

Abstract
Our laboratory has suggested that loss of tolerance to pyruvate dehydrogenase (PDC-E2) leads to an anti-mitochondrial antibody response and autoimmune cholangitis, similar to human primary biliary cirrhosis (PBC). We have suggested that this loss of tolerance can be induced either via chemical xenobiotic immunization or exposure to select bacteria. Our work has also highlighted the importance of genetic susceptibility. Using the non-obese diabetic (NOD) congenic strain 1101 (hereafter referred to as NOD.1101 mice), which has chromosome 3 regions from B6 introgressed onto a NOD background, we exposed animals to 2-octynoic acid (2OA) coupled to bovine serum albumin (BSA). 2OA has been demonstrated previously by a quantitative structural activity relationship to react as well as or better than lipoic acid to anti-mitochondrial antibodies. We demonstrate herein that NOD.1101 mice immunized with 2OA-BSA, but not with BSA alone, develop high titre anti-mitochondrial antibodies and histological features, including portal infiltrates enriched in CD8+ cells and liver granulomas, similar to human PBC. We believe this model will allow the rigorous dissection of early immunogenetic cause of biliary damage.
Keywords: AMAs, autoimmune cholangitis, NOD.1101 mice, PBC, xenobiotic agents
Introduction
Cellular and molecular events involved during the inductive phase of autoimmune diseases have proved difficult to define. This is well illustrated in the case of primary biliary cirrhosis (PBC), a female predominant autoimmune liver disease. PBC is characterized by the destruction of intrahepatic bile ducts associated with high titres of epitope-specific anti-mitochondrial antibodies (AMAs) directed against E2 subunits of enzymes of the 2-oxo-acid dehydrogenase complex (2-OADC) family [1,2]. Among these 2-OADC enzymes, the predominant reactivity is directed against the E2 of the pyruvate dehydrogenase (PDC-E2), with the major immunodominant epitope for both T and B cells identified as the lipoic acid binding domain of the E2 subunits [3–7]. Our premise is that exposure to agents that modify the lipoic acid binding (lipoyl) region of PDC-E2 could lead to loss of tolerance and the development of autoimmune cholangitis in genetically susceptible hosts [8]. This thesis is supported by the observation that AMAs from patients with PBC recognize a class of xenobiotic modified PDC-E2 peptides, often with a higher affinity than the native autoantigen [9].
Among the first xenobiotic agents identified was 6-bromohexanoate which, when coupled to bovine serum albumin (BSA), was shown to break tolerance and induce AMA in both rabbits and guinea pigs [10–13]. More recently, our laboratory has developed more sophisticated quantitative structure–activity relationship analysis and this has led to identification of 2-octynoic acid (2OA) as being a more highly reactive hapten recognized by AMAs [14–16]. 2OA is not found in nature but can be synthesized chemically. Importantly, however, the methyl and ethyl esters of this compound are used widely in consumer products, including perfume, lipstick, soap, detergents, cream, lotion and many common food flavourings [14,15,17]. Recently, we have demonstrated further that 2OA BSA-conjugate breaks tolerance and leads to the production of AMA and human PBC-like liver histopathology in C57BL/6 (B6) mice [18].
The non-obese diabetic (NOD) mouse is a widely used animal model of autoimmune type 1 diabetes (T1D), and the genetic basis of T1D in the NOD model has been characterized extensively through the development of congenic strains [19]. NOD congenic strains can develop other autoimmune syndromes, such as the autoimmune biliary disease seen in the NOD.c3c4 mouse [20]. The NOD.1101, which shares a portion of the B6-derived chromosome 3 congenic region with the NOD.c3c4 strain, does not develop spontaneously the PBC-like disease seen in NOD.c3c4 mice and is protected partially from diabetes compared with the NOD parental strain. NOD.1101 mice have been shown to be more susceptible than other strains to developing T cell-mediated autoimmunity directed against small bile ducts following infection with Novosphingobium aromaticivorans[21]. We therefore hypothesized that NOD.1101 mice would also have increased susceptibility to biliary disease induced chemically. This hypothesis was tested by immunizing NOD.1101 mice with 2OA coupled to BSA and compared the outcome with our previous immunizations of C57/BL6 mice.
Materials and methods
Mouse strain
The NOD.B6 Idd10/Idd18R2 mice, herein called NOD.1101, were obtained from Taconic, Inc. (Germantown, NY, USA). The genetic origin of these mice has been described in detail previously [21,22]. In NOD.1101 mice, NOD alleles at two insulin-dependent diabetes (Idd) genes, Idd10 and Idd18 on chromosome 3, are replaced by B6-derived resistance alleles (Fig. 1a), which partially protect these mice from developing diabetes. Mice are available through the Emerging Models programme as Line 7754 (for a detailed description of the B6-derived introgressed interval on distal chromosome 3, see http://www.t1dbase.org/cgi-bin/dispatcher.cgi/DrawStrains/display?taconic_line=7754). The NOD.1101 strain was found during the course of this study to harbour a 3·19 Mb B6-derived region on chromosome 18 having a centromeric ‘out’ boundary at rs6303064 (19·74 Mb in Ensembl, NCBI m36) and a telomeric ‘out’ boundary at rs13483251 (22·93 Mb). This region on chromosome 18 is not responsible for the protection from T1D observed in NOD.1101 mice [22]. All mice were maintained in individually ventilated cages under specific pathogen-free conditions.
