

Abstract
The proteasome inhibitor, bortezomib, has direct anti-tumour effects and has been demonstrated to sensitize tumour cells to tumour necrosis factor-related apoptosis-inducing ligand-mediated apoptosis. Natural killer (NK) cells are effective mediators of anti-tumour responses, both through cytotoxic granule killing and apoptosis-inducing pathways. We therefore investigated if bortezomib sensitized human breast cancer cells to killing by the human NK cell line, NK-92. Bortezomib was unable to sensitize MDA-231 breast cancer cells to NK cell-mediated killing in short-term in vitro assays. However, bortezomib did cause these cells to up-regulate apoptosis-related mRNA as well as death receptors on the cell surface. In a long-term in vitro tumour outgrowth assay that allows NK cells to use their full repertoire of killing pathways, bortezomib sensitized three breast cancer cell lines to NK cell-mediated killing, which led to greater anti-tumour effects than either treatment alone. We then used a xenogeneic mouse model in which CB-17 SCID mice were injected with human breast cancer cells. This model displayed the effectiveness of NK-92 cells, but the addition of bortezomib did not increase the survival further or reduce the number of lung metastases in tumour-bearing mice. However, while bortezomib was highly cytotoxic to NK-92 cells in vitro, bortezomib treatment in vivo did not decrease NK-92 function, suggesting that through alternative dosing or timing of bortezomib, greater efficacy may occur from combined therapy. These data demonstrate that combined treatment of human breast cancer with bortezomib and NK cells has the potential to generate superior anti-tumour responses than either therapy alone.
Keywords: breast cancer, natural killer cell, proteasome inhibition
Introduction
The proteasome is a multi-subunit protein complex which regulates proteolysis within the cell, removing misshapen or unneeded proteins. The proteasome maintains the degradation of critical cell cycle proteins which are vital to survival and proliferation [1]. The proteasome exists most commonly as a roughly 2000 kDa protein complex with a Svedberg sedimentation coefficient of 26 (26S) [2]. The 26S proteasome consists of a 20S core complex with two 19S regulatory complexes on either side [3]. Degradation of proteins occurs through three separate catalytic sites: the tryptic, chymotryptic and post-glutamyl peptide hydrolytic-like [4,5]. The proteasome degrades proteins which have been tagged by a series of covalently bonded ubiquitin molecules. These ubiquitin-tagged proteins are degraded in the proteasome with the ubiquitin molecules being recycled for future use. The proteasome can also function through an ubiquitin-independent pathway, typically in post-translational protein processing and in the degradation of inherently unstable proteins [6].
Bortezomib (formerly PS-341, Velcade®; Millennium Pharmaceuticals, Cambridge, MA, USA) is a proteasome inhibitor which received Food and Drug Administration approval in 2003 for the treatment of patients with multiple myeloma and mantle cell lymphoma who had received one previous treatment of chemotherapy [7,8]. Recent Phase I and Phase II clinical trials have also examined the use of bortezomib in solid tumours [9–11]. Bortezomib specifically inhibits the chymotryptic-like subunit of the 26S proteasome [4]. Proteasome inhibition is associated with decreased tumour proliferation and increased apoptosis [3]. Our laboratory has demonstrated previously that bortezomib can sensitize tumour cells to apoptosis through tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor ligation [12]. The death ligand TRAIL is expressed on T lymphocytes and natural killer (NK) cells and binds to the death receptors DR4 and DR5 [13]. Tumour cells are sensitized by bortezomib to TRAIL-mediated therapies in part because of a reduction in the survival protein, cellular Fas-associated death domain-like interleukin (IL)-1 converting enzyme (FLICE)-like inhibitory protein (c-FLIP), and an increase in the surface expression of death ligand receptors, including DR5 and Fas, on the target cells [12,14].
The NK cells are a subset of lymphocytes which are capable of mediating cytotoxicity against tumour cells and virally infected cells and constitute a key component of the innate immune system [15]. NK cells possess a repertoire of various killing pathways to kill target cells; these include the release of cytotoxic granules, the production of cytokines which can induce an immune response and the ability to engage death receptors on the surface of target cells [16]. The latter is of particular interest, as NK cells possess TRAIL and Fas ligand (FasL), both of which can trigger apoptosis in target cells. Killing via cytotoxic granules has been shown to be the primary mechanism by which NK cells kill in short-term in vitro assays [17], although these short-term assays do not reflect the engagement of death ligands on tumour targets. Therefore, it may be more informative to use longer-term assays in which NK cells can use their full complement of killing pathways; these conditions may be a better predictor of how effective the NK cells function in vivo.
The NK-92 is a human NK cell line first established in 1994 from a 50-year-old male patient with an aggressive NK cell lymphoma [18]. NK-92 cells are negative for the T cell surface markers CD3, CD4, and CD8 and express the CD56bright phenotype, which is found most abundantly on human NK cells that occur in the lymph nodes. NK-92 cells do not express the generally inhibitory killer cell immunoglobulin-like receptors which is thought to explain their high cytotoxic activity when compared with primary NK cells [19]. The NK-92 cell line has been examined clinically as a treatment for advanced sarcomas and leukaemias [20]. The parental NK-92 cell line is highly dependent on the cytokine IL-2 and therapies involving these cells in vivo require superphysiological amounts of IL-2. However, an IL-2 independent cell line, NK-92MI, which has been shown to be virtually identical to the parental cell line, may be a more appropriate choice for clinical therapies[21]. We used the parental NK-92 cell line in our murine in vivo studies because toxicity related to IL-2 was not observed. With their high cytotoxic activity and long-term growth potential, the NK-92 cell line is an attractive model to study the function of human NK cells.
