-3β and inactivates its activity. The major target of GSK-3β in controlling cytoskeleton dynamics is collapsin response mediator protein-2 (CRMP-2) (Yoshimura et al., 2005). Phosphorylation of CRMP-2 by GSK-3β decreases its ability to promote microtubule assembly. Inactivation of GSK-3β increases non-phosphorylated CRMP-2 which promotes microtubule assembly to enhance elongation of neuronal processes (Yoshimura et al., 2005).
Taken together, our results show that neuronal laminin may participate in neuritogenesis and neuronal migration through the integrin and AKT/GSK-3β signaling pathways during cerebral cortex development.
Supplementary Material
Figure S1: Intensity of laminin γ1 immunostaining in the EGFP expression areas of the mutant cortex is significantly lower than control cortex
Brain sections of E14.5 control or mutant embryos (also carry Z/EG reporter gene) were stained for laminin γ1. EGFP activity or laminin γ1 immunofluorescent staining was analyzed under a fluorescent microscope. The intensity of laminin γ1 immunofluorescent staining in the EGFP expression areas in the mutant cortex and that of the corresponding regions of the control cortex were analyzed by Image J. The differences between them were analyzed by Student's t-test.
Figure S2: Progressive decrease of laminin γ1 immunostaining intensity in mutant cortex during embryonic development stages
Brain sections of E14.5, E16.5 and E18.5 mutant (also carry Z/EG reporter gene) and control (floxed lamγ1) embryos were stained for laminin γ1. EGFP activity or laminin γ1 immunofluorescent staining was analyzed under a fluorescent microscope. The intensity of laminin γ1 immunofluorescent staining in the mutant cortex (including EGFP expression and non expression areas) and control cortex were analyzed by Image J. The decreases of laminin γ1 immunostaining in the mutant cortex relative to control cortex at the same development stages were analyzed. The differences of laminin γ1 immunostaining decreases among mutant cortices at different developmental stages were analyzed by One-way ANOVA. From E14.5 to E18.5, there were progressively more cells expressing EGFP (Cre) so the average intensity of laminin γ1 immunostaining further decreased relative to the control cortices at the same stages.
Figure S3: Mutant cortical organization disruption is correlated to regions that show EGFP expression in large groups of cells in localized areas
Brain sections of E16.5 mutant embryos (also carry Z/EG reporter gene) were stained for EGFP (red) and counter stained with DAPI. Cortical structure disruption occurred in a region that exhibit EGFP expression in a large group of cells in a localized area.
Acknowledgments
We would like to thank Dr. Erin H. Norris for reading the manuscript and members of the Strickland laboratory for useful discussions. This work was supported by grants from the NIH (NS035704 and NS050537) (SS), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (SS), the Muscular Dystrophy Association (MDA4066) (Z-LC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angevine JB, Sidman RL. Autoradiographic study of the cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–68. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- Arimura N, Kaibuchi K. Key regulators in neuronal polarity. Neuron. 2005;48:881–4. doi: 10.1016/j.neuron.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. 2003;40:501–14. doi: 10.1016/s0896-6273(03)00666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner J, O'Connor TP. The permissive cue laminin is essential for growth cone turning in vivo. J Neurosci. 2001;21:9782–91. doi: 10.1523/JNEUROSCI.21-24-09782.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzo C, Defilippi P, Silengo L, Tarone G. Role of tyrosine phosphorylation in matrix-induced neurite outgrowth in human neuroblastoma cells. Exp Cell Res. 1994;214:313–22. doi: 10.1006/excr.1994.1263. [DOI] [PubMed] [Google Scholar]
