

Abstract
Pattern recognition receptors (PRRs) are an integral part of the innate immune system and govern the early control of foreign microorganisms. Single nucleotide polymorphisms (SNPs) in the intracellular pattern recognition receptor nucleotide-binding oligomerization domain-containing protein (NOD2, nucleotide oligomerization domain 2) are associated with Crohn's disease (CD). We investigated the impact of NOD2 polymorphisms on cytokine secretion and proliferation of peripheral blood mononuclear cells (PBMCs) in response to Toll-like receptor (TLR) and NOD2 ligands. Based on NOD2 SNP analyses, 41 CD patients and 12 healthy controls were studied. PBMCs were stimulated with NOD2 and TLR ligands. After 18 h culture supernatants were measured using multiplex assays for the presence of human cytokines granulocyte–macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-1β and tumour necrosis factor (TNF)-α. In CD patients, TLR-induced GM-CSF secretion was impaired by both NOD2-dependent and -independent mechanisms. Moreover, TNF-α production was induced by a TLR-2 ligand, but a down-regulatory function by the NOD2 ligand, muramyl dipeptide, was impaired significantly in CD patients. Intracellular TLR ligands had minimal effect on GM-CSF, TNF-α and IL-1β secretion. CD patients with NOD2 mutations were able to secrete TNF-α, but not GM-CSF, upon stimulation with NOD2 and TLR-7 ligands. CD patients have impaired GM-CSF secretion via NOD2-dependent and -independent pathways and display an impaired NOD2-dependent down-regulation of TNF-α secretion. The defect in GM-CSF secretion suggests a hitherto unknown role of NOD2 in the pathogenesis of CD and is consistent with the hypothesis that impaired GM-CSF secretion in part constitutes a NOD2-dependent disease risk factor.
Keywords: Crohn's disease, cytokines, NOD2, TLR
Introduction
A susceptibility locus for Crohn's disease (CD) is mapped to the gene nucleotide-binding oligomerization domain containing protein 2 (NOD2)/caspase-activating and recruitment domain containing protein (CARD)15[1–4]. Nucleotide oligomerization domain 2 (NOD2) recognizes muramyl dipeptide (MDP), a fragment of peptidoglycan (PGN) [5,6], which is found in the cell walls of both Gram-positive and, to a lesser degree, of Gram-negative bacteria. NOD2 is composed of three domains: a C-terminal leucine-rich repeat domain (LRR) which recognizes MDP, a central nucleotide-binding domain and two N-terminal CARD [7]. Upon MDP recognition NOD2 engages the CARD-containing serine–threonine kinase RICK/RIP2/CARDIAK via homophilic CARD/CARD interaction, which leads subsequently to nuclear factor kappa B (NF-κB) activation [7] and production of cytokines, including interleukin (IL)-1β and IL-8 [8,9].
Three single nucleotide polymorphisms (SNPs) in NOD2 account for 80% of the genetic risk for CD conferred by the NOD2 region [10]. SNP13 is a cytosine insertion 3020insC (Leu1007Pro) resulting in a truncated NOD2, while SNP12 and SNP8 are amino acid substitutions in the LLR (2722G→C, Gly908Arg) and in the LRR adjacent region (2104C→T, Arg702Trp) [2,3]. The prevalence of CD patients with polymorphisms in NOD2 varies considerably between populations [2–4]. The relative risk for CD increases significantly in individuals having two or more NOD2 polymorphisms. Whereas NOD2 SNP heterozygous genotypes increase the risk approximately threefold, the presence of homozygous or compound heterozygous genotypes increases the risk of CD approximately 40-fold [2–4]. NOD2 polymorphisms have been associated with impaired MDP recognition [5,6], and peripheral blood mononuclear cells (PBMCs) from CD patients carrying two polymorphisms produce only minimal levels of IL-1β and IL-8 in response to MDP [9,11].
Recent studies have suggested that NOD2 interferes with Toll-like receptor (TLR) signalling events [12,13], and this regulatory mechanism may be impaired in CD patients with polymorphisms in NOD2[9,11,14]. TLRs recognize pathogen-associated molecular patterns, such as bacterial and viral products, through LRRs [15,16]. Synergistic effects of NOD2 and TLR and loss of synergism in CD patients with polymorphisms in NOD2 have been reported for TLR-2, TLR-3 and TLR-4 [13,17] as well as for TLR-7 [11] and TLR-9 [18]. Functional interactions between TLR-2 and NOD2 remain controversial, because synergistic interactions between NOD2 and TLR-2 were reported in humans [13] and in experimental inflammatory bowel disease models [19,20], whereas a negative regulation of TLR-2 by NOD2 was reported in a murine NOD2 knock-out model [14]. TLR-9 signalling seems to be involved critically in the intestinal epithelial barrier function [21,22] and may play a role in CD, because a TLR-9 polymorphism has been described as associated with CD [23].