In this study, we demonstrate that bortezomib treatment of human breast cancer cells increases the surface expression of apoptosis-related genes as well as the surface expression of death receptors. We also demonstrate that while bortezomib does not sensitize tumours to NK cell-mediated killing in short-term assays, long-term assays demonstrated a significant increase in NK cell-mediated cytotoxicity against tumour cells. These results demonstrate the potential for proteasome inhibition by bortezomib to increase the effectiveness of NK cell function and to improve current cancer therapies.
Methods
Mice
CB17 SCID mice were obtained from the Animal Production Area of the National Cancer Institute-Frederick (Frederick, MD, USA). All mice used were females between the ages of 2 and 4 months. Animal studies were performed at the University of Nevada, Reno animal facility under specific pathogen-free conditions in accordance with the Institutional Animal Care and Use Committees and procedures outlined by the National Institute of Health.
Cell lines and culture
The NK-92 human NK cell line along with human mammary carcinoma lines MDA-231, BT-20 and MCF-7 (adenocarcinoma) were obtained from the American Type Culture Collection (Rockville, MD, USA). NK-92 cells were maintained in 1× alpha-minimum essential medium supplemented with 12·5% fetal bovine serum (FBS) (Gemini Bio-Products, Woodland, CA, USA), 12·5% horse serum, 1·5 g/l sodium bicarbonate, 0·2 mM Myo-inositol, 0·1 mM 2-mercaptoethanol, 0·02 mM folic acid and 500 IU/ml recombinant human (rh) IL-2 (Developmental Therapeutics Program, NCI-Frederick). MDA-231 cells were maintained in RPMI-1640 media supplemented with 5% FBS (Gemini Bio-Products, West Sacramento, CA, USA), 2 mM l-glutamine, 100 U/ml penicillin/streptomycin. All in vitro experiments which required the co-culturing of NK-92 and MDA-231 cells contained 50% NK-92 growth medium and 50% MDA-231 growth medium.
Reagents
Bortezomib (PS-341, Velcade®) was provided by Millennium Pharmaceuticals. Stock solutions of bortezomib were made at 1 mg/ml in phosphate-buffered saline (PBS) and frozen at −70°C. Stock solutions were thawed and diluted to 1 µg/ml in RF-10 complete medium and used within 2 weeks of dilution. Bortezomib solutions were protected from light at all times. rh IL-2 [IL-2, TECIN (Teceleukin)] was provided by the National Cancer Institute. 5,6-carboxy-succinimidyl-fluorescein-ester (CFSE) labelling was performed using the Molecular Probes (Invitrogen, Carlsbad, CA, USA) Vybrant® CFDA SE Cell Tracer Kit according to the manufacturer's protocol.
Flow cytometry
Approximately 106 MDA-231 cells per sample were brought to a single-cell suspension and were washed with blocking buffer (1% FBS, 1% human antibody serum in PBS). Cells were labelled with phycoerythrin (PE)-anti-DR5 (DJR2-4), PE-anti-Fas (DX2) and PE-anti-human leucocyte antigen (HLA)-A, B, C (W6/32) (eBioscience, San Diego, CA, USA). Cellular labelling was performed at 4°C for 15 min. All samples were analysed on a Becton Dickinson fluorescence activated cell sorter (FACScan) flow cytometer using CellQuest software (BD Biosciences, San Jose, CA, USA).
RNase protection assay
Total RNA from bortezomib-treated MDA-231 cells was isolated with RNA STAT-60 (Tel-test, Friendswood, TX, USA) according to the manufacturer's protocol. mRNA levels were then determined using the Riboquant RNase Protection Assay (BD Biosciences) and the human hAPO-3d probe set (Pharmingen, San Diego, CA, USA), as described previously [22]. Data are presented as a ratio of mRNA signal normalized to the levels of the housekeeping gene L32.
Chromium-release assay
MDA-231 cells were cultured with the indicated amounts of bortezomib for 48 h then labelled with 100 µCi sodium chromate (51Cr; PerkinElmer, Boston, MA, USA) for 1 h. Two thousand labelled cells were incubated with NK-92 cells at the indicated effector-to-target (E : T) ratios in a 96-well, round-bottomed plate for 7 h. Cytotoxicity was determined by measuring the amount of radioactivity released into 100 µl of the media with a 1450 MicroBeta TriLux scintillation counter (PerkinElmer). The percentage lysis was calculated as follows: [(experimental lysis − spontaneous lysis)/(maximum lysis − spontaneous lysis)] × 100.
Tumour outgrowth assay
Target cells (1 × 105) which had been incubated with the indicated amounts of bortezomib for 48 h and CFSE-labelled were added to a six-well, flat-bottomed culture plate. The cells were washed and incubated for an additional 24 h after which NK-92 cells were added at an E : T ratio of 1:1. After 6 days, all cells were removed from the wells and analysed by flow cytometry for expression of CFSE and 7-amino-actinomycin D (7-AAD).
Proliferation assay
For proliferation assays, 105 NK-92 cells were cultured in NK-92 media alone (0 nM) or in the indicated amounts of bortezomib for 48 h prior to being pulsed with [3H]-thymidine (1 µCi per well) (Amersham Pharmacia Life Science, Amersham, Buckinghamshire, UK) 16–18 h before harvesting and counted in the presence of scintillation fluid on a Wallac β-plate reader (Perkin Elmer, Waltham, MA, USA). Three individual wells were analysed per data point.
In vivo tumour studies
For survival studies, four groups of seven mice were treated with 0·2 ml anti-asialo-GM1 rabbit anti-serum (Wako Chemicals, Dusseldorf, Germany) diluted 1:10 in PBS intravenously (i.v.) on day 0 to deplete host NK cells. On day 1, mice were injected with 5 × 105 MDA-231 cells i.v. One group received 12·5 µg bortezomib (in 0·2 ml PBS) on days 2, 4 and 6, and one group received 5 × 106 NK-92 cells i.v. on day 7 plus 2 × 105 IU rhIL-2 on days 7 and 8. Another group received both treatments. Mice were monitored for survival against a tumour-bearing control group.
