

Abstract
U18666A is a cholesterol transport-inhibiting agent that is used widely to mimic Niemann–Pick type C disease. The effect of U18666A on tumour necrosis factor (TNF)-α production in mouse macrophage cell line, RAW 264·7 cells and peritoneal macrophages was examined. U18666A induced TNF-α mRNA expression 48 h after the treatment, and TNF-α production 48 and 72 h after stimulation in RAW 264·7 cells. U18666A accumulated intracellular free cholesterol in the culture of normal medium but not cholesterol-free medium. U18666A also induced reactive oxygen species (ROS) generation in normal medium but much less in cholesterol-free medium. Anti-oxidant N-acetyl-L-cysteine (NAC) abolished U18666A-induced TNF-α production. U18666A led to the phosphorylation of p38 mitogen-activated protein kinase 24 and 48 h after the stimulation and the p38 activation was inhibited in presence of cholesterol-free medium or NAC. A p38 inhibitor reduced U18666A-induced TNF-α production. Taken together, U18666A was suggested to induce TNF-α production in RAW 264·7 cells via free cholesterol accumulation-mediated ROS generation.
Keywords: free cholesterol, Niemann–Pick C disease, p38, ROS, TNF-α, U18666A
Introduction
The pharmacological agent, U18666A (3-β-[2-(diethylamino)ethoxy]androst-5-en-17-one), is a cholesterol transport-inhibiting class-2 amphiphile, which is used widely to mimic Niemann–Pick disease type C (NPC) as a fatal neurovisceral lipid storage disease of autosomal inheritance [1–5]. U18666A induces dysfunction of lipid storage, and inhibition of cholesterol movement from the plasma membrane to the endoplasmic reticulum and from the lysosome to the plasma membrane [5]. Therefore, free cholesterol accumulates in late endosomes of the cells treated with U18666A [6–8] and leads to an impaired cell function similar to NPC disease.
Chronic exposure of primary cortical neurones to U18666A causes neuronal apoptosis [9]. U18666A increases generation of intracellular reactive oxygen radical species (ROS) [9] and the oxidative damages may lead to neuronal apoptosis. In NPC disease, the cell death in the brain also occurs through apoptosis and it seems to be mediated by the tumour necrosis factor (TNF) receptor superfamily pathway [10]. Further, the expression of TNF-α mRNA is reported to increase up to 30–50-fold in the cerebellum of 7–9-week-old NPC1-deficient mice compared with wild-type mice [10]. On the other hand, macrophages from mice treated with U18666A have a specific defect in transporting lipoprotein-derived cholesterol from late endosome to the endoplasmic reticulum, and are resistant to free cholesterol-induced apoptosis [11]. Recently, NPC1 is reported to regulate intracellular cholesterol trafficking and oxidative stress in macrophages [12]. The precise action of U18666A on macrophages is not known, although the action on neuronal cells is studied extensively. The effect of U18666A on the macrophage function should be clarified in order to understand the involvement of inflammatory response in NPC disease. In the present work we studied if and how U18666A induced TNF-α production in mouse macrophage cell line, RAW 264·7 cells. Here, we report the putative mechanism of U18666A-induced TNF-α production.
Materials and methods
Materials
U18666A was purchased from Biomol International (Plymouth Meeting, PA, USA). Oleic acid and N-acetyl-L-cysteine (NAC) were obtained from Sigma (St Louis, MO, USA); (±)-3-hydroxy-3-methyl-5-pentanolide (DL-mevalolactone) were purchased from Wako (Osaka, Japan). Antibodies to serine/threonine kinase/protein kinase B (Akt), p65 nuclear factor (NF)-κB, stress-activated protein kinase (SAPK)/Jun N-terminal kinase (JNK), p38, extracellular signal regulated kinase (ERK) 1/2 and their phosphorylated forms were purchased from Cell Signalling Technology (Beverly, MA, USA), and anti-β-actin antibody was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). SB203508 as a p38 inhibitor and PD98058 as an ERK1/2 inhibitor were obtained from Calbiochem (La Jolla, CA, USA).
