

Abstract
Pathogenetic mechanisms leading to chronic obstructive pulmonary disease (COPD) remain poorly understood. Because clonogenic T cells (CD4+CD28null) were shown to be increased in autoimmune diseases we hypothesized that CD4+CD28null T cells play a role in COPD. Here we describe that enhanced presence of CD4+CD28null cells is associated with impaired lung function. Sixty-four patients and controls were included. T cell phenotype was analysed using flow cytometry. Enzyme-linked immunosorbent assays were utilized to determine cytokines. Statistical evaluations were performed using non-parametric group comparisons and correlations. A logistic regression model was used to determine predictive values of CD4+CD28null in the diagnosis of COPD. Populations of CD4+ T cells lacking surface co-stimulatory CD28 were enlarged significantly in evaluated patients when compared with controls. Natural killer (NK)-like T cell receptors (CD94, 158) and intracellular perforin, granzyme B were increased in CD4+CD28null cells. Cytokine production after triggering of peripheral blood mononuclear cells (PBMCs) was elevated in patients at early disease stages. Receiver operating characteristic curve plotting revealed that presence of CD4+CD28null T cells has a diagnostic value. These CD4+CD28null T cells show increased expression of NK-like T cell receptors (CD94, 158) and intracellular perforin and granzyme B. Furthermore, triggering of PBMCs obtained from patients with mild COPD led to increased interferon-γ and tumour necrosis factor-α production in vitro compared with controls. Our finding of increased CD4+CD28null T cells in COPD indicates that chronic antigen exposure, e.g. through contents of smoke, leads to loss of CD28 and up-regulation of NK cell receptors expression on T cells in susceptible patients.
Keywords: CD4+CD28null, COPD, granzyme, perforin, smoking
Introduction
Chronic obstructive pulmonary disease (COPD) is a leading cause of death worldwide [1]. By 2020, only ischaemic heart disease and cerebrovascular disease will account for a higher mortality among the world's population [2]. Prevalence and hospitalization rates have increased significantly over the past years [3,4]. Several studies were able to show a strong correlation between tobacco abuse and the development of COPD. However, not every smoker develops clinical features of COPD [5,6]. Pathogenesis of the disease is characterized by irreversible airflow obstruction because of constant remodelling of the airways and chronic inflammatory responses [7]. The impairment of the immune system is not restricted to the lungs, as COPD patients are also at higher risk for systemic failures including cardiovascular diseases [8]. Diagnosis of airway obstruction according to the guidelines of the Global Initiative for Chronic Obstructive Lung Diseases (GOLD) requires the use of spirometry. A post-bronchodilator forced expiratory volume in 1 s(FEV1)/forced vital capacity ratio of less than 70% indicates an irreversible airflow obstruction, and is therefore considered to be the main parameter for the diagnosis of COPD [9].
Although smoking is accepted widely as the major risk factor for the development of the disease, descriptions of specific pathogenetic mechanisms remain vague. For decades, neutrophils and macrophages, as part of the innate immunity, were considered pivotal in the airway remodelling process occurring in patients with COPD. Recent reports have challenged this pathognomonic concept by demonstrating increased CD8+ and CD4+ T cells as part of the adaptive immune system in bronchoalveolar lavage and sputum analyses of COPD patients. These T lymphocytes contained higher levels of perforin and revealed cytotoxic activity compared with cells of healthy donors or non-COPD smokers [10,11]. Furthermore, Di Stefano et al. presented two papers in which they were able to show that stable mild/moderate COPD is associated with an active T helper 1 cell/type-1 cytotoxic T cell inflammatory process involving activation of signal transducer and activator of transcription 4 and interferon (IFN)-γ production and natural killer (NK) cells in COPD lung tissue and bronchoalveolar lavage [12,13]
Based on the data of Hodge et al., who described CD8+CD28null in COPD and Di Stefano et al.'s data, we hypothesized that a specific chronic inflammatory reaction of the adaptive immune system is occurring in patients with COPD. Antigenic stimulation causes a rapid expansion of antigen-specific T cells that increase to large clonal size. This physiological increment is counterbalanced by a pre-programmed clonal contraction. This process is robust and usually suffices to maintain a diverse memory T cell compartment [14,15]. Chronic antigen exposure because of infections with human immunodeficiency virus or cytomegalovirus [16,17] and advanced age [15] also leads to expansion of monoclonal T cell populations.
Replicatively stressed CD4+ T cells undergo multiple phenotypic and functional changes. The most widely acknowledged phenotypic change is the loss of the co-stimulatory surface marker CD28. Expansion of CD4+ T cells and loss of CD28 are presumably senescent (CD4+CD28null). This has been described in several autoimmune diseases, such as diabetes mellitus, rheumatoid arthritis, Wegener's granulomatosis, multiple sclerosis and ankylosing spondylitis [18–20]. CD4+CD28nullcells are clonally expanded and are known to include autoreactive T cells, implicating a direct role in autoimmune disease. These expanded CD4+ clonotypes are phenotypically distinct from the classic T helper cells. Because of a transcriptional block of the CD28 gene, clonally expanded CD4+ T cells lack surface expression of the major co-stimulatory molecule CD28. CD4+CD28null T cells release large amounts of IFN-γ and contain intracellular perforin and granzyme B, providing them with the ability to lyse target cells. Their outgrowth into large clonal populations may be attributed partially to a defect in down-regulating Bcl-2 when deprived of T cell growth factors. In the absence of the CD28 molecule, these unusual CD4+ T cells use alternative co-stimulatory pathways. Several of these functional features in CD4+CD28null T-cells are reminiscent of NK cells. Like NK cells, CD4+CD28null T cells are cytotoxic and can express NK cell receptors such as CD94 and CD158. NK cells are regulated closely by a family of polymorphic receptors that interact with major histocompatibility complex (MHC) class I molecules, resulting in signals that control NK-mediated cytotoxicity and cytokine production. MHC class I-mediated triggering of the full-length NK cell receptors transduces a dominant inhibitory signal that blocks the cytolytic activity and cytokine release of NK cells. These receptors also contain highly homologous members that have truncated cytoplasmatic domains and transmit activating signals [21–24].