- Brakebusch C, Fassler R. The integrin-actin connection, an eternal love affair. Embo J. 2003;22:2324–33. doi: 10.1093/emboj/cdg245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Turner CE, Romer LH. Tyrosine phosphorylation of paxillin and pp125FAK accompanies cell adhesion to extracellular matrix: a role in cytoskeletal assembly. J Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MJ, Morrison JH. Monoclonal antibody to neurofilament protein (SMI-32) labels a subpopulation of pyramidal neurons in the human and monkey neocortex. J Comp Neurol. 1989;282:191–205. doi: 10.1002/cne.902820204. [DOI] [PubMed] [Google Scholar]
- Chen ZL, Indyk JA, Strickland S. The hippocampal laminin matrix is dynamic and critical for neuronal survival. Mol Biol Cell. 2003;14:2665–76. doi: 10.1091/mbc.E02-12-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZL, Strickland S. Laminin gamma1 is critical for Schwann cell differentiation, axon myelination, and regeneration in the peripheral nerve. J Cell Biol. 2003;163:889–99. doi: 10.1083/jcb.200307068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZL, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Colognato H, Yurchenco PD. Form and function: the laminin family of heterotrimers. Dev Dynamics. 2000;218:213–234. doi: 10.1002/(SICI)1097-0177(200006)218:2<213::AID-DVDY1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fassler R. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–22. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Curtis I, Malanchini B. Integrin-mediated tyrosine phosphorylation and redistribution of paxillin during neuronal adhesion. Exp Cell Res. 1997;230:233–43. doi: 10.1006/excr.1996.3423. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Zeitlin S. CaMKIIalpha-Cre transgene expression and recombination patterns in the mouse brain. Genesis. 2000;26:133–5. doi: 10.1002/(sici)1526-968x(200002)26:2<133::aid-gene10>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Esch T, Lemmon V, Banker G. Local presentation of substrate molecules directs axon specification by cultured hippocampal neurons. J Neurosci. 1999;19:6417–26. doi: 10.1523/JNEUROSCI.19-15-06417.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alonso L, Fetter RD, Goodman CS. Genetic analysis of Laminin A in Drosophila: extracellular matrix containing laminin A is required for ocellar axon pathfinding. Development. 1996;122:2611–21. doi: 10.1242/dev.122.9.2611. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Mark M, Messaddeq N, Gansmuller A. Essential role of alpha 6 integrins in cortical and retinal lamination. Curr Biol. 1998;8:983–6. doi: 10.1016/s0960-9822(98)70402-6. [DOI] [PubMed] [Google Scholar]
- Gomez TM, Letourneau PC. Filopodia initiate choices made by sensory neuron growth cones at laminin/fibronectin borders in vitro. J Neurosci. 1994;14:5959–72. doi: 10.1523/JNEUROSCI.14-10-05959.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–79. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Grimpe B, Dong S, Doller C, Temple K, Malouf AT, Silver J. The critical role of basement membrane-independent laminin gamma 1 chain during axon regeneration in the CNS. J Neurosci. 2002;22:3144–60. doi: 10.1523/JNEUROSCI.22-08-03144.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halfter W, Dong S, Yip YP, Willem M, Mayer U. A critical function of the pial basement membrane in cortical histogenesis. J Neurosci. 2002;22:6029–40. doi: 10.1523/JNEUROSCI.22-14-06029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopker VH, Shewan D, Tessier-Lavigne M, Poo M, Holt C. Growth-cone attraction to netrin-1 is converted to repulsion by laminin-1. Nature. 1999;401:69–73. doi: 10.1038/43441. [DOI] [PubMed] [Google Scholar]
- Huang C, Borchers CH, Schaller MD, Jacobson K. Phosphorylation of paxillin by p38MAPK is involved in the neurite extension of PC-12 cells. J Cell Biol. 2004;164:593–602. doi: 10.1083/jcb.200307081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Damsky CH, Yamamoto T. Focal adhesion kinase: at the crossroads of signal transduction. J Cell Sci. 1997;110(Pt 4):401–7. doi: 10.1242/jcs.110.4.401. [DOI] [PubMed] [Google Scholar]