Cell culture
The murine macrophage cell line RAW 264·7 was obtained from Riken Cell Bank (Tsukuba, Japan) and maintained in RPMI-1640 medium containing 10% heat-inactivated fetal bovine serum (FBS) (Gibco-BRL, Gaithersburg, MD, USA) and antibiotics at 37°C under 5% CO2. The medium was denoted as medium X. RPMI-1640 supplemented with 3% delipidated FBS, 20 µM lovastatin, 50 mM oleic acid and 300 µM mevalonate was used for the cholesterol-free medium denoted as medium Y. Delipidated FBS was prepared as described elsewhere [13]. Medium Y inhibited cholesterol synthesis almost completely, as described elsewhere [14]. RAW 264·7 cells were adapted to medium Y by cultivation with RPMI-1640 containing 3% FBS, followed by 12 h cultivation with RPMI-1640 containing 3% delipidated FBS. Peritoneal cells were obtained by washing out the peritoneal cavity of BALB/c mice (Japan SLC, Hamamatsu, Japan) with RPMI-1640 medium. The experiment was carried out under the guide for care and use of laboratory animals, Aichi Medical University.
Determination of TNF-α production
The cell culture supernatant was collected from the cultures of RAW 264·7 cells stimulated with or without U18666A (1 µg/ml) for various times. The concentration of TNF-α was determined by enzyme linked immunosorbent assay kit (R&D Systems, Minneapolis, MN, USA). Experimental results are expressed as the mean of triplicates ± standard deviation (s.d.) in three independent experiments.
Real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (PCR) was performed essentially as described elsewhere [15]. RNA was extracted from cells using the RNeasy mini kit (Qiagen, Chatsworth, CA, USA). RNA was reverse-transcribed in ReverTra Ace (Toyobo, Osaka, Japan) with a three-step incubation according to the manufacturer's instructions, and quantitative PCR was carried out using SYBR green real-time PCR master mix (Toyobo) under the manufacturer's instructions. Mouse primers were designed as follows: TNF-α (sense: 5′-TGTTGCCTCCTCTTTTGCTT-3′, anti-sense: 5′-TGGTCACCAAAATCAGCGTTA-3′); glyceraldehydes-3-phosphate dehydrogenase (GAPDH) (sense: 5′-TGAAGCAGGCATCTGAGGG-3′, anti-sense: 5′-CGAAGGTGGAAGAGTGGGAG-3′) (Invitrogen, Carlsbad, CA, USA). PCR was performed with ABI PRISM 7700 sequence detection system (Applied Biosystems, Hamilton, New Zealand) and the PCR conditions were as follows: 95°C for 10 min and 40 cycles at 95°C for 30 s, 60°C for 1 min. The relative quantitative values of TNF-α expression in each case were normalized by the expression levels of reference gene GAPDH. The expression levels of TNF-α mRNA in each sample are presented as fold increase to the mean value of the control.
Determination of intracellular free cholesterol
Cellular lipid was extracted with hexane/isopropanol (3 : 2, v/v) for 30 min and the solvent was evaporated under a decicator. The amount of free cholesterol was determined with the Amplex red cholesterol assay kit (Molecular Probes, Invitrogen), according to the manufacturer's instructions. The reaction mixtures were incubated for 30 min at 37°C. Fluorescence intensity was measured at excitation wavelength 530 nm and emission wavelength 590 nm with a fluorescence microplate reader. Background fluorescence was subtracted from each value. The amount of cholesterol was calculated based on a standard curve with purified cholesterol.
Filipin staining
RAW 264·7 cells were cultured in an eight-well plastic plate with slides. On the following day U18666A (1 µg/ml) or vehicle control was added into the cultures and incubated for a further 24 h. The slides were washed with phosphate-buffered saline five times and used for the filipin staining with the cholesterol cell-based detection assay kit (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer's instructions. Digital images were acquired immediately after filipin labelling under fluorescence microscopy.