Because chronic antigen exposure because of history of smoking can be assumed in most COPD patients, we hypothesized that chronic stimulation of the adaptive immune system leads to increased levels of systemic clonogenic CD4+CD28null T cell populations. To prove this we included age- and sex-matched healthy non-smokers, smokers with a history of tobacco abuse and normal lung function and patients with diagnosed COPD according to GOLD classification (groups: mild, COPD I–II; severe, COPD III–IV respectively). We determined intracellular expression of cytolytic proteins perforin and granzyme B as well as surface expression of the NK cell receptors CD94 and CD158 on CD4+CD28null T cells. We further designed in vitro experiments to explore whether peripheral blood mononuclear cells (PBMCs) obtained from each study group secrete augmented levels of IFN-γ, tumour necrosis factor (TNF)-α and interleukin (IL)-12 after T cell triggering. Because systemic inflammation is associated with systemic proinflammatory cytokines in vivo, we correlated serum levels of IL-1β, TNF-α, IFN-γ and IL-10 with lung function parameters. We conclude in this work that patients with COPD show increased circulating clonogenic T cells that have diagnostic potential for detection of COPD according to the GOLD classification.
Methods
Patients
The study protocol was approved by the ethics committee of the Medical University of Vienna (EK no. 091/2006) and was performed in accordance with the Declaration of Helsinki and current revisions of the Good Clinical Practice Guidelines of the Medical University of Vienna. A total number of 64 volunteers, at least 40 years old, participated in this trial. Healthy non-smokers (n = 15), smokers (n = 14) and smokers meeting the GOLD diagnostic criteria for COPD I–II (n = 19) and COPD III–IV (n = 16) [25] were recruited. COPD patients with acute exacerbation as defined by the guidelines from the WHO and GOLD [9,26] within 14 days before study entry were excluded. Additional exclusion criteria were a history of asthma, autoimmune diseases or other relevant lung diseases (e.g. lung cancer, known α1-anti-trypsin deficiency). Furthermore, all patients were free from known coronary artery disease, peripheral artery disease and carotid artery disease. All patients provided written, informed consent before collection of blood samples and lung function. Height and weight (Seca; Vogel and Halke, Hamburg, Germany) were measured and body mass index was determined. Pulmonary function was measured using the same model spirometer (AutoboxV6200; SensorMedics, Vienna, Austria). Measurements were made before and – if criteria for airflow obstruction were met – 15–30 min after inhaling 200 µg salbutamol. Arterial blood gases (PaO2, PaCO2) were obtained at rest while breathing room air in a sitting position. Measurement of arterial blood gases was performed with an ABL 510 gas analyser (Radiometer, Copenhagen, Denmark). Results are expressed as absolute values and as percentages of predicted values for age, sex and height, according to the European Community for Steel and Coal prediction equations [27]. Predicted normal values were derived from the reference values of the Austrian Society of Pulmonary Medicine.
Flow cytometry analysis
Heparinized blood samples were incubated on ice with fluorochrome-labelled antibodies. Prior to antibody incubation, erythrocytes were lysed by addition of BD fluorescence activated cell sorter lysing solution (Becton Dickinson, Franklin Lakes, NJ, USA). Cells were then stained with fluorescein isothiocyanate-conjugated anti-CD4 (BD Biosciences Pharmingen, San Jose, CA, USA), phycoerythrin (PE)-labelled anti-CD158 (R&D Systems, Minneapolis, MN, USA), PE-Cy5-labelled anti-CD28 (Biolegend, San Diego, CA, USA) and PE-conjugated anti-CD94 (eBioscience, San Diego, CA, USA) at various combinations. Stained cells were analysed using a Cytomics FC 500 flow cytometer (Beckman Coulter, Fullerton, CA, USA). For intracellular staining, PE-conjugated antibodies directed against perforin and granzyme B (BD Biosciences Pharmingen; Serotec, Dusseldorf, Germany) were used and incubated with pre-stained cells after permeabilization of the cell membrane with saponin solution.
Enzyme-linked immunosorbent assays
The enzyme-linked immunosorbent assays (ELISA) technique (BenderMedSystems, Vienna, Austria) was used to quantify levels of IL-1β, TNF-α, IFN-γ and IL-10 in serum samples obtained after centrifugation of whole blood. Ninety-six-well plates were coated with a monoclonal antibody directed against the specific antigen and incubated overnight at 4°C. After a washing step, plates were blocked with assay buffer for 2 h. Following another washing step, samples and standards with defined concentrations of antigen were incubated as described by the manufacturer. Plates were then washed and incubated with enzyme-linked polyclonal antibodies. Tetramethylbenzidine substrate solution was applied after the appropriate incubation time and another washing step. Colour development was then monitored using a Wallac Multilabel counter 1420 (PerkinElmer, Boston, MA, USA). The optical density values obtained were compared with the standard curve calculated from optical density values of standards with known concentrations of antigen.
Stimulation of freshly prepared PBMCs
Freshly prepared PBMCs were separated by standard Ficoll densitiy gradient centrifugation. Cells were then washed twice in phosphate-buffered saline, counted and transferred to a 96-well flat-bottomed plate at 1 × 105 cells per well in 200 µl serum-free ultra culture medium (Cambrex Corp., East Rutherford, NJ, USA) containing 0·2% gentamycinsulphate (Sigma, St Louis, MO, USA) and 0·5% β-mercapto-ethanol (Sigma) 1% l-glutamine (Sigma). Anti-CD3 (CD3) (10 µg/ml) or phytohaemagglutinin (PHA) (7 µg/ml) were added and plates were transferred to a humidified atmosphere (5% CO2, 37°C) for 18 h. Supernatants were harvested and stored at −20°C.