In active CD, tissue levels of tumour necrosis factor (TNF)-α, IL-1β and IL-8 are elevated [24–26] and treatment with anti-TNF-α antibodies improves relapsing CD [27]. Furthermore, granulocyte–macrophage colony-stimulating factor (GM-CSF) may be important in the pathogenesis of CD, because GM-CSF treatment improved active CD [28]. GM-CSF is a cytokine that stimulates production of granulocytes and monocytes. It exerts a crucial function during normal inflammatory responses by recruiting granulocytes and monocytes, and it enhances further the differentiation of monocytes into macrophages, a process important for controlling infections. Nevertheless, the mechanisms by which GM-CSF may potentially improve active CD remains unclear.
While genetic variants in NOD2 are a risk factor for the development of CD, the majority of patients carry wild-type (WT) NOD2. This suggests that CD patients may have defects that are enhanced further by the presence of NOD2 polymorphisms [29]. We therefore investigated NOD2-dependent and -independent secretion of GM-CSF, IL-1β and TNF-α in CD patients in the presence or absence of combinations of TLR agonists and MDP stimulation.
Materials and methods
Patients
Patients were recruited from out-patient university clinics in Aarhus and Copenhagen, Denmark and had been diagnosed according to established clinical and histopathological criteria. A total of 41 patients and 12 healthy controls were included in this study. Patients known to carry NOD2 variants were invited to participate, while patients with NOD2 WT alleles were included consecutively in the study. Characteristics of patients are shown in the Supplementary material. The project was approved by the local Ethics Committee (j. no. 1998/4330 with amendment), and all participating individuals provided written informed consent. PBMCs were isolated from venous blood using Ficoll-Paque™ PLUS (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) according to the manufacturer's procedure.
Genotyping of NOD2 variants
Genomic DNA (gDNA) was extracted from cryopreserved PBMCs using the FlexiGene DNA kit (Qiagen, Hilden, Germany). PBMCs (1 × 106 cells) were lysed, treated with protease, and gDNA was isolated following isopropanol precipitation. The gDNA integrity was evaluated by 1% agarose gel electrophoresis, and concentration was determined from the optical density at 260 nm.
Genotyping was carried out in duplicate using a Taqman 5′ nuclease fluorogenic assay [4,30], in which a fluorescent dye-labelled probe specific for the target SNP is amplified by real-time reverse transcription–polymerase chain reaction. The frequencies of the three SNPs that have been associated with CD were determined separately: SNP8 (c.2104C > T/p.R702W), SNP12 (c.2722G > C/p.G908R) and SNP13 (c.3019_3020insC/p.Leu1007fsinsC). Patients who carried the WT alleles were categorized as WT (CD WT). Patients who carried one polymorphism were defined as single-mutated (CD 1mut), whereas patients who carried more than one polymorphism, i.e. compound heterozygotes and homozygotes, were defined as double-mutated (CD 2mut).
Nucleotide oligomerization domain 2 and TLR agonists
All NOD2 and TLR ligands were purchased from InvivoGen (San Diego, CA, USA). Agents were used at the following concentrations: NOD2 ligand, MDP (20 ng/ml); TLR-2 ligand, PGN from Staphylococcus aureus (10 µg/ml); TLR-3 ligand, polyinosine–polycytidylic acid [poly (I : C)] (25 µg/ml); TLR-4 ligand, lipopolysaccharide (LPS) ultra-pure Escherichia coli (1 µg/ml); TLR-7 ligand, loxoribine (400 µM); and TLR-9 ligand, cytosine-guanoside dinucleotides (CpG) DNA ODN M362 (1 µM).
Cell stimulation
The PBMCs (105 cells/200 µl/well) were added to flat-bottomed 96-well plates (Nunc A/S, Roskilde, Denmark) in complete medium in the presence or absence of MDP, or TLR ligands, or both. The cells were incubated at 37°C with 5% CO2. After 18 h, 50 µl culture supernatants from each well were transferred to 96-well Minisorp plates (Nunc A/S). Prior to transfer of supernatants, Minisorp plates were blocked with phosphate-buffered saline + 3% bovine serum albumin (Sigma-Aldrich Chemie GmbH, Steinheim, Germany) + 0·1% Tween® 20 (Sigma-Aldrich Chemie GmbH). Culture supernatants were stored at −70°C until analysis. All experiments were performed in triplicate and supernatants from each experiment were pooled prior to measurement of cytokine concentration.
Multiplex bead immunoassays
The culture supernatants were measured using multiplex beads kit for the presence of human cytokines GM-CSF, IL-1β and TNF-α (Biosource International, Inc., Camarillo, CA, USA). The beads were analysed using the Luminex 100™ instrument (Bio-Plex™ System, Luminex xMAP™ Technology, Bio-Rad Laboratories, Hercules, CA, USA) and the software program Bio-Plex Manager 4·0 (Bio-Rad).