Determination of intracellular ROS by 2¢,7¢-dichlorofluorescein diacetate
The amount of intracellular ROS was measured by using DCF-DA (Sigma). Briefly, RAW 264·7 cells were cultured at 2 × 105/well in a 96-well microplate for 12 h and stimulated with 1 µg/ml U18666A for various times. After removal of the culture medium, the cells were incubated further with the culture medium containing 100 µM DCF-DA at 37°C for 30 min. After twice washing, the fluorescence intensity was measured by a fluorescence spectrophotometer at excitation and emission wavelengths of 485 and 530 nm, respectively, at intervals of 30 min. The data represent one of the three experiments and the fluorescence intensity is expressed as means of triplicate ± s.d.
Immunoblotting
Immunoblotting were performed as described elsewhere [16]. In brief, cells were lysed by adding an equal volume of a twofold concentrated sample buffer and the cell lysates were subjected to sodium dodecyl sulphate (SDS)–polyacrylamide gel electrophoresis using 8–12% gradient gels. The proteins were transferred electrically to a polyvinylidene difluoride membrane and the membrane was treated with various antibodies, followed by horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse immunoglobulin G (Santa Cruz Biotechnology). The protein bands were visualized by a chemiluminescence reagent (Pierce, Rockford, IL, USA). For reprobing, membranes were stripped with the solution containing 2% SDS, 62·5 mM Tris, pH 6·8 and 100 mM 2-mercaptoethanol at 50°C for 30 min and treated with corresponding antibodies. The molecular sizes of the antigens were determined by comparison with a prestained protein size marker kit (Invitrogen).
Statistical analysis
Experimental data are expressed as the mean of triplicates ± s.d. in at least three independent experiments. Statistical analysis based on Student's t-test was performed for comparisons between two experiments. A value of P < 0·01 was considered statistically significant.
Results
U18666A induces production of TNF-α in RAW 264·7 cells
The effect of U18666A on TNF-α production in RAW 264·7 cells was studied. RAW 264·7 cells were incubated with U18666A (1 µg/ml) for 24, 48 or 72 h. There was no significant TNF-α production in U18666A-treated RAW 264·7 cells until 24 h after the treatment. Surprisingly, U18666A markedly induced TNF-α production at 48 and 72 h (Fig. 1a). There was a long time lag in U18666A-induced TNF-α production. The effect of U18666A on TNF-α production in mouse peritoneal cells was also examined. U18666A induced TNF-α production in peritoneal macrophages as well as RAW 264·7 cells 48 and 72 h after the treatment, although the level of TNF-α was lower than that of RAW 264·7 cells (Fig. 1a). Subsequently, the time–course of the TNF-α mRNA expression was examined by real-time PCR. The level of TNF-α mRNA increased significantly 48 h after U18666A treatment, although there was no significant increase in the TNF-α mRNA level within 24 h (Fig. 1b). The time–course of TNF-α mRNA expression corresponded to that of TNF-α production. In addition, the effect of U18666A on the cell viability of RAW 264·7 cells was determined by an assay with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium (MTT) (Chemicon, Temecula, CA, USA). The MTT assay demonstrated that U18666A exhibited no cytotoxic action on those cells 24, 48 and 72 h after U18666A treatment.
Fig. 1.
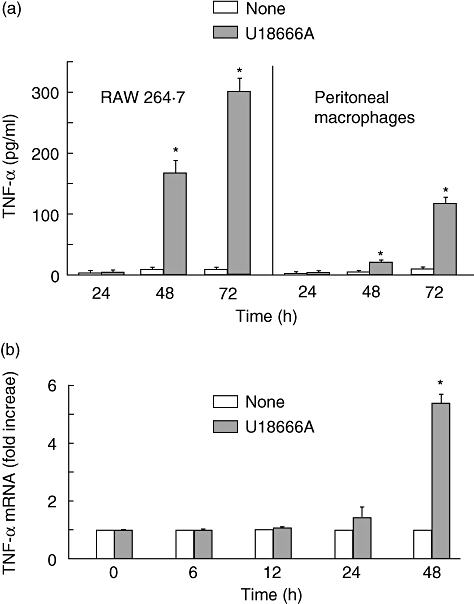
Tumour necrosis factor (TNF)-α production in U18666A-treated RAW 264·7 cells. RAW 264·7 cells and peritoneal macrophages were incubated with U18666A (1 µg/ml) for various hours. (a) TNF-α production was determined with enzyme-linked immunosorbent assay. *P < 0·01 versus none (vehicle control). (b) TNF-α mRNA expression was analysed with real-time polymerase chain reaction. *P < 0·01 versus 0 h.