Quantification of IFN-γ, TNF-α and IL-12 in supernatants
The ELISA technique (BenderMedSystems) was used to quantify levels of IFN-γ, TNF-α and IL-12 in supernatants of stimulated cells, as described above.
Statistical methods
Comparison of the primary end-point CD4+CD28null% of CD4+ and the second end-points (IFN-γ, TNF-α and IL-12 ex vivo CD3 and PHA, IL-1β, TNF-α, IFN-γ and IL-10 serum values) between healthy non-smokers, healthy smokers, COPD I–II and COPD III–IV patients was performed with the non-parametric Kruskal–Wallis test. Pairwise comparisons between groups were performed with Wilcoxon tests. For the six pairwise between-group comparisons of the primary end-point CD4+CD28null% of CD4+ additionally adjusted critical values, according to Shaffer (1986) [28], were applied to control the familywise error rate in the strong sense.
Parametric 95% confidence intervals (CI) for the mean CD4+CD28null percentages in each group were computed. Correlations of percentage of CD4+CD28null cells and serum cytokine levels with parameters of lung function were calculated using the Spearman's correlation coefficient. These correlations were performed for all patients, the subgroup of smokers and the subgroup of COPD patients.
The prevalence of perforin, granzyme B and expression of CD94 and CD158 was compared between CD4+CD28null and CD4+CD28+ cells using Wilcoxon's signed-rank tests. Additionally, parametric 95% CI for the mean percentages for each variable are given.
In the subgroup of smokers a logistic regression with dependent variable COPD (yes/no) and independent variable CD4+CD28null% was performed. To account for an outlying observation, the square root of the percentages was used in this analysis. To assess the predictive capacity of the percentage of CD4+CD28null a receiver operating characteristic (ROC) curve with its area under the curve (AUC) was computed.
Results
Demographic characteristics of study patients
Demographic characteristics of patients are depicted in Table 1. Healthy non-smokers, healthy smokers, GOLD-classified COPD I–II and COPD III–IV were included. In all groups a similar number of patients were included and age and sex were distributed equally.
Table 1.
Clinical characteristics. Severity of airflow obstruction was determined using lung function test in all subjects; chronic obstructive pulmonary disease (COPD) patients meeting the Global Initiative for Chronic Obstructive Lung Diseases diagnostic criteria for COPD.
| Subject category | Healthy | Healthy smoker | COPD GOLD I–IV | COPD GOLD I–II | COPD GOLD III–IV |
|---|---|---|---|---|---|
| N | 15 | 14 | 35 | 19 | 16 |
| Male/female | 10/5 | 7/7 | 20/15 | 10/9 | 10/6 |
| Age | 57·20 (12·50) | 56·64 (9·17) | 59·60 (8·01) | 60·68 (7·39) | 58·31 (8·75) |
| Lung function | |||||
| FVC (l) | 4·55 (0·94) | 3·84 (0·66) | 2·80 (1·08) | 3·33 (1·06) | 2·14 (0·70) |
| FEV1 (%) | 105·37 (17·11) | 94·40 (11·96) | 52·76 (23·71) | 70·21 (13·33) | 30·67 (12·66) |
| FEV1/VC (%) | 76·80 (7·85) | 75·95 (3·99) | 51·18 (16·83) | 61·74 (8·36) | 37·80 (15·33) |
| MEF50 (%) | 100·67 (28·92) | 87·64 (21·45) | 27·29 (18·68) | 39·42 (15·93) | 11·93 (6·60) |
| MEF25 (%) | 103·53 (33·89) | 75·71 (31·33) | 29·71 (15·31) | 37·37 (16·19) | 20·00 (5·94) |
| Smoking history | |||||
| Never-smoker | 15 | 0 | 0 | 0 | 0 |
| Ex-smoker | 0 | 3 | 7 | 4 | 3 |
| Current-smoker | 0 | 11 | 28 | 15 | 13 |
| Pack years | 0 | 34 (25·2) | 45·8 (30·6) | 47·3 (29·7) | 44·0 (32·6) |
| Body weight (kg) | 71·6 (13·9) | 76·4 (8·6) | 80·4 (21·6) | 79·7 (16·7) | 81·1 (27·2) |
| Body height (cm) | 172·7 (10·9) | 168·7 (8·1) | 169·2 (10·5) | 167·7 (12·1) | 171·2 (7·9) |
Data are given as mean (±standard deviation) if not otherwise stated. FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; MEF, maximum expiratory flow.
CD4+CD28null cells show increased occurrence in patients suffering from COPD
To test our hypothesis whether CD4+CD28null cells are increased in patients with COPD, we evaluated blood samples using multi-stain flow cytometry. Figure 1a and Table 2 illustrate percentages of CD4+CD28null cells of the total CD4+ cell population. The COPD III–IV group showed significantly increased values compared with the healthy non-smoker and smoker groups (Wilcoxon test: P = 0·012, P = 0·002, Kruskal–Wallis test for the overall comparison: P = 0·005). Additionally, we observed a significant difference between the COPD I–II group and the healthy smoker group (Wilcoxon test: P = 0·046). Applying the Shaffer (1986) [28] multiplicity adjusted critical values, only the differences between the COPD III–IV and the healthy groups remained significant.
Fig. 1.