For lung metastases studies, mice were injected with 0·2 ml anti-asialo-GM1 i.v. on day 0. On day 1, four groups of five mice were injected with 5 × 105 MDA-231 cells i.v. One group was left untreated, one group received 20 µg bortezomib i.v. on days 2 and 3, one group received 5 × 105 NK-92 cells i.v. on day 4 plus 2 × 105 IU rhIL-2 on days 4 and 5, and a final group received both treatments. Mice were killed on day 49 and the number of metastatic lesions in the lungs was counted.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 4 software (GraphPad, San Diego, CA, USA). One-way and two-way analyses of variance were used where appropriate.
Results
Bortezomib displays direct anti-tumour effects on MDA-231 breast cancer cells
Bortezomib has been shown previously to have direct effects in vitro on human breast cancer cells [23]. We sought to determine if the MDA-231 cell line would also display a sensitization to bortezomib in viability and proliferation (Fig. 1). In Fig. 1a, MDA-231 cells were exposed to varying concentrations of bortezomib for 24 h then assessed for apoptosis using annexin-V and propidium iodide. MDA-231 cells displayed a slight, but significant (P < 0·001) increase in apoptosis at or above 15 nM concentrations of bortezomib. Next, the effect of bortezomib on MDA-231 cell proliferation was determined (Fig. 1b) by performing cell counts after MDA-231 cells had been exposed to varying amounts of bortezomib for 24, 48 or 72 h. Cells incubated with 10 or 20 nM bortezomib displayed a significant decrease (P < 0·001) in proliferation after 48 h. These data demonstrate that the MDA-231 breast cancer cell line is tolerant to low levels of bortezomib treatment, but that at higher levels or longer treatment durations MDA-231 cells are susceptible to increased cytotoxicity.
Fig. 1.
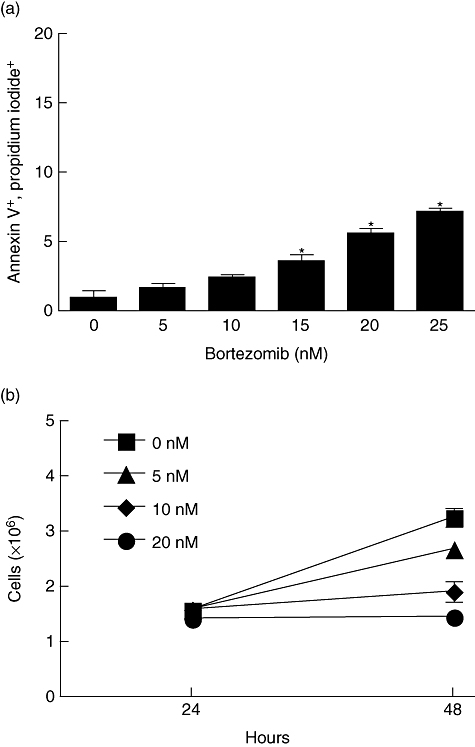
Bortezomib administration in vitro causes a slight but significant increase in apoptosis and a decrease in proliferation in MDA-231 breast cancer cells. (a) MDA-231 cells were incubated with varying concentrations of bortezomib for 24 h, then stained with annexin-V and propidium iodide to determine the percentage of apoptotic cells. (b) MDA-231 cells were exposed to 0, 5, 10 or 20 nM bortezomib. After 24 and 48 h, the number of viable cells was determined through trypan blue staining. *P < 0·01 as determined by one-way analysis of variance (anova) with Dunnett's post-test in (a), two-way anova with Bonferroni post-test in (b).
Short-term killing assays do not reflect an increase in the sensitization of MDA-231 cells by NK-92-mediated killing
We have demonstrated previously that bortezomib can sensitize tumour cells to TRAIL-mediated apoptosis [12] as well as murine NK cell-mediated killing [14]. We sought to expand our studies to determine if bortezomib could also sensitize tumour cells to human NK cell-mediated killing in an in vitro model (Fig. 2). MDA-231 human breast cancer cells were incubated with varying concentrations of bortezomib for 48 h. The highest dose of 20 nM was chosen because higher concentrations displayed cytotoxic effects on the MDA-231 cell line, while the 20 nM dose demonstrated growth inhibition, but minimal cytotoxic effects (Fig. 1a and b). The treated MDA-231 cells were then washed and entered in a short-term chromium-release assay using the indicated E : T ratios. No significant increase in NK-mediated lysis was observed in these short-term assays. This observation is consistent with our previous findings that bortezomib treatment has no effect on granule-mediated killing using murine tumours [14]. As the chromium-release assay is typically a measure of granule-mediated killing in NK cells, we hypothesized that in order to see the effects of other modes of NK cell cytotoxicity we would need to use a long-term assay.
Fig. 2.
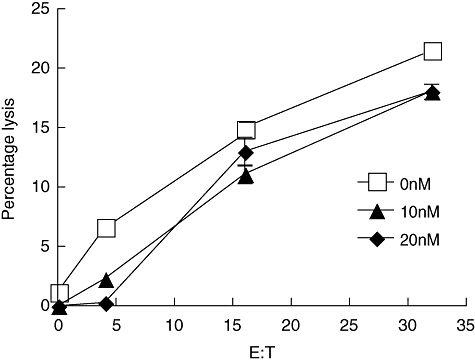
Sensitization of MDA-231 cells to natural killer (NK)-92 killing is not shown in short-term chromium release assays. MDA-231 cells were placed in a culture flask and treated with bortezomib at either 10 (▴) or 20 (♦) nM or left untreated (□) for 48 h. The MDA-231 cells were then washed twice, resuspended in media and used in a 7-h chromium-release assay. The NK-92 effector cells were added at the indicated effector-to-target (E : T) ratios, with a maximum E : T of 32:1. No significant difference was found between the treatments, as determined by a two-way analysis of variance with a Bonferroni post-test.