U18666A induces intracellular accumulation of free cholesterol in RAW 264·7 cells
U18666A is known to accumulate free cholesterol via inhibition of its intracellular transport [4]. Therefore, a possibility was raised that U18666A induced TNF-α production via intracellular accumulation of free cholesterol. The effect of U18666A on free cholesterol accumulation was examined by cultivation with medium X or cholesterol-free medium Y. U18666A increased the level of intracellular free cholesterol in the culture with medium X but not medium Y 24 h after the treatment (Fig. 2a). The level of intracellular free cholesterol increased gradually up to 72 h in the culture with medium X. Next, the localization of free cholesterol was examined with filipin III staining. U18666A led to the intense staining of filipin III at perinuclear regions and the reduced staining at plasma membranes 24 h after the treatment (Fig. 2b). On the other hand, only the plasma membrane and subcellular organelles were stained positively in untreated control cells.
Fig. 2.
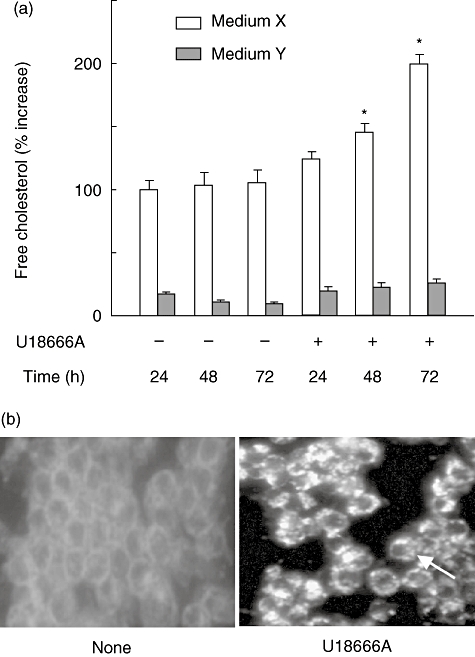
Intracellular free cholesterol accumulation in U18666A-treated RAW 264·7 cells. (a) RAW 264·7 cells were incubated with U18666A (1 µg/ml) for various hours in the culture with medium X or medium Y. *P < 0·01 versus none (vehicle control). (b) RAW 264·7 cells were incubated with U18666A (1 µg/ml) for 24 h and stained with filipin. Original magnification × 40.
U18666A induces the generation of ROS in RAW 264·7 cells
Cholesterol is an initial source of oxidative stress and triggers oxidative stress response via generation of ROS [17]. The effect of U18666A on ROS generation in RAW 264·7 cells was examined. U18666A markedly caused the ROS generation 24 h after the treatment. The ROS generation at 48 h was more than that at 24 h (Fig. 3). To study the involvement of free cholesterol accumulation in the ROS generation, we examined U18666A-induced ROS generation in the culture with medium X or cholesterol-free medium Y. U18666A-induced ROS generation was markedly prevented in the culture with medium Y 24 and 48 h after the treatment.
Fig. 3.
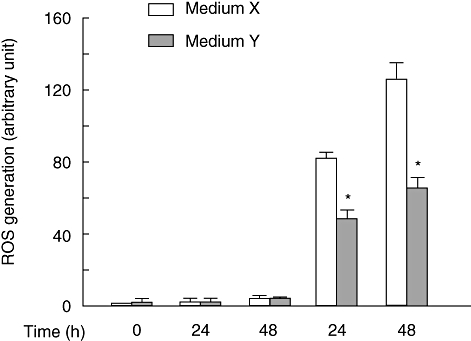
Reactive oxygen species generation in U18666A-treated RAW 264·7 cells. (a) RAW 264·7 cells were incubated with U18666A (1 µg/ml) for various hours in the culture with medium X or medium Y. *P < 0·01 versus medium X.