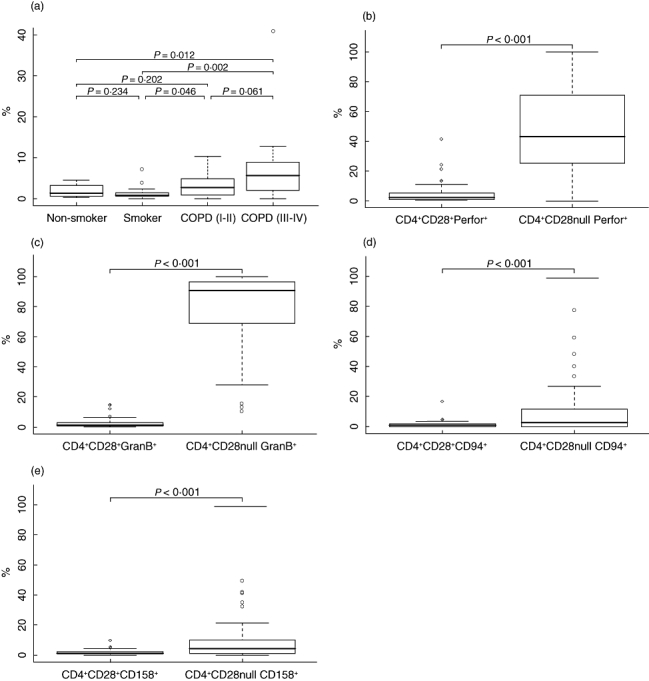
(a) Boxplot showing percentage of CD4+CD28null cells in the peripheral blood flow. (b,c) Subset of CD4+ T cells lacking co-stimulatory CD28 contained intracellular cytolytic proteins perforin and granzyme B. (d,e) CD4+CD28null cells showed significantly increased surface expression of natural killer (NK) cell receptors CD94 and CD158. Bars indicate medians; solid boxes show span between 25th and 75th percentiles; whiskers illustrate lowest and highest values. Outliers are marked as open circles.
Table 2.
Percentage of CD4+CD28null cells in the peripheral blood flow.
| Subject category | Healthy | Healthy smoker | COPD GOLD I–II | COPD GOLD III–IV |
|---|---|---|---|---|
| CD4+CD28null% of CD4+ | 1·96 (1·07–2·84) | 1·5 (0·41–2·59) | 3·22 (1·83–4·62) | 7·53 (2·67–12·39) |
| IFN-γ CD3 (pg/ml) | 272 (188–356) | 240 (178–301) | 440 (286–594) | 328 (214–442) |
| IFN-γ PHA (pg/ml) | 116 (83–149) | 91 (53–129) | 375 (135–615) | 134 (1–266) |
| TNF-α CD3 (pg/ml) | 922 (368–1476) | 731 (333–1128) | 1234 (793–1674) | 1508 (860–2157) |
| TNF-α PHA (pg/ml) | 1096 (551–1641) | 777 (411–1143) | 2465 (1532–3398) | 1144 (387–1901) |
| IL-12 CD3 (pg/ml) | 93 (46–139) | 63 (34–92) | 72 (36–108) | 42 (13–71) |
| IL-12 PHA (pg/ml) | 44 (8–80) | 33 (19–47) | 78 (31–125) | 17 (8–25) |
Furthermore, cytokine expression in supernatants of peripheral blood mononuclear cells stimulated with either anti-CD3 or phytohaemagglutinin (PHA) is described. All date are given as mean (95% confidence interval). COPD, chronic obstructive pulmonary disease; GOLD, Global Initiative for Chronic Obstructive Lung Disease; IFN-γ, interferon-γ; IL, interleukin; TNF-α, tumour necrosis factor-α.
Unstimulated CD4+CD28null cells contain cytolytic proteins perforin and granzyme B
To evaluate the intra-cytoplasmic content of cytolytic proteins perforin and granzyme B in CD4+ cells, flow cytometric analysis of blood samples was performed after co-incubation with saponin solution and intracellular staining. Content of perforin was more prevalent in CD4+CD28null cells compared with CD4+CD28+ cells (Fig. 1b) [46·13% (39·34–52·91) versus 4·68% (3·04–6·32), P < 0·001; all means (95% CI)]. Positive staining for intracellular granzyme B in CD4+CD28null cells was more frequent than in CD4+CD28+ cells (Fig. 1c) [78·63% (72·65–84·61) versus 2·36% (1·63–3·11), P < 0·001; all means (95% CI)].
Increased prevalence of NK cell receptors on CD4+CD28null cells
Flow cytometry analysis was used to evaluate expression of CD94 and CD158 on the surface of CD4+ cells. Figure 1d and e shows increased expression of surface antigens CD94 and CD158 on CD4+CD28null cells [CD94, 10·00% (6·04–13·97) versus 1·41% (0·85–1·97), P < 0·001; CD158, 9·35% (6·22–12·47) versus 2·00% (1·61–2·39), P < 0·001; all means (95% CI)].
Percentage of CD4+CD28null cells correlates negatively with routine parameters of spirometry
For verification of our flow cytometry data with routine clinical data, we correlated the percentage of CD4+CD28null with FEV1% of vital capacity, 50% maximum expiratory flow (MEF50%) of predicted value and MEF25% of predicted value. All parameters showed a statistically significant negative correlation with percentage of CD4+CD28null cells (Spearman's correlation coefficients: FEV1%, R = −0·49, P < 0·001; MEF50%, R = −0·40, P = 0·001; MEF25%, −0·38, P = 0·002; Fig. 2a–c). Similarly, we observed significant correlations in the subgroup of smokers (Spearman's correlation coefficients: FEV1%, R = −0·52, P < 0·001; MEF50%, R = −0·48, P = 0·001; MEF25%, R = −0·40, P = 0·004). In the subgroup of COPD patients marginally significant correlations with FEV1% and MEF50% (Spearman's correlation coefficients: FEV1%, R = −0·32, P = 0·068; MEF50%, R = −0·36, P = 0·04) and no significant correlation with MEF25% (Spearman's correlation coefficient: MEF25%, R = −0·15, P = 0·38) were found.
Fig. 2.