Bortezomib treatment increases the mRNA levels of death receptors in MDA-231 cells
Our previous studies have demonstrated that bortezomib increases the surface expression of death ligands on the surface of mouse leukaemia cells [14]. We sought to determine if bortezomib treatment would increase the amount of death receptors and other apoptosis-related genes in the human mammary carcinoma line, MDA-231 (Fig. 3). MDA-231 cells were exposed to varying amounts of bortezomib for 24 h and then mRNA levels were determined by RNase protection assay analysis. With incremental bortezomib concentrations, MDA-231 cells displayed an insignificant but marked increase in the levels of Fas, DR5, DR4, the TNF receptor (TNFR)1-associated death domain and caspase 8. However, there was a significant (P < 0·05) increase in the ratio of DR3, the p55 TNF receptor and the receptor-interacting protein. The increase in the mRNA of the aforementioned apoptosis-related genes suggests that MDA-231 cells treated with bortezomib may be sensitized to Fas and TRAIL-mediated killing. These data indicated that at low bortezomib doses, bortezomib treatment results in increased expression of numerous extrinsic pro-apoptotic genes. However, increasing death receptors and other components of extrinsic apoptosis does not explain apparently increased intrinsic apoptosis, as shown in Fig. 1. Changes in intrinsic pro-apoptotic proteins (PUMA, Bax, etc.) or anti-apoptotic proteins (Bcl-2, etc.) are probably responsible.
Fig. 3.
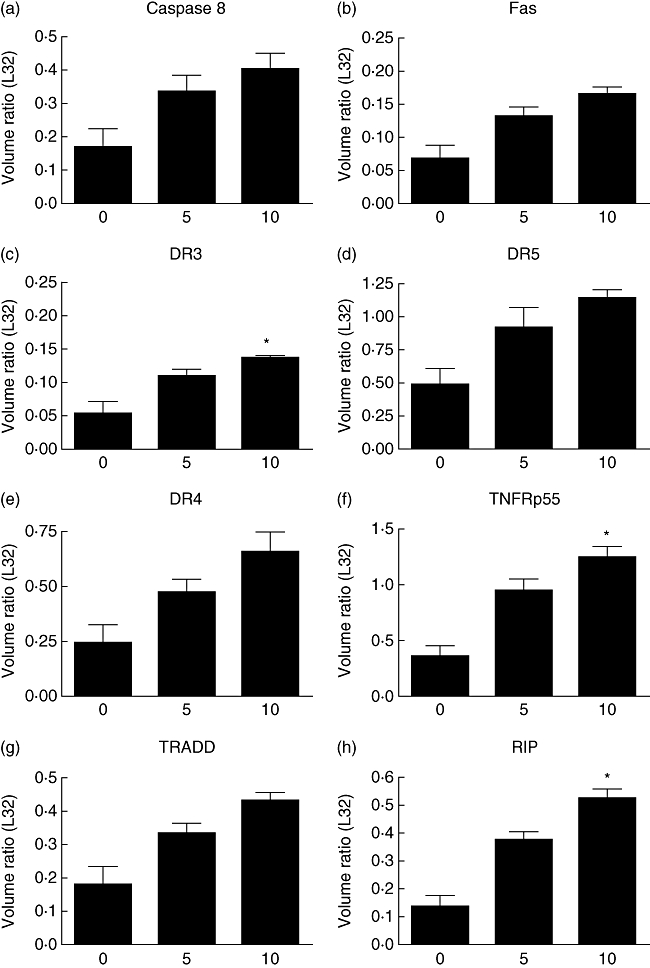
Bortezomib pretreatment on MDA-231 cells increases the mRNA of proteins associated with apoptosis. MDA-231 cells were incubated with 0, 5 or 10 nM of bortezomib for 24 h. RNA was then isolated and a ribonuclease protection assay as performed to determine the amount of mRNA present from apoptosis-related proteins. The ratio of caspase 8 (a), Fas (b), death receptor 3 (DR3) (c), DR5 (d), DR4 (e), the p55 tumour necrosis factor (TNF) receptor (f), TNF receptor-associated death domain TRADD (g) and receptor-interacting protein (RIP) (h) was determined through comparison with the L32 ribosomal protein. Although all proteins examined displayed a marked increase, only the increases in the indicated panels proved to be significant. *P < 0·05 by one-way analysis of variance with Bonferroni post-test.
Bortezomib treatment increases the surface expression of death ligands and decreases the expression HLA-A, B, C
To determine if the increase in death receptor mRNA correlated with an increase in cell surface expression, we used flow cytometry to determine the surface levels of DR5 and Fas with and without bortezomib treatment (Fig. 4). MDA-231 cells were maintained in normal culture condition or treated with 20 nM bortezomib for 24 h before labelling with PE-conjugated anti-DR5, anti-Fas and anti-HLA-A, B, C. The surface expression of both DR5 (Fig. 4a) and Fas (Fig. 4b) was up-regulated on the cells that received bortezomib treatment. These data correspond with the increase in death receptor mRNA expression displayed in Fig. 3. Interestingly, bortezomib treatment also decreased the expression of HLA-A, B and C (Fig. 4c). This may have been due to proteasome inhibition preventing proteolysis, thus preventing proteins from becoming suitable peptide antigens to be displayed on the major histocompatibility complex (MHC) class I molecule [24]. Without a suitable peptide, the MHC protein does not persist on the cell surface. This bortezomib-mediated decrease in MHC class I expression may result in increased susceptibility to NK cell-mediated killing, as demonstrated previously [25]. These data demonstrate that bortezomib treatment increases the surface expression of death receptors DR5 and Fas, as results in a decrease in the expression of MHC class I molecules. These combined effects may present a mechanism by which bortezomib increases the susceptibility of tumour cells to NK cell-mediated killing.
Fig. 4.