Proliferation assay
Proliferation of the stimulated cells was measured after 4 and 8 days as the amount of [3H]-thymidine incorporated into DNA. The cells were incubated for 16–18 h with 1 µCi [methyl-3H]-thymidine (GE Healthcare Bio-Sciences AB) prior to harvesting onto filtermats (printed filtermats 1450–421; PerkinElmer, Inc.) using a Mach manual harvester (Tomtec, Hamden, CT, USA). Filters were dried and placed in sample bags with 3·5 Betaplate Scintillation buffer (PerkinElmer, Inc., Wellesley, MA, USA). Filters were placed in 1450 MicroBeta® Wallac TriLux Liquid Scintillation and Luminescence Counter (PerkinElmer, Inc.) and the incorporated [methyl-3H]-thymidine was measured as count per minutes and analysed using the software Wallac MicroBeta Windows Workstation version 3.00.005 (PerkinElmer, Inc.).
Statistical analysis
Evaluation of data demonstrated that normal distribution and equality of variances could not be assumed. We therefore used non-parametric statistics for the analysis. Differences between two groups were analysed by Mann–Whitney U-test. The Wilcoxon signed-ranks test was used to analyse MDP dependency. Trends across categories were tested using Spearman's rank correlation. P levels below 0·05 were considered statistically significant. Continuous data are presented as mean ± standard error of the mean (s.e.m.). All statistical analyses were carried out using the spss version 11.0 software package (SPSS Inc., Chicago, IL, USA).
Results
Impaired cytokine responses are correlated with the number of NOD2 polymorphisms
Genotyping of NOD2 regarding SNP8, SNP12 and SNP13 in a selected patient cohort of 224 patients led to the identification of 22 patients carrying CD-associated NOD2 variants. One patient was homozygous for SNP8 polymorphism and one for SNP13 polymorphism. Four patients were compound heterozygous: one for SNP8/12/13 polymorphism and three for SNP8/13. Sixteen patients were heterozygous; six for SNP8 and 10 for SNP13. In addition, 19 patients and 12 healthy controls, which were homozygous for the WT allele, were included.
To elucidate functional differences associated with NOD2 polymorphisms, we compared the cytokine responses in PBMCs following stimulation with the NOD2 ligand MDP. In the presence of 20 ng/ml MDP, the secretion of GM-CSF, TNF-α and IL-1β increased significantly (P < 0·01) in healthy controls by 10-fold, 20-fold and fourfold respectively. MDP failed to induce proliferation (data not shown).
Compared with the healthy controls, the GM-CSF secretion induced by MDP was impaired in CD patients, and this impairment correlated with the number of polymorphisms in NOD2. When both alleles of NOD2 had polymorphisms, GM-CSF secretion was reduced to the level of unstimulated PBMCs (Fig. 1). Similarly, the response to TNF-α and IL-1β decreased significantly (P < 0·01) with NOD2 polymorphisms (Fig. 1). The negative association between the number of NOD2 polymorphisms and cytokine production was independent of disease activity, disease location, steroid treatment, 5-aminosalicylic acid treatment and biological treatment (anti-TNF-α antibodies) (data not shown). Thus, these findings demonstrate a NOD2-dependent decrease in GM-CSF, TNF-α and IL-1β secretion in CD patients following MDP stimulation.
Fig. 1.
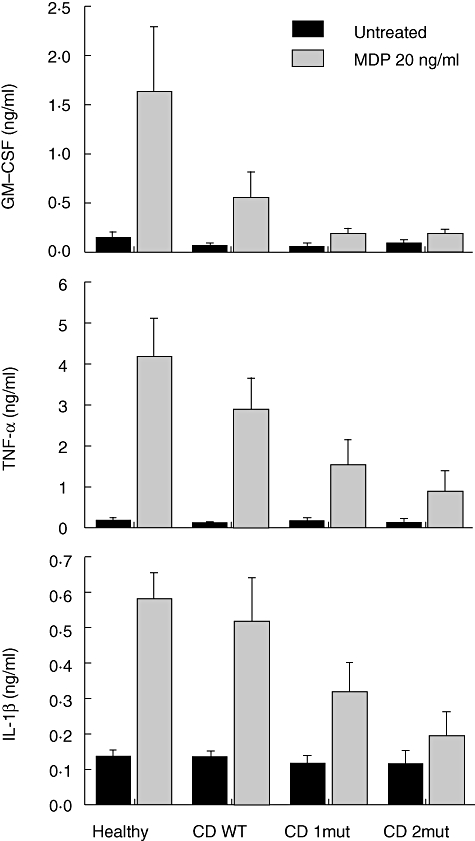
Cytokine responses to muramyl dipeptide (MDP) are correlated negatively with the number of common nucleotide oligomerization domain 2 (NOD2) polymorphisms in Crohn's disease (CD). Peripheral blood mononuclear cells (PBMCs) from healthy controls and CD patients were stimulated in presence or absence of 20 ng/ml MDP for 18 h. Supernatants were assessed for granulocyte–macrophage colony-stimulating factor (GM-CSF), tumour necrosis factor (TNF)-α and interleukin (IL)-1β as described in the methods. Healthy: healthy controls carrying none of the three common NOD2 polymorphisms; CD WT: patients carrying none of the three common NOD2 polymorphisms; CD 1mut: patients carrying one of the three NOD2 polymorphisms; CD 2mut: patients carrying two or more of the three NOD2 polymorphisms. Error bars indicate standard error of the mean. See text for statistically significant associations.