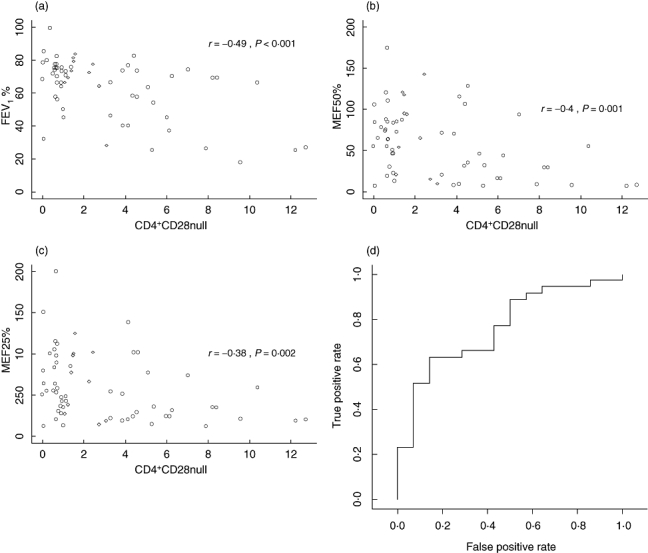
(a,b,c) Scatterplots showing correlations of CD4+CD28null% of CD4+ and Forced expiratory volume in 1 s, maximum expiratory flow (MEF50%), and MEF25%, Spearman's correlation coefficients and P-values are given. (d) Receiver operating characteristic curve for the prediction of chronic obstructive pulmonary disease in the subgroup of smokers based on the CD4+CD28null% measurement.
Prediction capacity of the percentage of CD4+CD28null cells for COPD in smokers
In the logistic regression analysis for the subset of smokers the independent variable percentage of CD4+CD28null cells showed a significant association with COPD (P = 0·012). The corresponding ROC curve (Fig. 2d) has an AUC = 0·76.
Correlations of serum cytokine concentrations (IL-1β, TNF-α, IFN-γ and IL-10) with FEV1%, MEF50% and MEF25%
Table 3 embraces the results of non-parametric correlations of serum cytokines IL-1β, TNF-α, IFN-γ and IL-10 with routine lung function parameters.
Table 3.
Correlations of serum cytokine levels with parameters of lung function test.
| All patients |
All smokers |
All COPD |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Correlation | n | Coeff. | P-value | n | Coeff. | P-value | n | Coeff. | P-value |
| CD4+CD28null-FEV1% | 62 | −0·485 | <0·001 | 48 | −0·517 | <0·001 | 34 | −0·317 | 0·068 |
| CD4+CD28null-MEF50% | 62 | −0·404 | 0·001 | 48 | −0·479 | <0·001 | 34 | −0·355 | 0·04 |
| CD4+CD28null-MEF25% | 62 | −0·38 | 0·002 | 48 | −0·403 | 0·004 | 34 | −0·154 | 0·384 |
| IFN-γ-FEV1% | 62 | 0·461 | <0·001 | 48 | 0·613 | <0·001 | 34 | 0·491 | 0·003 |
| IFN-γ-MEF50% | 62 | 0·556 | <0·001 | 48 | 0·645 | <0·001 | 34 | 0·541 | <0·001 |
| IFN-γ-MEF25% | 62 | 0·489 | <0·001 | 48 | 0·618 | <0·001 | 34 | 0·492 | 0·003 |
| TNF-α-FEV1% | 62 | 0·374 | 0·003 | 48 | 0·336 | 0·019 | 34 | 0·226 | 0·198 |
| TNF-α-MEF50% | 62 | 0·337 | 0·007 | 48 | 0·275 | 0·058 | 34 | 0·123 | 0·489 |
| TNF-α-MEF25% | 62 | 0·309 | 0·014 | 48 | 0·249 | 0·087 | 34 | 0·039 | 0·828 |
| IL-1β-FEV1% | 62 | 0·344 | 0·006 | 48 | 0·287 | 0·048 | 34 | 0·066 | 0·709 |
| IL-1β-MEF50% | 62 | 0·282 | 0·026 | 48 | 0·256 | 0·079 | 34 | 0·078 | 0·663 |
| IL-1β-MEF25% | 62 | 0·266 | 0·037 | 48 | 0·22 | 0·133 | 34 | 0·002 | 0·993 |
| IL-10-FEV1% | 62 | 0·256 | 0·044 | 48 | 0·178 | 0·226 | 34 | 0·096 | 0·587 |
| IL-10-MEF50% | 62 | 0·328 | 0·009 | 48 | 0·278 | 0·055 | 34 | 0·329 | 0·058 |
| IL-10-MEF25% | 62 | 0·3 | 0·018 | 48 | 0·226 | 0·122 | 34 | 0·214 | 0·223 |
FEV1, forced expiratory volume in 1 s; IFN-γ, interferon-γ; IL, interleukin; MEF, maximum expiratory flow; TNF-α, tumour necrosis factor-α.
Stimulated PBMCs of patients suffering from early-stage COPD produce increased levels of IFN-γ and TNF-αex vivo
To verify the functional activity of PBMCs we performed blastogenesis assays using lymphocyte-specific anti-CD3 and PHA. This analysis was performed for seven patients per group (except for the COPD III–IV group, where only five patients were included). Groupwise means and 95% CI are given in Table 2. Supernatants of patients with COPD I–II showed increased levels of IFN-γ compared with healthy smokers (Wilcoxon test: CD3 P = 0·026, PHA: P = 0·038); however, the differences failed to reach significance after correcting for multiple testing (Kruskal–Wallis test: CD3: P = 0·06; PHA: P = 0·09). Concentrations of the healthy group and of patients with COPD III–IV were lower but showed no significant difference to the COPD I–II group. None of the remaining pairwise comparisons was statistically significant. Significant difference of TNF-α (PHA) levels between groups were observed (Kruskal–Wallis test: P = 0·007). The COPD I–II group showed significantly elevated levels of TNF-α (PHA) levels compared with healthy smokers (Wilcoxon test P = 0·001) and non-smokers (Wilcoxon test: P = 0·007) and marginally significant elevated levels compared with COPD III–IV patients (Wilcoxon test: P = 0·03). None of the remaining pairwise comparisons was statistically significant. For TNF-α (CD3) no significant differences between groups were observed. For IL-12 (PHA) we observed marginally significant between-group differences (Kruskal–Wallis test: P = 0·048). There were higher IL-12 (PHA) levels in the COPD I–II group compared with the other groups. However, only the difference to the COPD III–IV group reached statistical significance (Wilcoxon test: P = 0·018). Additionally, the difference between non-smokers and COPD III–IV patients was marginally significant (Wilcoxon test: P = 0·048). Concentrations of IL-12 (CD3) showed no significant between group differences.