- Indyk JA, Chen ZL, Strickland S. Laminin chain expression suggests that laminin-10 is a major isoform in the mouse hippocampus and is degraded by the tPA/plasmin system during excitotoxic injury. Neurosci. 2003;116:359–371. doi: 10.1016/s0306-4522(02)00704-2. [DOI] [PubMed] [Google Scholar]
- Ivankovic-Dikic I, Gronroos E, Blaukat A, Barth BU, Dikic I. Pyk2 and FAK regulate neurite outgrowth induced by growth factors and integrins. Nat Cell Biol. 2000;2:574–81. doi: 10.1038/35023515. [DOI] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123–35. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Kafitz KW, Greer CA. Role of laminin in axonal extension from olfactory receptor cells. J Neurobiol. 1997;32:298–310. [PubMed] [Google Scholar]
- Kiryushko D, Berezin V, Bock E. Regulators of neurite outgrowth: role of cell adhesion molecules. Ann N Y Acad Sci. 2004;1014:140–54. doi: 10.1196/annals.1294.015. [DOI] [PubMed] [Google Scholar]
- Kuhn TB, Schmidt MF, Kater SB. Laminin and fibronectin guideposts signal sustained but opposite effects to passing growth cones. Neuron. 1995;14:275–85. doi: 10.1016/0896-6273(95)90285-6. [DOI] [PubMed] [Google Scholar]
- Letourneau PC, Madsen AM, Palm SL, Furcht LT. Immunoreactivity for laminin in the developing ventral longitudinal pathway of the brain. Dev Biol. 1988;125:135–44. doi: 10.1016/0012-1606(88)90066-8. [DOI] [PubMed] [Google Scholar]
- Liesi P. Do neurons in the vertebrate CNS migrate on laminin? Embo J. 1985;4:1163–70. doi: 10.1002/j.1460-2075.1985.tb03755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesi P. Extracellular matrix and neuronal movement. Experientia. 1990;46:900–7. doi: 10.1007/BF01939382. [DOI] [PubMed] [Google Scholar]
- Liesi P. Neuronal migration on laminin involves neuronal contact formation followed by nuclear movement inside a preformed process. Exp Neurol. 1992;117:103–13. doi: 10.1016/0014-4886(92)90119-b. [DOI] [PubMed] [Google Scholar]
- Liesi P, Fried G, Stewart RR. Neurons and glial cells of the embryonic human brain and spinal cord express multiple and distinct isoforms of laminin. J Neurosci Res. 2001;64:144–67. doi: 10.1002/jnr.1061. [DOI] [PubMed] [Google Scholar]
- Liesi P, Hager G, Dodt HU, Seppala I, Zieglgansberger W. Domain-specific antibodies against the B2 chain of laminin inhibit neuronal migration in the neonatal rat cerebellum. J Neurosci Res. 1995;40:199–206. doi: 10.1002/jnr.490400208. [DOI] [PubMed] [Google Scholar]
- Liesi P, Seppala I, Trenkner E. Neuronal migration in cerebellar microcultures is inhibited by antibodies against a neurite outgrowth domain of laminin. J Neurosci Res. 1992;33:170–6. doi: 10.1002/jnr.490330122. [DOI] [PubMed] [Google Scholar]
- Luckenbill-Edds L. Laminin and the mechanism of neuronal outgrowth. Brain Res Brain Res Rev. 1997;23:1–27. doi: 10.1016/s0165-0173(96)00013-6. [DOI] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–83. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- McLoon SC, McLoon LK, Palm SL, Furcht LT. Transient expression of laminin in the optic nerve of the developing rat. J Neurosci. 1988;8:1981–90. doi: 10.1523/JNEUROSCI.08-06-01981.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiners S, Mercado ML. Functional peptide sequences derived from extracellular matrix glycoproteins and their receptors: strategies to improve neuronal regeneration. Mol Neurobiol. 2003;27:177–96. doi: 10.1385/MN:27:2:177. [DOI] [PubMed] [Google Scholar]
- Menager C, Arimura N, Fukata Y, Kaibuchi K. PIP3 is involved in neuronal polarization and axon formation. J Neurochem. 2004;89:109–18. doi: 10.1046/j.1471-4159.2004.02302.x. [DOI] [PubMed] [Google Scholar]
- Miner JH, Cunningham J, Sanes JR. Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol. 1998;143:1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell KJ, Pinson KI, Kelly OG, Brennan J, Zupicich J, Scherz P, Leighton PA, Goodrich LV, Lu X, Avery BJ, Tate P, Dill K, Pangilinan E, Wakenight P, Tessier-Lavigne M, Skarnes WC. Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat Genet. 2001;28:241–9. doi: 10.1038/90074. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, Hoshi T, Campbell KP. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–5. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–50. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Parnavelas JG. Modes of neuronal migration in the developing cerebral cortex. Nat Rev Neurosci. 2002;3:423–32. doi: 10.1038/nrn845. [DOI] [PubMed] [Google Scholar]