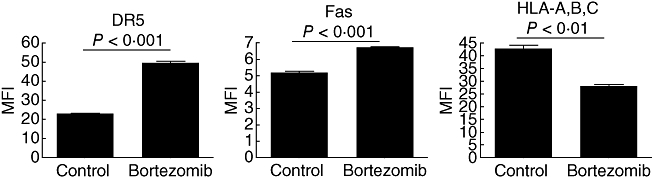
Bortezomib pretreatment increases the surface expression of death receptor 5 (DR5) and Fas and decreases the expression of human leucocyte antigen (HLA) molecules. MDA-231 cells were treated with 20 nM bortezomib or left untreated for 24 h. Cells were stained with fluorescent-conjugated anti-DR5, anti-Fas and anti-HLA-A, B, C and analysed by flow cytometry. The mean fluorescent intensity was calculated in order to determine the relative amount of the indicated surface protein. The P-values shown were calculated by a two-tailed Student's t-test.
Bortezomib treated MDA-231 cells are sensitized to NK-92 killing in a tumour outgrowth assay
In earlier results, we did not observe an increase in NK cell killing of MDA-231 cells after bortezomib sensitization in a short-term cytotoxicity assay (Fig. 2) because the mechanism of killing in short-term cytotoxicity assays is primarily granule-mediated. Therefore, we hypothesized that bortezomib sensitization would increase if we used a longer-term assay. To address this, we used a tumour outgrowth assay (Fig. 5). The tumour outgrowth assay consisted of a 20 nM bortezomib pretreatment of BT-20 (Fig. 5a) and a 10 nM pretreatment of MCF-7 (Fig. 5b) and MDA-231 (Fig. 5c) breast cancer cells for 48 h. The cells were then counted, equal numbers were plated, and the cells were allowed to adhere to plates for 24 h. Tumour cells were labelled with CFSE, washed and incubated with NK-92 cells at a 1:1 E : T ratio for 6 days. All cells were then removed from the plates and analysed by flow cytometry for CFSE and 7-AAD expression. The 6-day period in which the tumour cells were exposed to the NK-92 cells allowed for killing through multiple mechanisms to occur, including through the ligation of death receptors. The bortezomib pretreatment resulted in a decreased number of live tumour cells after the 6-day period in MCF-7 and MDA-231, but not BT-20. NK-92 cells effectively decreased the number of live tumour cells in all groups. However, the combination of NK-92 cells and bortezomib pretreatment resulted in a decrease in proliferation that was lower than each treatment alone. These results indicate that the enhanced anti-tumour effects may be detectable only by using long-term assays.
Fig. 5.
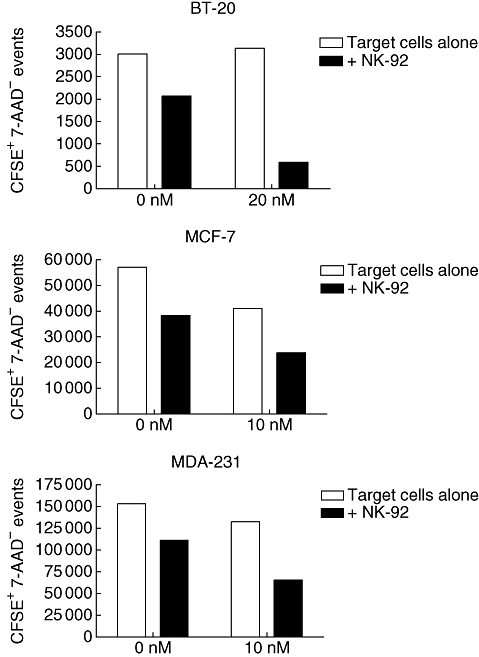
A long-term tumour outgrowth assay displays a synergistic increase in natural killer (NK)-mediated killing of MDA-231 cells after bortezomib pretreatment; 5 × 105 BT-20 (a), MCF-7 (a) or MDA-231 (c) cells were incubated with bortezomib (BT-20, 20 nM; MCF-7 and MDA-231, 10 nM) for 48 h. Target cells were then washed, labelled with 5,6-carboxy-succinimidyl-fluorescein-ester (CFSE), and allowed another 24 h to adhere to wells. In some wells, NK-92 cells were then added at a 1:1 effector-to-target ratio. Plates were incubated for an additional 6 days then analysed by flow cytometry for CFSE and 7-amino-actinomycin D (7-AAD). The mean fluorescence intensity of 7-AAD was determined on CFSE-positive cells.
The NK-92 cells are sensitive to bortezomib treatment in vitro
Having demonstrated that bortezomib has direct effects on MDA-231 cells in vitro and can sensitize them to NK cell-mediated killing in long-term assays, we decided to examine the effects of bortezomib on the NK-92 cells themselves (Fig. 6). NK-92 cells were treated with 0, 5, 10 or 20 nM bortezomib for 48 h, then pulsed with [3H]-thymidine for 24 h to determine proliferation. NK-92 cells were highly sensitive to bortezomib, displaying a significant decrease (P < 0·01) in proliferation in all treated groups. A previous study demonstrated that human NK cells were not induced into apoptosis by bortezomib exposure (10 nM for 24 h) [26]. These data demonstrate that NK-92 cells are highly sensitive to bortezomib. Therefore, therapies designed to use both NK-92 cells and bortezomib should be designed to utilize administration schedules which minimize NK-92 cell exposure to bortezomib.
Fig. 6.