Patients with severe COPD (GOLD III–IV) patients show decreased serum levels of IFN-γ
Significant differences of IFN-γ serum levels between groups have been observed (Kruskal–Wallis test: P = 0·002). COPD III–IV patients showed lower IFN-γ serum levels than healthy smokers (Wilcoxon test: P < 0·001) and healthy non-smoker (Wilcoxon test: P = 0·002) patients. Additionally, marginally significantly lower values were observed in the COPD I–II group compared with the healthy smoker group. Note that in 94% of COPD III–IV and 74% in COPD I–II patients (compared with 40% in healthy controls and 21% in healthy smokers) no serum IFN-γ could be detected. For serum TNF-α, serum IL-10 and serum IL-1β no significant between-group differences were found.
Discussion
The total number of lymphocytes circulating in the blood and their subset distribution is under strict homeostatic control. We report for the first time that patients with COPD show a profound change in the representation of functionally and phenotypically distinct subsets of CD4+ T cells. We propose that clonogenic CD4+ T cells with characterized loss of co-stimulatory CD28 and intracellular storage of the cytolytic proteins granzyme B and perforin might be causal for continuing systemic inflammatory state in COPD patients. The basic mechanisms causing replacement of other CD4+ T cells by CD4+CD28null clonotypes are incompletely understood. However, phenotypic and functional analyses of CD4+CD28null T cells have suggested that they are related to NK cells and represent a population of NK-like T cells [29]. In support of this hypothesis, we found that CD4+CD28null T cells express MHC class I-recognizing receptors of the immunoglobulin superfamily (CD94, CD158) [30,31]. Our data corroborate the concept that CD4+CD28null T cells share multiple features with NK cells and may combine functional properties of innate and adaptive immunity in COPD patients.
To prove relevant immune functions we separated PBMCs of the study groups and activated them via specific and unspecific T cell stimulation in vitro. We were able to show that systemic white blood cells derived from COPD GOLD I–II secreted augmented levels of IFN-γ and TNF-α– cytokines that are known to increase macrophage and dendritic cell activity – compared with controls and severe COPD (GOLD III–IV). This observation is particularly interesting, as this in vitro phenomenon was observed only in patients at the initial stage of COPD progression (GOLD stages I–II), indicating a specific role of NK-like T cells in triggering initial lung tissue destruction. Our data confirm and corroborate the pathophysiological speculation by Hodge et al. [11] and Di Stefano et al. [12, 13], who argued that T cell activation is leading to enhanced secretion of IFN-γ, a cytokine that activates macrophages and enhances innate immunity, and is thus causing tissue destruction in COPD-susceptible patients. [32, 33]
This in vitro finding led us to explore whether systemic serum levels of IL-1β, TNF-α, IFN-γ and IL-10 were elevated in COPD patients without recent exacerbation of COPD disease. Contrary to our assumption, the level of inflammatory cytokine IFN-γ correlated negatively with spirometric parameters. This finding underlines the importance of a local interaction of cell-based immune system and lung tissue interphase in the presence of T cell-triggering noxious substances (e.g. inhaled smoke). In a final attempt we investigated whether systemic presence of clonogenic CD4+CD28null T cells is relevant for diagnosing COPD by means of flow cytometry analysis ex vivo. We performed a logistic regression analysis and were able to show that presence of systemic CD4+CD28null T cells was highly predictive for diagnosing COPD. Because of these data we are currently designing a clinical trial to evaluate whether systemic determination of CD4+CD28null by means of flow cytometry analysis is an appropriate tool to identify COPD patients at risk.
Clinical perspective in comparison with other aetiologies
Whatever competing mechanism is causative for COPD, the presence of systemic chronic inflammation in COPD has been associated with a variety of co-morbidities including cachexia [34], osteoporosis [35] and cardiovascular diseases. The relationship between COPD and cardiovascular diseases is especially germane, as more than half of patients with COPD die of cardiovascular causes [36–38]. Nakajima et al. demonstrated that patients with acute ischaemic heart disease are characterized by a perturbation of functional T cell repertoire (CD4+CD28null) with a bias towards increased IFN-γ production compared with controls [39,40]. Of particular importance is a study by Pingiotti et al. They were able to show that patients with rheumatoid arthritis (RA) show increased circulating CD4+CD28null T cells that are related directly to pre-clinical atherosclerotic changes, such as arterial endothelial dysfunction and carotid artery wall thickening [41]. Our observation of T cell pool perturbation in COPD might be relevant in explaining the previously observed long-term cardiovascular risk in this disease entity. However, it remains unclear whether the higher percentage of CD4+CD28null T cells is the result of the inflammatory process, i.e. prematurely senescent CD4+ cells that are unable to go into cell death but still secrete cytokines, or if it represents a subset of COPD subjects whose pathogenetic process includes generation of this T cell subset at an early stage of the disease.
If we interpret our data correctly, a detailed picture is emerging. Chronic antigen exposure, e.g. through contents of tobacco smoke, leads to loss of CD28 and up-regulation of NK cell receptors expression on T cells in potentially genetically susceptible patients. This induced immunological ‘senescence’ is accompanied by a dysregulation of apoptosis-inducing signals, e.g. Bcl-2, fostering longevity of cytotoxic T cells and increased secretion of IFN-γ and TNF-α upon T cell triggering [15]. In conclusion, we believe that the appearance of clonogenic T cells in COPD patients is partially causative for the progressive cell-based inflammatory process in lung tissue irrespective of smoking status.