- Nakagami Y, Abe K, Nishiyama N, Matsuki N. Laminin degradation by plasmin regulates long-term potentiation. J Neurosci. 2000;20:2003–2010. doi: 10.1523/JNEUROSCI.20-05-02003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–55. [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Parsons JT, Martin KH, Slack JK, Taylor JM, Weed SA. Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19:5606–13. doi: 10.1038/sj.onc.1203877. [DOI] [PubMed] [Google Scholar]
- Previtali SC, Nodari A, Taveggia C, Pardini C, Dina G, Villa A, Wrabetz L, Quattrini A, Feltri ML. Expression of laminin receptors in schwann cell differentiation: evidence for distinct roles. J Neurosci. 2003;23:5520–30. doi: 10.1523/JNEUROSCI.23-13-05520.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. Principles of neural cell migration. Experientia. 1990;46:882–91. doi: 10.1007/BF01939380. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Edson KJ, Letourneau PC, McLoon SC. Distribution of laminin in the developing peripheral nervous system of the chick. Dev Biol. 1986;113:429–35. doi: 10.1016/0012-1606(86)90177-6. [DOI] [PubMed] [Google Scholar]
- Schaller MD. Paxillin: a focal adhesion-associated adaptor protein. Oncogene. 2001;20:6459–72. doi: 10.1038/sj.onc.1204786. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Anton ES. Role of integrins in the development of the cerebral cortex. Cereb Cortex. 2003;13:219–24. doi: 10.1093/cercor/13.3.219. [DOI] [PubMed] [Google Scholar]
- Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 2003;112:63–75. doi: 10.1016/s0092-8674(02)01249-7. [DOI] [PubMed] [Google Scholar]
- Siao CJ, Tsirka SE. Tissue plasminogen activator mediates microglial activation via its finger domain through annexin II. J Neurosci. 2002;22:3352–8. doi: 10.1523/JNEUROSCI.22-09-03352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidman RL, Rakic P. Neuronal migration, with special reference to developing human brain: a review. Brain Res. 1973;62:1–35. doi: 10.1016/0006-8993(73)90617-3. [DOI] [PubMed] [Google Scholar]
- Sievers J, Pehlemann FW, Gude S, Berry M. Meningeal cells organize the superficial glia limitans of the cerebellum and produce components of both the interstitial matrix and the basement membrane. J Neurocytol. 1994;23:135–49. doi: 10.1007/BF01183867. [DOI] [PubMed] [Google Scholar]
- Skarnes WC, Moss JE, Hurtley SM, Beddington RS. Capturing genes encoding membrane and secreted proteins important for mouse development. Proc Natl Acad Sci U S A. 1995;92:6592–6. doi: 10.1073/pnas.92.14.6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth N, Vatansever HS, Murray P, Meyer M, Frie C, Paulsson M, Edgar D. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol. 1999;144:151–60. doi: 10.1083/jcb.144.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JW, Cooley MA, Broome JM, Salgia R, Griffin JD, Lombardo CR, Schaller MD. The role of focal adhesion kinase binding in the regulation of tyrosine phosphorylation of paxillin. J Biol Chem. 1999;274:36684–92. doi: 10.1074/jbc.274.51.36684. [DOI] [PubMed] [Google Scholar]
- Timpl R. Macromolecular organization of basement membranes. Curr Opin Cell Biol. 1996;8:618–624. doi: 10.1016/s0955-0674(96)80102-5. [DOI] [PubMed] [Google Scholar]
- Timpl R, Brown JC. The laminins. Matrix Biology. 1994;14:275–281. doi: 10.1016/0945-053x(94)90192-9. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Gleeson JG. Nucleokinesis in neuronal migration. Neuron. 2005;46:383–8. doi: 10.1016/j.neuron.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Turner CE. Paxillin and focal adhesion signalling. Nat Cell Biol. 2000;2:E231–6. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- Turner CE, West KA, Brown MC. Paxillin-ARF GAP signaling and the cytoskeleton. Curr Opin Cell Biol. 2001;13:593–9. doi: 10.1016/s0955-0674(00)00256-8. [DOI] [PubMed] [Google Scholar]