The NAC inhibits U18666A-induced TNF-α production
In the preceding section, the free cholesterol accumulation was suggested to trigger the ROS generation in RAW 264·7 cells. A possibility was raised that in U18666A-treated RAW 264·7 cells the ROS generation triggered by free cholesterol accumulation might lead to TNF-α production. The effect of NAC on TNF-α production in U18666A-treated RAW 264·7 cells was examined in medium X and cholesterol-free medium Y. The U18666A-induced TNF-α was abolished completely by NAC in medium X and Y (Fig. 4), suggesting the involvement of ROS in U18666A-induced TNF-α production.
Fig. 4.
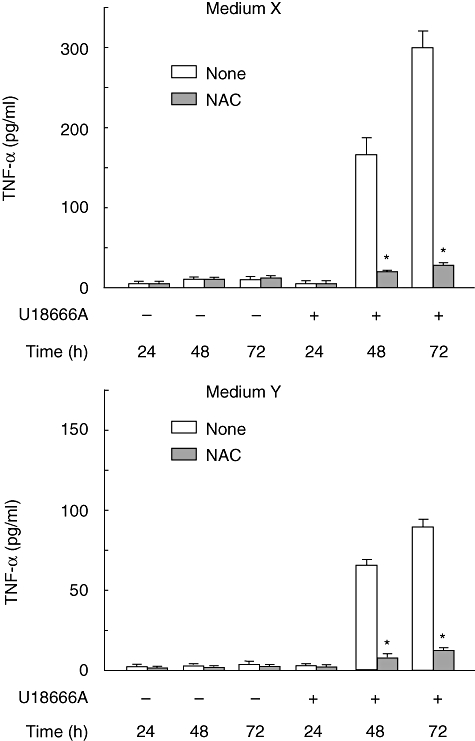
Tumour necrosis factor (TNF)-α production in U18666A-treated RAW 264·7 cells in medium X and Y. RAW 264·7 cells were incubated with U18666A (1 µg/ml) for various hours in the presence or absence of N-acetyl-L-cysteine (5 mM) in medium X and Y. *P < 0·01 versus none (vehicle control).
U18666A activates p38 mitogen-activated protein kinase at the late stage after the treatment
The effect of U18666A on a series of signal transduction on RAW 264·7 cells was examined to clarify the signal pathway initiating TNF-α production (Fig. 5a). First, we examined NF-κB signalling on which TNF-α production is mainly dependent. p65 NF-κB phosphorylation occurred 1 h after U18666A treatment and increased gradually up to 12 h. The phosphorylation of p65 was undetectable at 24 and 48 h. The NF-κB-related AKT phosphorylation occurred at 1 h. The highest phosphorylation was seen at 4 h and thereafter it waned. Next, the effect of U18666A on p38, SAPK/JNK and ERK1/2 signalling was examined. The activation of p38 was seen from 1 h to 8 h after U18666A treatment and disappeared temporally at 12 h. The phosphorylated p38 band again appeared at 24 h and then continued up to 48 h. The ERK1/2 phosphorylation was detected at 4 h and 12 h after U18666A treatment. On the other hand, SAPK/JNK continued from 1 h to 12 h but the phosphorylation at 24 h and 48 h was lower compared with that at 12 h. Moreover, the effect of NAC or cholesterol-free medium Y on the p38 phosphorylation was examined (Fig. 5b). Anti-oxidant NAC and medium Y inhibited the U18666A-induced p38 activation, especially 48 h after U18666A treatment.
Fig. 5.