Toll-like receptor-2 and TLR-4 agonists induce GM-CSF secretion through NOD2-dependent and -independent pathways
We next asked whether polymorphisms in NOD2 impaired the responses through TLR-2 and TLR-4. PBMCs incubated with 10 µg/ml PGN (TLR-2 ligand) or 1 µg/ml LPS (TLR-4 ligand) generated a strong secretion of GM-CSF, TNF-α and IL-1β compared with the secretion induced by 20 ng/ml MDP (Fig. 2a). However, the GM-CSF secretion stimulated via TLR-2 and TLR-4 was impaired in CD and the impairment increased with the number of polymorphisms. Importantly, there were no differences in TLR-2- and TLR-4-induced TNF-α and IL-1β secretions in healthy controls and CD patients (Fig. 2a). Thus, secretion of GM-CSF was impaired in CD patients by both NOD2-dependent and -independent pathways.
Fig. 2.
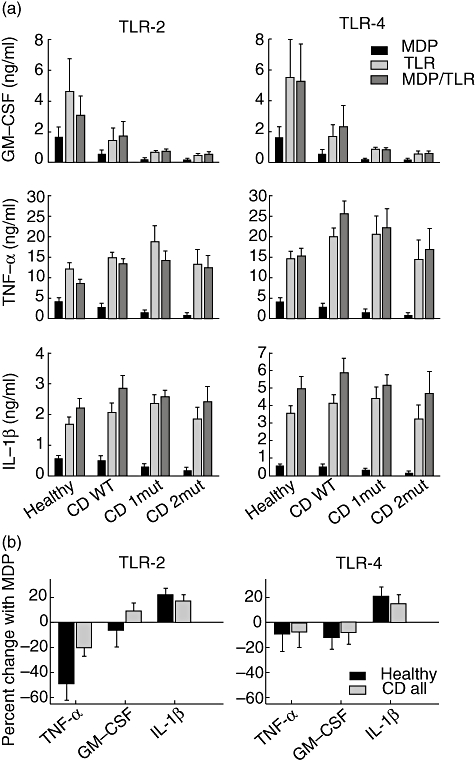
Toll-like receptor (TLR)-2 and TLR-4 agonists induce granulocyte–macrophage colony-stimulating factor (GM-CSF) secretion through nucleotide oligomerization domain 2 (NOD2)-dependent and -independent pathways. (a) Peripheral blood mononuclear cells (PBMCs) from healthy controls and Crohn's disease (CD) patients were stimulated in presence of either 20 ng/ml muramyl dipeptide (MDP) or 10 µg/ml peptidoglycan (PGN) or 1 µg/ml lipopolysaccharide (LPS) or both MDP and a TLR agonist for 18 h. Supernatants were assessed for GM-CSF, tumour necrosis factor (TNF)-α and interleukin (IL)-1β, as described in the Methods. (b) To calculate the inhibitory effect by MDP on TLR-induced TNF-α production, the individual contribution by MDP and TLR were subtracted from the combined contribution by MDP/TLR. This TNF-α production was expressed as percentage of the production obtained with the individual MDP and TLR stimulations. Healthy: healthy controls carrying none of the three common NOD2 polymorphisms; CD WT: patients carrying none of the three common NOD2 polymorphisms; CD 1mut: patients carrying one of the three NOD2 polymorphisms; CD 2mut: patients carrying two or more of the three NOD2 polymorphisms. Error bars indicate standard error of the mean. See text for statistically significant associations.
Altered regulation of TNF-α secretion in CD patients
The impact of NOD2 on TLR-2 signalling is controversial. To address this issue further, we compared cytokine secretion after combined stimulation of NOD2 and TLR-2 with secretion obtained after stimulation of either NOD2 or TLR-2 alone.
After combined stimulation with PGN and MDP, TNF-α secretion in PBMCs from healthy controls was reduced significantly (P = 0·006) compared with TNF-α secretion induced through TLR-2 alone (Fig. 2a). This suggested that NOD2 inhibited TLR-2-induced TNF-α secretion in healthy individuals. In CD patients, however, PGN-induced TNF-α secretion was not reduced significantly in combination with MDP (Fig. 2a). MDP inhibited PGN-induced TNF-α secretion by 5% in healthy controls, whereas TNF-α levels were only reduced by 20% in the total group of CD patients (Fig. 2b). This demonstrates that CD patients had an impaired down-regulation of TNF-α secretion (P = 0·04). Proliferation induced through TLR-2 was also down-regulated by NOD2, and this effect was not seen in CD patients with two NOD2 polymorphisms (data not shown). TLR-4-induced TNF-α secretion was not regulated negatively by MDP, nor was proliferation in any of the tested groups (Fig. 2a and b).
In contrast, we did not find a significant difference in the NOD2 regulation of TLR-2-induced secretion of GM-CSF and IL-1β (Fig. 2a). Although not statistically significant, MDP reduced GM-CSF secretion in healthy controls when used in combination with PGN, whereas the opposite was seen in CD patients (Fig. 2a and b).