Fig. 1.
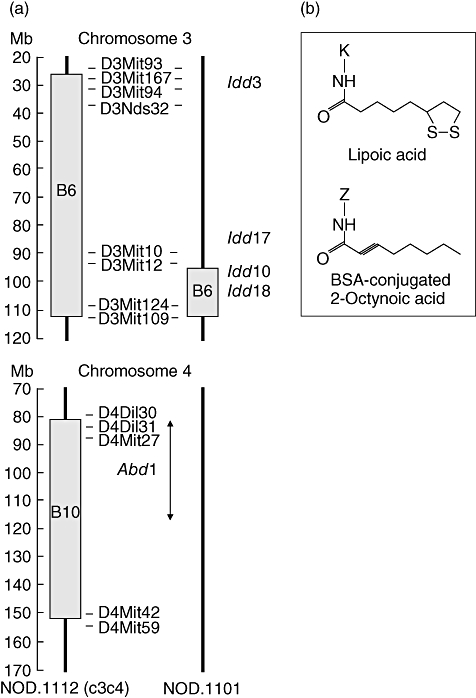
(a) Genetic map of chromosomes 3 and 4 in non-obese diabetic (NOD).1101 and NOD.c3c4 mice. The NOD.1101 strain is free from spontaneous autoimmune biliary disease and includes a portion of the B6-derived chromosome 3 region present in NOD.c3c4 mice but not the B10-derived chromosome 4 segment (labelled Abd1). (b) The structures of lipoic acid and 2-octynoic acid (2OA)-bovine serum albumin (BSA). 2OA was conjugated with BSA and used for immunization. ‘Z’ represents the lysine residue of BSA.
Preparation of immunogen
2-octynoic acid was purchased from (St Louis, MO, USA) and was conjugated with BSA (EMD Chemicals, Gibbstown, NJ, USA), as described previously [18]: 2OA was dissolved in dry dimethyl ether. N-hydroxysuccinimide (NHS) was then added and the solution was cooled to 0°C and stirred for 20 min. Dicyclohexylcarbodiimide was then added and the mixture was allowed to warm to ambient temperature overnight. The solution was filtered, concentrated by roto-evaporation under reduced pressure, re-dissolved with ethyl ether, washed with water, NaHCO3 (1M), brine, dried under magnesium sulphate, filtered and concentrated. The product was then purified using flash chromatography (30% ethyl acetate/hexane). NHS-activated 2OA was dissolved in dimethyl sulphoxide and then coupled to the lysine residues of BSA (Fig. 1b). The solution was allowed to react for 3 h followed by dialysis [phosphate-buffered saline (PBS)].
Immunization
Male and female mice at 6 weeks of age were maintained individually in ventilated cages under specific pathogen-free conditions and immunized with either 2OA conjugated to BSA (2OA-BSA) at 100 µg/25 µl per animal intraperitoneally in the presence of complete Freund's adjuvant (CFA) (Sigma-Aldrich, 25 µl/animal) containing 10 mg/ml of Mycobacterium tuberculosis strain H37Ra or BSA alone in CFA. All animals were given booster immunizations every 2 weeks with either the compound mixture or BSA in incomplete Freund's adjuvant (IFA) (Sigma-Aldrich). Sera were collected before immunization and serially thereafter for assays of AMA. Animals were killed 12 or 24 weeks after immunization and liver tissues collected for histological evaluation or spleen tissues collected for cytokine production assay. All animal experiments were performed after receiving approval of the Institutional Animal Care and Use Committee of the University of California at Davis.
Detection of AMA reactivity
Serum AMA was quantified using an enzyme-linked immunosorbent assay (ELISA) with recombinant human PDC-E2, as described previously [23]. Briefly, purified recombinant antigen at 10 µg/ml in carbonate buffer (pH 9·6) were coated onto 96-well ELISA plates at 4°C overnight, washed five times with PBS-T, and blocked with 3% skimmed milk in PBS for 1 h. One hundred µl of the diluted sera (1:500) was added to the wells and incubated for 1 h at room temperature (RT) followed by PBS-T washes; 100 µl of horseradish peroxidase-conjugated anti-mouse immunoglobulin (Ig)G, IgA or IgM (1:2000) (Zymed, San Francisco, CA, USA) was added to each well for 1 h at RT followed by another set of PBS-T washes. Immunoreactivity was determined by measuring the optical density (OD) at 450 nm after incubation with 100 µl of 3,3′,5,5′-tetramethylbenzidine (BD Biosciences, San Jose, CA, USA) for 30 min.