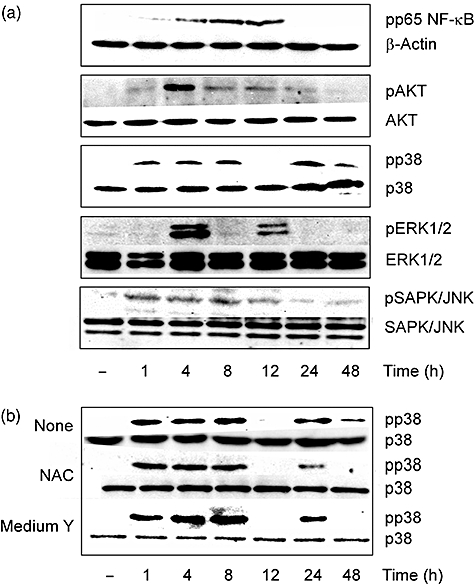
Oxidative stress-responsive signal transduction in U18666A-treated RAW 264·7 cells. (a) RAW 264·7 cells were incubated with U18666A (1 µg/ml) for various hours. (b) RAW 264·7 cells were incubated with U18666A (1 µg/ml) for various hours in the presence of N-acetyl-L-cysteine (5 mM) or medium Y. The phosphorylation was detected by immunoblotting.
The effect of a p38 or ERK 1/2 inhibitor on U18666A-induced TNF-α production was examined. SB203580 as a p38 inhibitor completely prevented U18666A-induced TNF-α production, whereas PD98058 as an ERK1/2 inhibitor did not affect it (Fig. 6). The addition of SB203580 12 h after U18666A treatment markedly inhibited the TNF-α production and the 12 h post-treatment of SB203580 was effective for TNF-α inhibition.
Fig. 6.
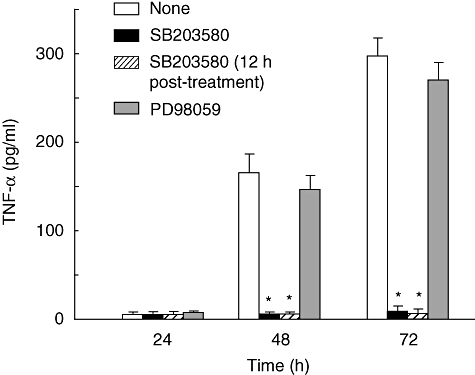
Participation of p38 in U18666A-induced tumour necrosis factor (TNF)-α production. RAW 264·7 cells were incubated with U18666A (1 µg/ml) in the presence of SB203580 (10 µM) or PD98058 (10 µM). In one of the experimental groups SB203580 was added into the culture 12 h after U18666A treatment. TNF-α production was determined with enzyme-linked immunosorbent assay. *P < 0·01 versus none (vehicle control).
Discussion
In the present study we demonstrate that U18666A may induce TNF-α production in RAW 264·7 macrophage cells via free cholesterol accumulation-mediated ROS generation. U18666A is a cholesterol transport-inhibiting class-2 amphiphile, which is used widely to mimic NPC disease [1–5]. Therefore, U18666A inhibits the intracellular transport of free cholesterol and accumulates intracellular free cholesterol. The free cholesterol accumulation may cause ROS generation and the ROS generation triggers oxidative stress-related p38 activation [18]. The p38 activation is reported to trigger TNF-α production [18,19]. Thus, free cholesterol accumulation as the primary action of U18666A is possible to induce TNF-α production via ROS generation. This is the first report on U18666A-induced TNF-α production.
How does free cholesterol accumulation cause the ROS generation? It is well established that elevated cholesterol levels are associated with the intensity of oxidative stress [20]. Further, altered cholesterol metabolism causes the production of 24s-hydroxycholesterol that may be involved in ROS generation [12]. Furthermore, U18666A reduces significantly the intracellular glutathione level, which is the most abundant intracellular anti-oxidant [21]. The free cholesterol accumulation by U18666A is possible to cause ROS generation and further oxidative stress. The inhibition of ROS generation by cholesterol-free medium Y also suggests that accumulated free cholesterol is a key factor of U18666A-induced ROS generation.
U18666A-induced TNF-α production occurs at a late stage (48–72 h) after the treatment and requires a long time lag before the production. Considering that TNF-α is produced 1–2 h after lipopolysaccharide stimulation, the long time lag is a characteristic of U18666A-induced TNF-α production. The delay may exclude the possibility that U18666A activates the signalling initiating TNF-α production directly. Rather, it suggests that U18666A-induced TNF-α production is induced secondarily by the primary action of U18666A, which inhibits the intracellular free cholesterol transfer. The delay in U18666A-induced TNF-α production may be dependent upon the time-period for free cholesterol accumulation and subsequent ROS generation.