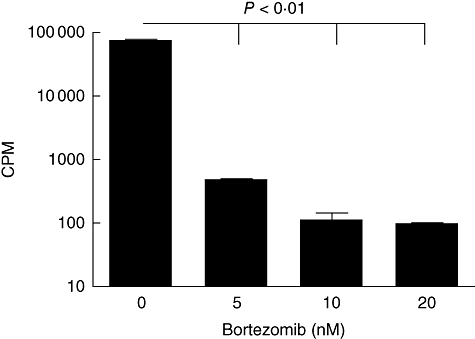
Natural killer (NK)-92 cells are sensitive to bortezomib in vitro. 1 × 105 NK-92 were exposed to 0, 5, 10 or 20 nM bortezomib for 48 h, then pulsed with [3H]-thymidine to determine proliferation. The doses of 5, 10 and 20 nM each displayed a significant decrease in proliferation when compared with the untreated control cells (P < 0·001) as determined by one-way analysis of variance with Dunnett's post-test.
Administration of NK-92 cells prolongs survival and reduces the number of lung metastases in breast cancer-bearing SCID mice
Having established that bortezomib sensitizes tumours to NK cell-mediated killing in in vitro assays, we sought to expand our regimen to an in vivo model. In Fig. 7, CB-17 SCID mice with an established MDA-231 tumour burden were treated with bortezomib, NK-92 cells or a combination of both. The treatment of mice with bortezomib did not significantly alter the survival of tumour-bearing mice (Fig. 7a). Treatment of mice with NK-92 cells and IL-2 led to significant increases in survival compared with control groups (P < 0·01). However, the mice treated with both bortezomib and NK-92 cells did not offer any advantage compared with mice treated with NK-92 cells alone. This may be due to improper timing and dosing of the bortezomib in the xenogeneic model, as others have shown previously a positive effect of bortezomib in breast cancer as well as other solid tumours clinically and in mouse models [27]. It is important to note that administration of bortezomib did not reduce the efficacy of NK-92 cells in vivo, suggesting that the bortezomib administration was not affecting the NK cells adversely as we observed in in vitro studies. We also examined the role of NK-92 cells and bortezomib to inhibit the growth of metastatic lesions in the lungs (Fig. 7b). Tumour-bearing CB-17 SCID mice were treated with bortezomib and NK-92 cells, as described in Methods, prior to being killed on day 49 and enumerating the metastatic lesions in the lungs. The administration of NK-92 cells reduced (P < 0·001) the lung metastases significantly by nearly two-thirds, whereas bortezomib administration showed no difference in the number of lung metastases. While combined treatment did not offer any advantage compared with NK-92 cells alone, these data suggest that more detailed studies altering the timing or dosing of bortezomib may be needed to observe enhancement of the killing by NK-92 cells, as observed in our in vitro data.
Fig. 7.
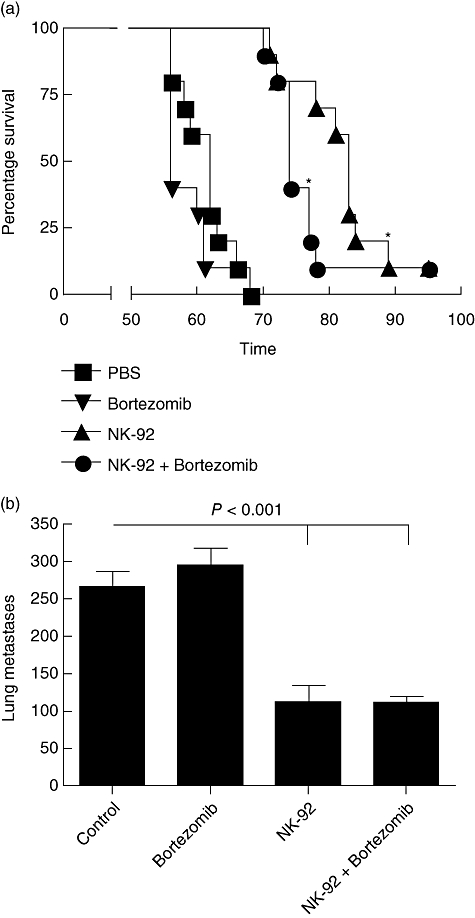
Administration of natural killer (NK)-92 cells increases survival and decreases lung metastases in xenogeneic breast cancer studies while bortezomib displayed no effect. (a) MDA-231 cells were administered intravenously in SCID mice prior to treatment with bortezomib, NK-92 cells and interleukin (IL)-2, or a combination of both as described in Methods. Mice were monitored for survival and the experiment was terminated on day 95. (b) An MDA-231 tumour burden was established in SCID mice with treatments of bortezomib, NK-92 and IL-2, or a combination of both, as described in Methods. Mice were killed on day 49 and lungs were examined to determine the number of tumour metastases. *P < 0·01 by log-rank test compared with MDA-231 alone.
Discussion
Molecular targeting of cancers offers great promise in attacking tumour cells selectively. Recent studies have examined the capacity of these agents to predispose cancers to other methods of immunotherapy [28]. In particular, augmenting NK cell-mediated attack is an attractive strategy to increase the efficacy of immunotherapies. Treatment of multiple myeloma with bortezomib has powerful direct anti-tumour effects [29]. We have demonstrated previously that bortezomib also sensitizes tumour cells to TRAIL-mediated killing [12]. Our subsequent studies, using a murine model, demonstrated that bortezomib could also augment NK cell-mediated killing of tumours [14]. Interestingly, the augmentation of NK cells in that study was not observed in short-term, granule-mediated killing assays, but only in long-term tumour outgrowth assays. In this report, we further developed our studies by examining the augmentation of the anti-tumour responses mediated by a human NK cell line. Herein we demonstrate that treatment of a human breast cancer cell line with bortezomib leads to a dose-dependent increase in the transcription of numerous pro-apoptotic genes. These data also correlated with an increase in the surface expression of death ligand receptors DR5 and Fas. We also demonstrated, in agreement with our previous study [14], that the killing of bortezomib-treated targets in short-term killing assays was not augmented, and that enhancement was observed only in long-term tumour outgrowth studies. We also observed that the human NK cell line NK-92 was highly sensitive to bortezomib in vitro. Finally, using an in vivo model in which CB-17 SCID mice bearing established human breast cancer were treated with bortezomib and NK cells, we did not see enhanced survival compared with NK cell treatment alone. Importantly, we did not observe any detrimental effects of bortezomib treatment on NK cell-responses in vivo, suggesting that in vivo treatment may be possible with better timing or dosing of the bortezomib and NK cells. These data extend our earlier studies which examined the effects of bortezomib on NK cell killing in a murine model [14].