Acknowledgments
Drs Lambers and Hacker contributed equally to this manuscript. Dr Lambers was responsible for clincial data evaluation. Drs Hacker, Hoetzenecker, Pollreisz and Lichtenauer performed laboratory work. Dr Hacker and Dr Lichtenauer helped to edit the paper. Professor Klepetko provided infrastructure support. Professor Posch was responsible for statistical analysis. This study was supported by FOLAB Chirurgie, private funding (H. J. A.) and the Medical University of Vienna. Dr Hacker was awarded the poster award of the Austrian Society of Pulmonary Medicine (ÖGP) 2008. Dr Ankersmit edited the manuscript and designed and co-ordinated the study. We are thankful to all participants who supported our investigation voluntarily.
Disclosures
The Medical University of Vienna claims financial interest.
References
- 1.Murray CJ, Lopez AD. Mortality by cause for eight regions of the world: Global Burden of Disease study. Lancet. 1997;349:1269–76. doi: 10.1016/S0140-6736(96)07493-4. [DOI] [PubMed] [Google Scholar]
- 2.Murray CJ, Lopez AD. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet. 1997;349:1498–504. doi: 10.1016/S0140-6736(96)07492-2. [DOI] [PubMed] [Google Scholar]
- 3.Hurd S. The impact of COPD on lung health worldwide: epidemiology and incidence. Chest. 2000;117:1S–4S. doi: 10.1378/chest.117.2_suppl.1s. [DOI] [PubMed] [Google Scholar]
- 4.Mannino DM. COPD: epidemiology, prevalence, morbidity and mortality, and disease heterogeneity. Chest. 2002;121:121S–6S. doi: 10.1378/chest.121.5_suppl.121s. [DOI] [PubMed] [Google Scholar]
- 5.Lange P, Groth S, Nyboe GJ, et al. Effects of smoking and changes in smoking habits on the decline of FEV1. Eur Respir J. 1989;2:811–6. [PubMed] [Google Scholar]
- 6.Higenbottam T, Clark TJ, Shipley MJ, Rose G. Lung function and symptoms of cigarette smokers related to tar yield and number of cigarettes smoked. Lancet. 1980;1:409–11. doi: 10.1016/s0140-6736(80)90955-1. [DOI] [PubMed] [Google Scholar]
- 7.Mercer PF, Shute JK, Bhowmik A, Donaldson GC, Wedzicha JA, Warner JA. MMP-9, TIMP-1 and inflammatory cells in sputum from COPD patients during exacerbation. Respir Res. 2005;6:151. doi: 10.1186/1465-9921-6-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sin DD, Man SF. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation. 2003;107:1514–9. doi: 10.1161/01.cir.0000056767.69054.b3. [DOI] [PubMed] [Google Scholar]
- 9.Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2007. Available at: http://www.goldcopd.org.
- 10.Hodge S, Hodge G, Nairn J, Holmes M, Reynolds PN. Increased airway granzyme b and perforin in current and ex-smoking COPD subjects. COPD. 2006;3:179–87. doi: 10.1080/15412550600976868. [DOI] [PubMed] [Google Scholar]
- 11.Hodge G, Nairn J, Holmes M, Reynolds PN, Hodge S. Increased intracellular T helper 1 proinflammatory cytokine production in peripheral blood, bronchoalveolar lavage and intraepithelial T cells of COPD subjects. Clin Exp Immunol. 2007;150:22–9. doi: 10.1111/j.1365-2249.2007.03451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Stefano A, Caramori G, Capelli A, et al. STAT4 activation in smokers and patients with chronic obstructive pulmonary disease. Eur Respir J. 2004;24:78–85. doi: 10.1183/09031936.04.00080303. [DOI] [PubMed] [Google Scholar]
- 13.Di Stefano A, Capelli A, Lusuardi M, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Crit Care Med. 1998;158:1277–85. doi: 10.1164/ajrccm.158.4.9802078. [DOI] [PubMed] [Google Scholar]
- 14.Campisi J. Replicative senescence: an old lives’ tale? Cell. 1996;84:497–500. doi: 10.1016/s0092-8674(00)81023-5. [DOI] [PubMed] [Google Scholar]
- 15.Goronzy JJ, Weyand CM. Aging, autoimmunity and arthritis: T-cell senescence and contraction of T-cell repertoire diversity – catalysts of autoimmunity and chronic inflammation. Arthritis Res Ther. 2003;5:225–34. doi: 10.1186/ar974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choremi-Papadopoulou H, Viglis V, Gargalianos P, Kordossis T, Iniotaki-Theodoraki A, Kosmidis J. Downregulation of CD28 surface antigen on CD4+ and CD8+ T lymphocytes during HIV-1 infection. J Acquir Immune Defic Syndr. 1994;7:245–53. [PubMed] [Google Scholar]
- 17.Hooper M, Kallas EG, Coffin D, Campbell D, Evans TG, Looney RJ. Cytomegalovirus seropositivity is associated with the expansion of CD4+CD28- and CD8+CD28- T cells in rheumatoid arthritis. J Rheumatol. 1999;26:1452–7. [PubMed] [Google Scholar]
- 18.Moosig F, Csernok E, Wang G, Gross WL. Costimulatory molecules in Wegener's granulomatosis (WG): lack of expression of CD28 and preferential up-regulation of its ligands B7-1 (CD80) and B7-2 (CD86) on T cells. Clin Exp Immunol. 1998;114:113–8. doi: 10.1046/j.1365-2249.1998.00695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markovic-Plese S, Cortese I, Wandinger KP, McFarland HF, Martin R. CD4+CD28- costimulation-independent T cells in multiple sclerosis. J Clin Invest. 2001;108:1185–94. doi: 10.1172/JCI12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schirmer M, Goldberger C, Wurzner R, et al. Circulating cytotoxic CD8(+) CD28(-) T cells in ankylosing spondylitis. Arthritis Res. 2002;4:71–6. doi: 10.1186/ar386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffiths GM, Alpert S, Lambert E, McGuire J, Weissman IL. Perforin and granzyme A expression identifying cytolytic lymphocytes in rheumatoid arthritis. Proc Natl Acad Sci USA. 1992;89:549–53. doi: 10.1073/pnas.89.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schirmer M, Vallejo AN, Weyand CM, Goronzy JJ. Resistance to apoptosis and elevated expression of Bcl-2 in clonally expanded CD4+CD28- T cells from rheumatoid arthritis patients. J Immunol. 1998;161:1018–25. [PubMed] [Google Scholar]
- 23.Lopez-Botet M, Bellon T. Natural killer cell activation and inhibition by receptors for MHC class I. Curr Opin Immunol. 1999;11:301–7. doi: 10.1016/s0952-7915(99)80048-x. [DOI] [PubMed] [Google Scholar]
- 24.Lanier LL. NK cell receptors. Annu Rev Immunol. 1998;16:359–93. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- 25.Global strategy for the diagnosis, management and prevention of chronic pulmonary disease (GOLD) 2005. NIH Publication no. 2701. National Heart, Lung, and Blood Institute/World Health Organization. Available at: http://www.goldcopd.org.