- Weisenhorn DM, Prieto EW, Celio MR. Localization of calretinin in cells of layer I (Cajal-Retzius cells) of the developing cortex of the rat. Brain Res Dev Brain Res. 1994;82:293–7. doi: 10.1016/0165-3806(94)90171-6. [DOI] [PubMed] [Google Scholar]
- Willem M, Miosge N, Halfter W, Smyth N, Jannetti I, Burghart E, Timpl R, Mayer U. Specific ablation of the nidogen-binding site in the laminin gamma1 chain interferes with kidney and lung development. Development. 2002;129:2711–22. doi: 10.1242/dev.129.11.2711. [DOI] [PubMed] [Google Scholar]
- Yamauchi J, Miyamoto Y, Sanbe A, Tanoue A. JNK phosphorylation of paxillin, acting through the Rac1 and Cdc42 signaling cascade, mediates neurite extension in N1E-115 cells. Exp Cell Res. 2006;312:2954–61. doi: 10.1016/j.yexcr.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Yang D, Bierman J, Tarumi YS, Zhong YP, Rangwala R, Proctor TM, Miyagoe-Suzuki Y, Takeda S, Miner JH, Sherman LS, Gold BG, Patton BL. Coordinate control of axon defasciculation and myelination by laminin-2 and -8. J Cell Biol. 2005;168:655–66. doi: 10.1083/jcb.200411158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Kikkawa Y, Mudd JL, Skarnes WC, Sanes JR, Miner JH. Expression of laminin chains by central neurons: analysis with gene and protein trapping techniques. Genesis. 2003;36:114–27. doi: 10.1002/gene.10206. [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–49. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Yu WM, Feltri ML, Wrabetz L, Strickland S, Chen ZL. Schwann cell-specific ablation of laminin gamma1 causes apoptosis and prevents proliferation. J Neurosci. 2005;25:4463–72. doi: 10.1523/JNEUROSCI.5032-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Walzer M, Wu YH, Zhou J, Dedhar S, Snider WD. Neurotrophins support regenerative axon assembly over CSPGs by an ECM-integrin-independent mechanism. J Cell Sci. 2006;119:2787–96. doi: 10.1242/jcs.03016. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3beta and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Intensity of laminin γ1 immunostaining in the EGFP expression areas of the mutant cortex is significantly lower than control cortex
Brain sections of E14.5 control or mutant embryos (also carry Z/EG reporter gene) were stained for laminin γ1. EGFP activity or laminin γ1 immunofluorescent staining was analyzed under a fluorescent microscope. The intensity of laminin γ1 immunofluorescent staining in the EGFP expression areas in the mutant cortex and that of the corresponding regions of the control cortex were analyzed by Image J. The differences between them were analyzed by Student's t-test.
Figure S2: Progressive decrease of laminin γ1 immunostaining intensity in mutant cortex during embryonic development stages
Brain sections of E14.5, E16.5 and E18.5 mutant (also carry Z/EG reporter gene) and control (floxed lamγ1) embryos were stained for laminin γ1. EGFP activity or laminin γ1 immunofluorescent staining was analyzed under a fluorescent microscope. The intensity of laminin γ1 immunofluorescent staining in the mutant cortex (including EGFP expression and non expression areas) and control cortex were analyzed by Image J. The decreases of laminin γ1 immunostaining in the mutant cortex relative to control cortex at the same development stages were analyzed. The differences of laminin γ1 immunostaining decreases among mutant cortices at different developmental stages were analyzed by One-way ANOVA. From E14.5 to E18.5, there were progressively more cells expressing EGFP (Cre) so the average intensity of laminin γ1 immunostaining further decreased relative to the control cortices at the same stages.
Figure S3: Mutant cortical organization disruption is correlated to regions that show EGFP expression in large groups of cells in localized areas
Brain sections of E16.5 mutant embryos (also carry Z/EG reporter gene) were stained for EGFP (red) and counter stained with DAPI. Cortical structure disruption occurred in a region that exhibit EGFP expression in a large group of cells in a localized area.