Interestingly, p38 is phosphorylated twice at 1–8 h and 24–48 h after U18666A treatment. The late p38 phosphorylation is prevented in medium Y, indicating that it is mediated by free cholesterol accumulation induced by U18666A. The late p38 phosphorylation is also inhibited by anti-oxidant NAC, suggesting that it is also dependent upon ROS production. Based on these findings, the late p38 activation might be critical for U186666A-induced TNF-α production. It is also supported by the finding that the addition of SB203580 12 h after U1866A treatment inhibits TNF-α production markedly. In addition, the early p38 activation might be required for free cholesterol accumulation and/or ROS generation. There is no late activation of NF-κB and SAPK/JNK 48 h after U18666A stimulation, suggesting that NF-κB and SAPK/JNK is not involved in U18666A-induced TNF-α production. It is still unclear the reason why oxidative stress-responsive NF-κB and SAPK/JNK are not activated in U18666A-stimulated cells.
U18666A is used widely for the experimental model of NPC disease as an endosome/lysosomal free cholesterol storage disorder [4,22,23] and causes neuronal apoptosis [24]. In NPC disease, the apoptotic cell death in the brain is mediated by the TNF receptor superfamily pathway [10]. The expression of TNF-α mRNA is reported to increase up to 30–50-fold in the cerebellum of 7–9-week-old NPC1-deficient mice compared with wild-type mice [10]. Therefore, U186666A-induced TNF-α production might be useful to clarify the pathogenesis of NPC disease.
Acknowledgments
This work was supported by in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan. We are grateful to K. Takahashi and A. Morikawa for technical assistance.
References
- 1.Mohammadi A, Perry RJ, Storey MK, et al. Golgi localization and phosphorylation of oxysterol binding protein in Niemann–Pick C and U18666a-treated cells. J Lipid Res. 2001;42:1062–71. [PubMed] [Google Scholar]
- 2.Pol A, Luetterforst R, Lindsay M, Heino S, Ikonen E, Parton RG. A caveolin dominant negative mutant associates with lipid bodies and induces intracellular cholesterol imbalance. J Cell Biol. 2001;152:1057–70. doi: 10.1083/jcb.152.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Runz H, Rietdorf J, Tomic I, et al. Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J Neurosci. 2002;22:1679–89. doi: 10.1523/JNEUROSCI.22-05-01679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liscum L, Faust JR. The intracellular transport of low density lipoprotein-derived cholesterol is inhibited in Chinese hamster ovary cells cultured with 3-β-[2-(diethylamino)ethoxy]androst-5-en-17-one. J Biol Chem. 1989;264:11796–06. [PubMed] [Google Scholar]
- 5.Hall AM, Krishnamoorthy L, Orlow SJ. Accumulation of tyrosinase in the endolysosomal compartment is induced by U18666A. Pigment Cell Res. 2003;16:149–58. doi: 10.1034/j.1600-0749.2003.00027.x. [DOI] [PubMed] [Google Scholar]
- 6.Feng B, Yao PM, Li Y, et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–92. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 7.Chang TY, Chang CCY, Ohgami N, et al. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–57. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- 8.Kellner-Weibel GK, Geng YJ, Rothblat GH. Cytotoxic cholesterol is generated by the hydrolysis of cytoplasmic cholesteryl ester and transported to the plasma membrane. Atherosclerosis. 1999;146:309–19. doi: 10.1016/s0021-9150(99)00155-0. [DOI] [PubMed] [Google Scholar]