Synergism between TLR-7 agonists and MDP
We next analysed whether CD patients responded normally to agonists of TLR-3 (poly I : C), TLR-7 (loxoribine) and TLR-9 (CpG), and whether NOD2 affected these pathways. In general, a weak cytokine secretion, if any, was induced in healthy controls and CD patients in response to TLR-3, TLR-7 and TLR-9 agonists (Fig. 3a), although these agonists induced significant proliferative responses (data not shown).
Fig. 3.
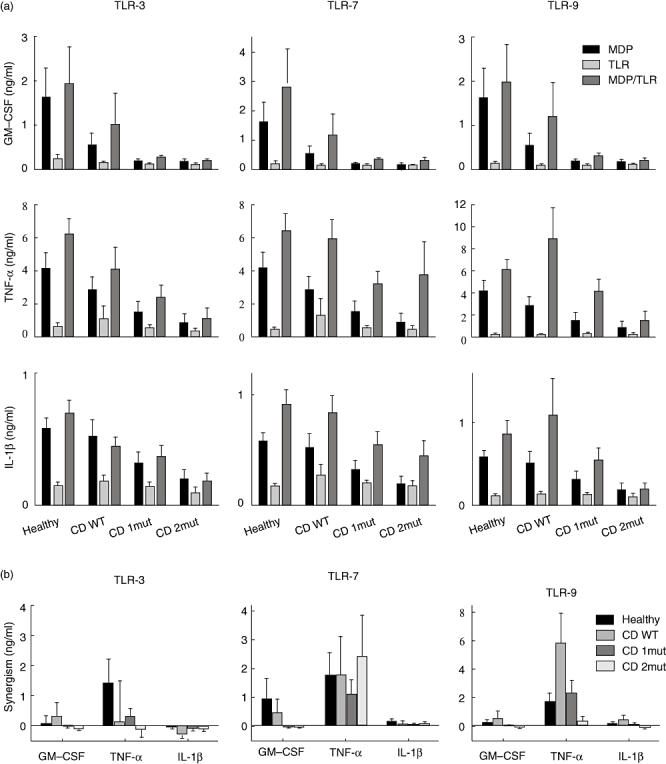
Toll-like receptor (TLR) agonists and muramyl dipeptide (MDP) induce cytokines synergistically. (a) Peripheral blood mononuclear cells (PBMCs) from healthy controls and Crohn's disease (CD) patients were stimulated in presence of either 20 ng/ml MDP, 25 µg/ml polyinosine–polycytidylic acid [poly (I : C)], 400 µM loxoribine or 1 µM cytosine-guanoside dinucleotides (CpG) or both MDP and a TLR agonist for 18 h. Supernatants were assessed for granulocyte–macrophage colony-stimulating factor (GM-CSF), tumour necrosis factor (TNF)-α and interleukin (IL)-1β, as described in the Methods. (b) The synergism was calculated by subtraction of baseline secretions induced by MDP and TLR agonists from secretions induced by combined treatment. Healthy: healthy controls carrying none of the three common nucleotide oligomerization domain 2 (NOD2) polymorphisms; CD WT: patients carrying none of the three common NOD2 polymorphisms; CD 1mut: patients carrying two of the three NOD2 polymorphisms; CD 2mut: patients carrying one or more of the three NOD2 polymorphisms. Error bars indicate standard error of the mean. See text for statistically significant associations.
However, agonists of TLR-3, TLR-7 or TLR-9 induced TNF-α secretion synergistically in healthy controls when combined with the NOD2 agonist MDP (Fig. 3b). In contrast to TLR-3 and TLR-9, a synergistic induction of TNF-α secretion via TLR-7 was maintained in CD patients despite polymorphisms in NOD2 (Fig. 3b). Hence, TNF-α secretion was restored partially in PBMCs from patient with NOD2 polymorphisms when stimulated with TLR-7 agonist and MDP together. This was not seen for GM-CSF secretion, which was impaired significantly in all CD patients (Fig. 3a). The TLR-3 agonist and MDP had a synergistic effect on proliferation in healthy controls, but not in CD patients. In both healthy individuals and CD patients the TLR-9 agonist induced very strong proliferation after 4 days, with a negative effect of simultaneous stimulation by MDP (data not shown).
Discussion
Genetic variants in NOD2 are associated with CD. At present, there are three main hypotheses of how NOD2 is involved in the pathogenesis of CD (reviewed by Strober and colleagues [31] and Schreiber et al.[32]). One suggests that NOD2 functions as a negative regulator of proinflammatory pathways, e.g. by inhibiting PGN (TLR-2)-induced IL-12 production [14]. The second suggests that NOD2 polymorphisms lead to loss-of-function with increased susceptibility to CD as a consequence of impaired host defence and α-defensin production [19], and the last hypothesis explains the association between NOD2 polymorphisms and CD as gain-of-function mutations where a defective NOD2 leads to hyperactivation of NF-κB and excessive IL-1β secretion [20].