The predominant killing pathway attributed to NK cells is granule-mediated killing through perforin and granzymes [17]. In short-term killing assays, granule exocytosis is the primary killing pathway available to NK cells, while death-ligands do not have sufficient time to mediate apoptotic effects. As bortezomib has demonstrated enhancement of TRAIL or FasL-mediated killing previously, it corroborates the lack of enhancement in short-term killing assays. In the same way, the augmentation of NK cell responses by bortezomib in long-term assays allows for engagement of death ligand receptors and induction of apoptosis.
We and others have demonstrated that treatment of tumours with bortezomib led to a reduction in the anti-apoptotic protein c-FLIP [12]. The physiological role of c-FLIP is to interact with the intracellular portion of death receptors such as DR5, interfering with the engagement of the Fas-associated death domain, which transduces death signals resulting in apoptosis [30]. A recent study demonstrated that pretreatment of tumours with bortezomib increased sensitivity to TRAIL-mediated NK cell killing, although no increase in FasL-mediated cytotoxicity was observed [26]. This study also did not observe any decrease in the expression of MHC class I or Fas. In contrast, our study demonstrates that bortezomib treatment of a human breast cancer cell line resulted in the up-regulation of Fas and DR5, as well as down-regulation in MHC class I, in agreement with our previously published report [14]. These discrepancies may be due to timing or to the tumour lines studied.
Another study demonstrated that TNF-α, but not perforin, FasL or TRAIL, was responsible for the increase in bortezomib-mediated sensitization of the B16 melanoma cell line [31]. Again, these data may reflect differences in the cell lines studied. Our study demonstrated that treatment of cells with bortezomib induces the expression of numerous pro-apoptotic genes which may be affected by any of the TNF-family of death ligands, suggesting that bortezomib-treated targets could be sensitized through any of the TNF-family receptors including Fas, TNFRI or DR5.
Finally, a recent study demonstrated that bortezomib-mediated reduction in MHC class I expression on tumours resulted in the enhanced killing of that tumour by freshly isolated human NK cells in short-term assays [32]. This report, similar to the current study, demonstrates that bortezomib results in a decrease in MHC class I as well as an increase in DR5 expression. However, the report by Shi et al. observed that the decrease in MHC class I was correlated with an increase in NK cell-mediated cytolysis in short-term chromium release assays. Neither our current study using NK-92 cells nor our previous study using murine NK cells [14] observed an increase in short-term killing assays, despite similar decreases in MHC class I expression. These differences may be due to the NK cells used, as our studies using NK-92 cells and murine NK cells may respond differently to reduced MHC class I expression compared with freshly isolated human NK cells.
Our study demonstrates that bortezomib enhances the anti-tumour ability of NK-92 cells in long-term, but not short-term, in vitro assays. Tumour survival studies demonstrated that NK-92 cells promoted powerful anti-tumour responses; however, co-treatment of tumour-bearing mice with bortezomib failed to show enhanced survival compared with NK-92 cells alone. Importantly, the cytotoxic effects of bortezomib on NK cells observed in vitro was not observed in vivo, as co-treatment did not reduce NK cell function. These data suggest that bortezomib can be utilized to sensitize solid tumours to NK cell-mediated killing and improve current cancer therapies, but more studies on dosing and timing may be needed given the sensitivity of the activated effector cells to proteasome inhibition.
Acknowledgments
This publication has been funded all or in part by National Institutes of Health grant R01CA95327-02 and R01CA102282-01A and by a Department of Defense Breast Cancer Concept Award to W. J. M. The authors would like to acknowledge Chris Rowan for assistance with experiments and Doug Redelman, Danice Wilkins and Maite Álvarez for carefully reviewing the manuscript.
References
- 1.Adams J. Proteasome inhibition: a novel approach to cancer therapy. Trends Mol Med. 2002;8(Suppl.)(4):S49–54. doi: 10.1016/s1471-4914(02)02315-8. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, Yu X. Regulation of apoptosis: the ubiquitous way. FASEB J. 2003;17:790–9. doi: 10.1096/fj.02-0654rev. [DOI] [PubMed] [Google Scholar]
- 3.Voorhees PM, Orlowski RZ. The proteasome and proteasome inhibitors in cancer therapy. Annu Rev Pharmacol Toxicol. 2006;46:189–213. doi: 10.1146/annurev.pharmtox.46.120604.141300. [DOI] [PubMed] [Google Scholar]
- 4.Richardson PG, Mitsiades C, Hideshima T, Anderson KC. Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu Rev Med. 2006;57:33–47. doi: 10.1146/annurev.med.57.042905.122625. [DOI] [PubMed] [Google Scholar]
- 5.Glickman MH, Ciechanover A. The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 6.Asher G, Reuven N, Shaul Y. 20S proteasomes and protein degradation ‘by default’. Bioessays. 2006;28:844–9. doi: 10.1002/bies.20447. [DOI] [PubMed] [Google Scholar]
- 7.Kane RC, Dagher R, Farrell A, et al. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2007;13:5291–4. doi: 10.1158/1078-0432.CCR-07-0871. [DOI] [PubMed] [Google Scholar]