- 9.Koh CH, Cheung NS. Cellular mechanism of U18666A-mediated apoptosis in cultured murine cortical neurons: bridging Niemann–Pick disease type C and Alzheimer's disease. Cell Signal. 2006;18:1844–53. doi: 10.1016/j.cellsig.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Wu YP, Mizukami H, Matsuda J, et al. Apoptosis accompanied by up-regulation by TNF-alpha death pathway genes in the brain of Niemann–Pick type C disease. Mol Genet Metab. 2005;84:9–17. doi: 10.1016/j.ymgme.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 11.Feng B, Zhang D, Kuriakose G, et al. Niemann–Pick C heterozygosity confers resistance to lesional necrosis and macrophage apoptosis in murine atherosclerosis. Proc Natl Acad Sci USA. 2003;100:10423–8. doi: 10.1073/pnas.1732494100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang JR, Coleman T, Langmade SJ, et al. Niemann–Pick C1 protects against atherosclerosis in mice via regulation of macrophage intracellular cholesterol trafficking. J Clin Invest. 2008;118:2281–90. doi: 10.1172/JCI32561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chain BE, Knowles BR. A solvent system for delipidation of plasma or serum without protein precipitation. J Lipid Res. 1976;17:176–81. [PubMed] [Google Scholar]
- 14.Cadigan KM, Spillane DM, Chang TY. Isolation and characterization of Chinese hamster ovary cell mutants defective in intracellular low density lipoprotein-cholesterol trafficking. J Cell Biol. 1990;110:295–08. doi: 10.1083/jcb.110.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naiki Y, Michelsen KS, Zhanq W, et al. Transforming growth factor-β differentially inhibits MyD88-dependent, but not TRAM- and TRIF-dependent, lipopolysaccharide-induced TLR4 signaling. J Biol Chem. 2005;280:5491–5. doi: 10.1074/jbc.C400503200. [DOI] [PubMed] [Google Scholar]
- 16.Chakravortty D, Kato Y, Sugiyama T, et al. The inhibitory action of sodium arsenite on lipopolysaccharide-induced nitric oxide production in RAW 267·4 macrophage cells: a role of Raf-1 in lipopolysaccharide signaling. J Immunol. 2003;166:2011–17. doi: 10.4049/jimmunol.166.3.2011. [DOI] [PubMed] [Google Scholar]
- 17.Pappolla MA, Smith MA, Thomas TB, et al. Cholesterol, oxidative stress, and Alzheimer's disease: expanding the horizons of pathogenesis. Free Radic Biol Med. 2002;33:73–81. doi: 10.1016/s0891-5849(02)00841-9. [DOI] [PubMed] [Google Scholar]
- 18.Matsuzawa A, Saegusa K, Noguchi T, et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–92. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- 19.Yamakawa T, Eguchi S, Matsumoto T, et al. Intracellular signaling in rat cultured vascular smooth muscle cells: roles of nuclear factor-kappaB and p38 mitogen-activated protein kinase on tumor necrosis factor production. Endocrinology. 1999;140:3562–72. doi: 10.1210/endo.140.8.6914. [DOI] [PubMed] [Google Scholar]
- 20.Prasad K, Kalra J. Oxygen free radicals and hypercholesterolemic atherosclerosis: effect of vitamin E. Am Heart J. 1993;125:958–73. doi: 10.1016/0002-8703(93)90102-f. [DOI] [PubMed] [Google Scholar]
- 21.Koh CH, Whiteman M, Li QJ, et al. Chronic exposure to U18666A is associated with oxidative stress in cultured murine cortical neurons. J Neurochem. 2006;98:1278–89. doi: 10.1111/j.1471-4159.2006.03958.x. [DOI] [PubMed] [Google Scholar]
- 22.Lange Y, Ye J, Steck TL. Circulation of cholesterol between lysosomes and the plasma membrane. J Biol Chem. 1998;273:18915–22. doi: 10.1074/jbc.273.30.18915. [DOI] [PubMed] [Google Scholar]
- 23.Underwood KW, Jacobs NL, Howley A, et al. Evidence for a cholesterol transport pathway from lysosomes to endoplasmic reticulum that is independent of the plasma membrane. J Biol Chem. 1998;73:4266–74. doi: 10.1074/jbc.273.7.4266. [DOI] [PubMed] [Google Scholar]
- 24.Bi X, Liu J, Yao Y, et al. Deregulation of the phosphatidylinositol-3 kinase signaling cascade is associated with neurodegeneration in Npc1−/− mouse brain. Am J Pathol. 2005;167:1081–92. doi: 10.1016/S0002-9440(10)61197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]