In our study, the NOD2 agonist MDP induced secretion of GM-CSF, TNF-α and IL-1β that was decreased significantly in CD patients, in particular but not exclusively in those carrying two NOD2 polymorphisms. Although these observations are consistent with previous findings [2,3,5,6,9,11], they also demonstrate that NOD2 polymorphisms result in loss-of-function, which aggravates functional defects already present in CD patients. In addition, we find that CD patients have a NOD2-independent loss-of-function in GM-CSF secretion. Importantly, this loss-of-function is aggravated further by NOD2 polymorphisms, indicating that GM-CSF secretion is induced by both NOD2-dependent and -independent pathways. This suggests strongly a hitherto unrecognized role of GM-CSF in the pathogenesis of CD, and this may help to explain why these polymorphisms are risk factors, although they are not necessary for developing the disease.
Previously, we have demonstrated an increased GM-CSF production in cultured colonic CD4+ T cells from patients with active CD compared with healthy volunteers [33]. Direct comparison of PBMCs and tissue-derived T cells may be difficult, because migration into peripheral tissues alters the functional maturation/activation of the T cells meeting local antigens. Others have suggested that increased GM-CSF in CD mucosal lesion leads to inflammation [34]. However, the opposite may also be true, i.e. GM-CSF in active disease is increased insufficiently compared with inflammatory controls, a mechanism which has been shown for regulatory T cells [35]. The necessity of competent GM-CSF to control inflammation is supported by a recent study demonstrating that GM-CSF treatment decreased disease severity in active CD and improved quality of life [36]. GM-CSF receptors are expressed by CD4+ T cells and Paneth cells of the intestinal epithelium and by both myeloid and intestinal epithelial cells throughout the gastrointestinal tract [37,38] although, to a different extent, all these cells have been shown to express NOD2 [7,39,40]. This suggests that an important future goal in CD research is to understand in more detail the significance of GM-CSF in the regulation of gut immunity.
We found that CD patients were impaired in their regulation of the proinflammatory cytokine TNF-α. Whereas PGN-induced TNF-α secretion was regulated negatively by MDP in healthy controls, the combined group of CD patients was impaired in this regulation. Although statistically significant for the combined group, the individual CD groups had too-small sample numbers to reach statistical significance. Nevertheless, the negative regulation of TLR-2 by NOD2 was also demonstrated in proliferation assays. Our findings, using human primary cells derived from CD patients and controls, support murine NOD2 knock-out models, which demonstrated overactive TLR-2 signalling and overproduction of IL-12 in the absence of functional NOD2 [14]. In the study by Watanabe and co-workers a concentration of 100 µg/ml of MDP was used, which is a higher concentration than can be expected in vivo. We used 0·02 µg/ml of MDP, which is more similar to concentrations used in other studies and in vivo concentrations. Importantly, we did not find a negative regulatory effect on TNF-α secretion when using 2 µg/ml of MDP (data not shown). This is in agreement with a recent report, where a high concentration of MDP (100 µg/ml) but not an intermediary concentration (1–25 µg/ml) exerted the negative regulatory effect on TNF-α[41]. However, other groups have reported opposing results with a synergistic effect between NOD2 and TLR-2 [8,11,13,19]. It is unclear how these opposing results may be reconciled, but it may be hypothesized that the net result of the cross-talk between NOD2 and TLR-2 signalling depends critically on a delicate balance of ligand concentration, receptor expression levels and the chronology of the receptor stimulation.
Importantly, an exception to the above regulation was observed with TNF-α secretion induced by TLR-7 and NOD2. The combined stimulation through TLR-7 and NOD2 virtually restored this cytokine secretion in CD patients with one or two NOD2 variants. This was surprising, because we did not expect an impact of MDP on the patients with NOD2 polymorphisms. However, we cannot rule out potential NOD2-independent effects of MDP. Although a similar trend for IL-1β secretion was observed, NOD2-independent effects were not seen for TNF-α secretion via TLR-3 and TLR-9 or for TLR-7/NOD2-induced GM-CSF secretion.
Synergism between NOD2 and intracellular TLRs is controversial. Whereas synergistic interactions of NOD2 and TLR-3 [12,13] or TLR-9 [8,18] have been reported, others failed to demonstrate synergisms between NOD2 and TLR-7 and TLR-9 [13]. As presented in Fig. 3b, we found that MDP and poly I : C, loxoribine or CpG induced TNF-α synergistically in healthy controls and the synergism was impaired in CD patients with two polymorphisms in NOD2 with regard to TLR-3 and TLR-9 but intact for TLR-7, and thus independent of NOD2 polymorphisms. We did not observe synergistic induction of GM-CSF and IL-1β. These data support the findings by van Heel and colleagues of synergistic secretion of TNF-α induced by MDP and CpG in healthy controls and the impairment in CD patients with two or more polymorphisms [18]. However, we do not know why the TNF-α response is less dependent on NOD2 when stimulated through TLR-7, as opposed to stimulation through TLR-3 or TLR-9.