- 26.Rabe KF, Beghe B, Luppi F, Fabbri LM. Update in chronic obstructive pulmonary disease 2006. Am J Respir Crit Care Med. 2007;175:1222–32. doi: 10.1164/rccm.200704-586UP. [DOI] [PubMed] [Google Scholar]
- 27.Quanjer PH, Tammeling GJ, Cotes JE, Pedersen OF, Peslin R, Yernault JC. Lung volumes and forced ventilatory flows. Report of the Working Party Standardization of Lung Function Tests, European Community for Steel and Coal. Official Statement of the European Respiratory Society. Eur Respir J Suppl. 1993;16:5–40. [PubMed] [Google Scholar]
- 28.Shaffer JP. Modified sequentially rejective multiple test procedures. J Am Stat Assoc. 1986;81:826–31. [Google Scholar]
- 29.Lanier LL. Back to the future – defining NK cells and T cells. Eur J Immunol. 2007;37:1424–6. doi: 10.1002/eji.200737418. [DOI] [PubMed] [Google Scholar]
- 30.Warrington KJ, Takemura S, Goronzy JJ, Weyand CM. CD4+,CD28- T cells in rheumatoid arthritis patients combine features of the innate and adaptive immune systems. Arthritis Rheum. 2001;44:13–20. doi: 10.1002/1529-0131(200101)44:1<13::AID-ANR3>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 31.Speiser DE, Valmori D, Rimoldi D, et al. CD28-negative cytolytic effector T cells frequently express NK receptors and are present at variable proportions in circulating lymphocytes from healthy donors and melanoma patients. Eur J Immunol. 1999;29:1990–9. doi: 10.1002/(SICI)1521-4141(199906)29:06<1990::AID-IMMU1990>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 32.Gallin JI, Farber JM, Holland SM, et al. Interferon-gamma in the management of infectious diseases. Ann Intern Med. 1995;123:216–24. doi: 10.7326/0003-4819-123-3-199508010-00009. [DOI] [PubMed] [Google Scholar]
- 33.Antoniou KM, Ferdoutsis E, Bouros D. Interferons and their application in the diseases in the lung. Chest. 2003;123:209–16. doi: 10.1378/chest.123.1.209. [DOI] [PubMed] [Google Scholar]
- 34.Agusti AG. Systemic effects of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:367–70. doi: 10.1513/pats.200504-026SR. discussion 71–2. [DOI] [PubMed] [Google Scholar]
- 35.Ionescu AA, Schoon E. Osteoporosis in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2003;46:64s–75s. doi: 10.1183/09031936.03.00004609. [DOI] [PubMed] [Google Scholar]
- 36.Calverley PM, Scott S. Is airway inflammation in chronic obstructive pulmonary disease (COPD) a risk factor for cardiovascular events? COPD. 2006;3:233–42. doi: 10.1080/15412550600977544. [DOI] [PubMed] [Google Scholar]
- 37.Friedman GD, Klatsky AL, Siegelaub AB. Lung function and risk of myocardial infarction and sudden cardiac death. N Engl J Med. 1976;294:1071–5. doi: 10.1056/NEJM197605132942001. [DOI] [PubMed] [Google Scholar]
- 38.Camilli AE, Robbins DR, Lebowitz MD. Death certificate reporting of confirmed airways obstructive disease. Am J Epidemiol. 1991;133:795–800. doi: 10.1093/oxfordjournals.aje.a115958. [DOI] [PubMed] [Google Scholar]
- 39.Nakajima T, Schulte S, Warrington KJ, et al. T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation. 2002;105:570–5. doi: 10.1161/hc0502.103348. [DOI] [PubMed] [Google Scholar]
- 40.Nakajima T, Goek O, Zhang X, et al. De novo expression of killer immunoglobulin-like receptors and signaling proteins regulates the cytotoxic function of CD4 T cells in acute coronary syndromes. Circ Res. 2003;93:106–13. doi: 10.1161/01.RES.0000082333.58263.58. [DOI] [PubMed] [Google Scholar]
- 41.Pingiotti E, Cipriani P, Marrelli A, et al. Surface expression of fractalkine receptor (CX3CR1) on CD4+/CD28 T cells in RA patients and correlation with atherosclerotic damage. Ann NY Acad Sci. 2007;1107:32–41. doi: 10.1196/annals.1381.004. [DOI] [PubMed] [Google Scholar]