- 8.Richardson PG, Mitsiades C, Schlossman R, Munshi N, Anderson K. New drugs for myeloma. Oncologist. 2007;12:664–89. doi: 10.1634/theoncologist.12-6-664. [DOI] [PubMed] [Google Scholar]
- 9.Dreicer R, Petrylak D, Agus D, Webb I, Roth B. Phase I/II study of bortezomib plus docetaxel in patients with advanced androgen-independent prostate cancer. Clin Cancer Res. 2007;13:1208–15. doi: 10.1158/1078-0432.CCR-06-2046. [DOI] [PubMed] [Google Scholar]
- 10.Lara PN, Jr, Koczywas M, Quinn DI, et al. Bortezomib plus docetaxel in advanced non-small cell lung cancer and other solid tumors: a Phase I California Cancer Consortium trial. J Thorac Oncol. 2006;1:126–34. [PubMed] [Google Scholar]
- 11.Ryan DP, O'Neil BH, Supko JG, et al. A Phase I study of bortezomib plus irinotecan in patients with advanced solid tumors. Cancer. 2006;107:2688–97. doi: 10.1002/cncr.22280. [DOI] [PubMed] [Google Scholar]
- 12.Sayers TJ, Brooks AD, Koh CY, et al. The proteasome inhibitor PS-341 sensitizes neoplastic cells to TRAIL-mediated apoptosis by reducing levels of c-FLIP. Blood. 2003;102:303–10. doi: 10.1182/blood-2002-09-2975. [DOI] [PubMed] [Google Scholar]
- 13.Takeda K, Stagg J, Yagita H, Okumura K, Smyth MJ. Targeting death-inducing receptors in cancer therapy. Oncogene. 2007;26:3745–57. doi: 10.1038/sj.onc.1210374. [DOI] [PubMed] [Google Scholar]
- 14.Hallett WH, Ames E, Motarjemi M, et al. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008;180:163–70. doi: 10.4049/jimmunol.180.1.163. [DOI] [PubMed] [Google Scholar]
- 15.Herberman RB, Ortaldo JR. Natural killer cells: their roles in defenses against disease. Science. 1981;214:24–30. doi: 10.1126/science.7025208. [DOI] [PubMed] [Google Scholar]
- 16.Hallett WH, Murphy WJ. Positive and negative regulation of natural killer cells: therapeutic implications. Semin Cancer Biol. 2006;16:367–82. doi: 10.1016/j.semcancer.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Ortaldo JR, Winkler-Pickett RT, Nagashima K, Yagita H, Okumura K. Direct evidence for release of pore-forming protein during NK cellular lysis. J Leukoc Biol. 1992;52:483–8. doi: 10.1002/jlb.52.5.483. [DOI] [PubMed] [Google Scholar]
- 18.Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–8. [PubMed] [Google Scholar]
- 19.Suck G. Novel approaches using natural killer cells in cancer therapy. Semin Cancer Biol. 2006;16:412–8. doi: 10.1016/j.semcancer.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res. 2001;10:535–44. doi: 10.1089/15258160152509145. [DOI] [PubMed] [Google Scholar]
- 21.Tam YK, Maki G, Miyagawa B, Hennemann B, Tonn T, Klingemann HG. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum Gene Ther. 1999;10:1359–73. doi: 10.1089/10430349950018030. [DOI] [PubMed] [Google Scholar]
- 22.Sun K, Wilkins DE, Anver MR, et al. Differential effects of proteasome inhibition by bortezomib on murine acute graft-versus-host disease (GVHD): delayed administration of bortezomib results in increased GVHD-dependent gastrointestinal toxicity. Blood. 2005;106:3293–9. doi: 10.1182/blood-2004-11-4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Codony-Servat J, Tapia MA, Bosch M, et al. Differential cellular and molecular effects of bortezomib, a proteasome inhibitor, in human breast cancer cells. Mol Cancer Ther. 2006;5:665–75. doi: 10.1158/1535-7163.MCT-05-0147. [DOI] [PubMed] [Google Scholar]
- 24.Strehl B, Seifert U, Kruger E, Heink S, Kuckelkorn U, Kloetzel PM. Interferon-gamma, the functional plasticity of the ubiquitin–proteasome system, and MHC class I antigen processing. Immunol Rev. 2005;207:19–30. doi: 10.1111/j.0105-2896.2005.00308.x. [DOI] [PubMed] [Google Scholar]
- 25.Shi J, Tricot GJ, Garg TK, et al. Bortezomib down-regulates the cell surface expression of HLA-class I and enhances natural killer cell-mediated lysis of myeloma. Blood. 2007;111:1309–17. doi: 10.1182/blood-2007-03-078535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lundqvist A, Abrams SI, Schrump DS, et al. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: a novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006;66:7317–25. doi: 10.1158/0008-5472.CAN-06-0680. [DOI] [PubMed] [Google Scholar]
- 27.Lenz HJ. Clinical update: proteasome inhibitors in solid tumors. Cancer Treat Rev. 2003;29(Suppl.)(1):41–8. doi: 10.1016/s0305-7372(03)00082-3. [DOI] [PubMed] [Google Scholar]
- 28.Marsoni S, Damia G. Molecular targeting: new therapeutic strategies to improve tumour apoptosis. Ann Oncol. 2004;15(Suppl.)(4):iv229–31. doi: 10.1093/annonc/mdh931. [DOI] [PubMed] [Google Scholar]
- 29.Hideshima T, Richardson P, Chauhan D, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071–6. [PubMed] [Google Scholar]
- 30.Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–44. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Schumacher LY, Vo DD, Garban HJ, et al. Immunosensitization of tumor cells to dendritic cell-activated immune responses with the proteasome inhibitor bortezomib (PS-341, Velcade) J Immunol. 2006;176:4757–65. doi: 10.4049/jimmunol.176.8.4757. [DOI] [PubMed] [Google Scholar]
- 32.Shi J, Tricot GJ, Garg TK, et al. Bortezomib down-regulates the cell-surface expression of HLA class I and enhances natural killer cell-mediated lysis of myeloma. Blood. 2008;111:1309–17. doi: 10.1182/blood-2007-03-078535. [DOI] [PMC free article] [PubMed] [Google Scholar]