In summary, our data suggest a significant role of GM-CSF in the pathogenesis of CD. Upon TLR-2/NOD2 stimulation, the secretion of GM-CSF was reduced in all CD patients, and was almost abolished in CD patients with NOD2 polymorphisms. This is in contrast to TNF-α secretion, which was down-regulated by NOD2, but more so in healthy individuals than in CD patients. This argues for both NOD2-dependent and NOD2-independent pathways in GM-CSF secretion. The NOD2-independent GM-CSF secretion may be mediated via p38 mitogen-activated protein kinase (MAPK) and subsequent extracellular regulated kinase (ERK-1/2) phosphorylation [42]. The mechanism by which NF-κB-independent reduced GM-CSF production may contribute to disease pathogenesis remains to be investigated, but might be related to diminished innate immune reactivity.
Acknowledgments
We thank Bettina Bundgaard, Rikke Andersen and Tanja Kaacksteen for technical assistance. We thank Dr Mogens Erlandsen, Department of Biostatistics, University of Aarhus, for assistance with statistical analysis. This study was funded by grants from the Danish Colitis and Crohn Foundation, the Danish Medical Research Council, the German Research Council (SFB415 and 617) and the German Ministry for Education and Research (BMBF, NGFN Pathway Mapping).
Supporting information
Additional Supporting Information may be found in the online version of this article:
Table S1. Brosbøl-Ravnborg et al. Toll-like receptor (TLR)induced granulocyte–macrophage colony-stimulating factor (GM-CSF) secretion.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Hugot JP, Laurent-Puig P, Gower-Rousseau C, et al. Mapping of a susceptibility locus for Crohn's disease on chromosome 16. Nature. 1996;379:821–3. doi: 10.1038/379821a0. [DOI] [PubMed] [Google Scholar]
- 2.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 3.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 4.Hampe J, Cuthbert A, Croucher PJ, et al. Association between insertion mutation in NOD2 gene and Crohn's disease in German and British populations. Lancet. 2001;357:1925–8. doi: 10.1016/S0140-6736(00)05063-7. [DOI] [PubMed] [Google Scholar]
- 5.Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. 2003;278:5509–12. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 6.Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–72. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 7.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappa B. J Biol Chem. 2001;276:4812–18. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 8.Uehara A, Yang S, Fujimoto Y, et al. Muramyldipeptide and diaminopimelic acid-containing desmuramylpeptides in combination with chemically synthesized Toll-like receptor agonists synergistically induced production of interleukin-8 in a NOD2- and NOD1-dependent manner, respectively, in human monocytic cells in culture. Cell Microbiol. 2005;7:53–61. doi: 10.1111/j.1462-5822.2004.00433.x. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Moran T, Swanson E, et al. Regulation of IL-8 and IL-1beta expression in Crohn's disease associated NOD2/CARD15 mutations. Hum Mol Genet. 2004;13:1715–25. doi: 10.1093/hmg/ddh182. [DOI] [PubMed] [Google Scholar]
- 10.Lesage S, Zouali H, Cezard JP, et al. CARD15/NOD2 mutational analysis and genotype–phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70:845–57. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Heel DA, Ghosh S, Butler M, et al. Muramyl dipeptide and Toll-like receptor sensitivity in NOD2-associated Crohn's disease. Lancet. 2005;365:1794–6. doi: 10.1016/S0140-6736(05)66582-8. [DOI] [PubMed] [Google Scholar]
- 12.Tada H, Aiba S, Shibata K, Ohteki T, Takada H. Synergistic effect of Nod1 and Nod2 agonists with Toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infect Immun. 2005;73:7967–76. doi: 10.1128/IAI.73.12.7967-7976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Netea MG, Ferwerda G, de Jong DJ, et al. Nucleotide-binding oligomerization domain-2 modulates specific TLR pathways for the induction of cytokine release. J Immunol. 2005;174:6518–23. doi: 10.4049/jimmunol.174.10.6518. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–8. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 15.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–80. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 16.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 17.Rosenstiel P, Sina C, End C, et al. Regulation of DMBT1 via NOD2 and TLR4 in intestinal epithelial cells modulates bacterial recognition and invasion. J Immunol. 2007;178:8203–11. doi: 10.4049/jimmunol.178.12.8203. [DOI] [PubMed] [Google Scholar]
- 18.van Heel DA, Ghosh S, Hunt KA, et al. Synergy between TLR9 and NOD2 innate immune responses is lost in genetic Crohn's disease. Gut. 2005;54:1553–7. doi: 10.1136/gut.2005.065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi KS, Chamaillard M, Ogura Y, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–4. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 20.Maeda S, Hsu LC, Liu H, et al. Nod2 mutation in Crohn's disease potentiates NF-{kappa}B activity and IL-1{beta} processing. Science. 2005;307:734–8. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 21.Lee J, Mo JH, Katakura K, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol. 2006;8:1327–36. doi: 10.1038/ncb1500. [DOI] [PubMed] [Google Scholar]
- 22.Lee J, Mo JH, Shen C, Rucker AN, Raz E. Toll-like receptor signaling in intestinal epithelial cells contributes to colonic homoeostasis. Curr Opin Gastroenterol. 2007;23:27–31. doi: 10.1097/MOG.0b013e3280118272. [DOI] [PubMed] [Google Scholar]
- 23.Torok HP, Glas J, Tonenchi L, Bruennler G, Folwaczny M, Folwaczny C. Crohn's disease is associated with a Toll-like receptor-9 polymorphism. Gastroenterology. 2004;127:365–6. doi: 10.1053/j.gastro.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 24.Ligumsky M, Simon PL, Karmeli F, Rachmilewitz D. Role of interleukin 1 in inflammatory bowel disease – enhanced production during active disease. Gut. 1990;31:686–9. doi: 10.1136/gut.31.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daig R, Andus T, Aschenbrenner E, Falk W, Scholmerich J, Gross V. Increased interleukin 8 expression in the colon mucosa of patients with inflammatory bowel disease. Gut. 1996;38:216–22. doi: 10.1136/gut.38.2.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacDonald TT, Hutchings P, Choy MY, Murch S, Cooke A. Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin Exp Immunol. 1990;81:301–5. doi: 10.1111/j.1365-2249.1990.tb03334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braat H, Peppelenbosch MP, Hommes DW. Immunology of Crohn's disease. Ann NY Acad Sci. 2006;1072:135–54. doi: 10.1196/annals.1326.039. [DOI] [PubMed] [Google Scholar]
- 28.Dieckgraefe BK, Korzenik JR. Treatment of active Crohn's disease with recombinant human granulocyte–macrophage colony-stimulating factor. Lancet. 2002;360:1478–80. doi: 10.1016/S0140-6736(02)11437-1. [DOI] [PubMed] [Google Scholar]
- 29.Marks DJ, Harbord MW, MacAllister R, et al. Defective acute inflammation in Crohn's disease: a clinical investigation. Lancet. 2006;367:668–78. doi: 10.1016/S0140-6736(06)68265-2. [DOI] [PubMed] [Google Scholar]
- 30.Livak KJ. Allelic discrimination using fluorogenic probes and the 5′ nuclease assay. Genet Anal. 1999;14:143–9. doi: 10.1016/s1050-3862(98)00019-9. [DOI] [PubMed] [Google Scholar]
- 31.Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol. 2006;6:9–20. doi: 10.1038/nri1747. [DOI] [PubMed] [Google Scholar]
- 32.Schreiber S, Rosenstiel P, Albrecht M, Hampe J, Krawczak M. Genetics of Crohn disease, an archetypal inflammatory barrier disease. Nat Rev Genet. 2005;6:376–88. doi: 10.1038/nrg1607. [DOI] [PubMed] [Google Scholar]
- 33.Agnholt J, Kelsen J, Brandsborg B, Jakobsen NO, Dahlerup JF. Increased production of granulocyte–macrophage colony-stimulating factor in Crohn's disease – possible target for infliximab treatment. Eur J Gastroenterol Hepatol. 2004;16:649–55. doi: 10.1097/01.meg.0000108344.41221.8b. [DOI] [PubMed] [Google Scholar]
- 34.Noguchi M, Hiwatashi N, Liu ZX, Toyota T. Increased secretion of granulocyte–macrophage colony-stimulating factor in mucosal lesions of inflammatory bowel disease. Digestion. 2001;63(Suppl)(1):32–6. doi: 10.1159/000051908. [DOI] [PubMed] [Google Scholar]
- 35.Maul J, Loddenkemper C, Mundt P, et al. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology. 2005;128:1868–78. doi: 10.1053/j.gastro.2005.03.043. [DOI] [PubMed] [Google Scholar]
- 36.Korzenik JR, Dieckgraefe BK, Valentine JF, Hausman DF, Gilbert MJ. Sargramostim for active Crohn's disease. N Engl J Med. 2005;352:2193–201. doi: 10.1056/NEJMoa041109. [DOI] [PubMed] [Google Scholar]
- 37.Armitage JO. Emerging applications of recombinant human granulocyte–macrophage colony-stimulating factor. Blood. 1998;92:4491–508. [PubMed] [Google Scholar]
- 38.Fukuzawa H, Sawada M, Kayahara T, et al. Identification of GM-CSF in Paneth cells using single-cell RT-PCR. Biochem Biophys Res Commun. 2003;312:897–902. doi: 10.1016/j.bbrc.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 39.Oh HM, Lee HJ, Seo GS, et al. Induction and localization of NOD2 protein in human endothelial cells. Cell Immunol. 2005;237:37–44. doi: 10.1016/j.cellimm.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 40.Rosenstiel P, Fantini M, Brautigam K, et al. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology. 2003;124:1001–9. doi: 10.1053/gast.2003.50157. [DOI] [PubMed] [Google Scholar]
- 41.Borm ME, van Bodegraven AA, Mulder CJ, Kraal G, Bouma G. The effect of NOD2 activation on TLR2-mediated cytokine responses is dependent on activation dose and NOD2 genotype. Genes Immun. 2008;9:274–8. doi: 10.1038/gene.2008.9. [DOI] [PubMed] [Google Scholar]
- 42.Meja KK, Seldon PM, Nasuhara Y, et al. p38 MAP kinase and MKK-1 co-operate in the generation of GM-CSF from LPS-stimulated human monocytes by an NF-kappa B-independent mechanism. Br J Pharmacol. 2000;131:1143–53. doi: 10.1038/sj.bjp.0703684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.