Flow cytometry
Immediately after the animals were killed, livers and spleens were collected and livers perfused with PBS containing 0·2% BSA, passed through a nylon mesh and re-suspended in PBS/0·2% BSA. Hepatocytes were removed as pellets after centrifugation at 700 g for 1 min and the remaining cells collected. Splenic tissue was disrupted between two glass slides and suspended in PBS/0·2% BSA. Lymphocytes from suspended liver and spleen cells were isolated using Accu-Paque (density: 1·086; Accurate Chemical & Scientific Corp., Westbury, NY, USA) gradient. After centrifugation, cells at the interface were washed with PBS/0·2% BSA and the viability of cells confirmed by trypan blue dye (Invitrogen, Carlsbad, CA, USA) exclusion. Cell preparations were incubated with monoclonal antibody 2·4G2 for FcR blocking (BioLegend, San Diego, CA, USA) and then exposed at 4°C to a combination of fluorochrome-conjugated antibodies that included anti-CD4 fluorescein isothiocyanate (eBiosciences, San Diego, CA, USA), anti-T cell receptor β phycoerythrin (PE) (eBiosciences), anti-CD8a PE-Cy5 (eBiosciences), anti-CD49b (DX5) Alexa Fluor 647 (BioLegend), anti-CD19 Alexa Fluor 647 (eBiosciences), anti-Gr-1 PE (BioLegend), anti-CD44 allophycocyanin (APC) (BioLegend) and anti-CD4 APC/Cy7 (BioLegend). Multi-colour flow analyses were performed using a fluorescence activated cell sorter (FACScan) flow cytometer (BD Immunocytometry System, San Jose, CA, USA) upgraded by Cytec Development (Fremont, CA, USA) to allow for five-colour analysis. Acquired data were analysed using cellquest software (BD Biosciences).
Cytokine analysis
Enriched populations of CD4+ or CD8+ splenic T cells were prepared using MS columns (magnetic affinity cell sorter) and a magnetic cell sorting separator (Miltenyi Biotech Inc., Miltenyi Biotec, Auburn, CA, USA). Cells were counted in a haemocytometer and viability determined using trypan blue exclusion and re-suspended at 5 × 105 cells of media. Media consisted of RPMI-1640 culture medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), 100 µg/ml of streptomycin, 100 U/ml of penicillin (Invitrogen). The cell suspension was dispensed in a volume of 0·2 ml into individual wells of a 96-well round-bottomed microtitre plate containing either media alone (control) or 0·5 µg/ml of anti-mouse CD3 antibody (BioLegend) and 2·5 µg/ml of anti-mouse CD28 antibody (BioLegend). The microtitre plate was cultured for 72 h at 37°C in a 5% CO2 humidified atmosphere. After 72 h culture, supernatant fluids of the culture media were collected and stored at −70°C. The levels of interferon (IFN)-γ, tumour necrosis factor (TNF)-α, interleukin (IL)-6 and IL-10 in the supernatant were measured using the cytometric bead array (CBA) kit (Mouse Inflammation Cytokine Kit; BD Biosciences). Samples were diluted 1:2 with assay diluent provided with the kit. Following the preparation, samples were analysed using a FACScan flow cytometer and BD CBA software.
Immunohistochemistry
Phenotypic analysis of the hepatic tissue cell infiltrates was performed utilizing rat anti-mouse Gr-1, anti-mouse CD4, anti-mouse CD8, anti-mouse TNF-α and anti-mouse IFN-γ antibodies (1:50 dilution, BioLegend, San Diego, CA, USA), as described previously [24]. Tissue sections were cut at 4 µm from representative tissue blocks. After de-paraffinization, sections were soaked in target retrieval-buffered saline (pH 6·1; Dako Cytomation, Carpinteria, CA, USA) in a non-metal-containing plastic-made pressure cooker and irradiated in a microwave for 10 min (maximum 500 W). After irradiation, sections were rinsed well under running water for 2 min, soaked in 3% H2O2 methanol solution for 5 min, and then soaked in 5% BSA for 1 min. Primary antibodies were diluted to a previously determined optimal concentration in PBS containing 5% BSA. The diluted antibodies were applied in a moist chamber and irradiated intermittently for 10 min (250 W, 4 s on, 3 s off). After three washes with Tris-buffered saline containing 1% Tween (TBS-T) for 1 min, peroxidase-conjugated Envision kit (Envision-PO, Envision System; Dako Cytomation) was applied on the specimens in the moist chamber. Irradiation was then performed intermittently for 10 min, as described above. After washing ×5 with TBS-T, the sections were immersed in 3′3′-diaminobenzidine solution (Sigma-Aldrich) with H2O2 and counterstained with haematoxylin (Dako Cytomation) and mounted under coverslips.
Statistical analysis
Statistical analysis was performed utilizing the unpaired t-test to compare data derived from the 2OA-BSA and BSA-immunized mice. P-values less than 0·05 were considered statistically significant.
Results
The AMA production
A positive AMA was defined as an OD reading that was 3 standard deviations (s.d.) above the mean of ELISA values of sera at week 0. Using these criteria, 100% of the sera from NOD.1101 mice immunized with 2OA-BSA were positive against PDC-E2 at 2 weeks after immunization. In contrast, none of the sera from mice immunized with BSA showed detectable reactivity against PDC-E2 (Fig. 2). At 2 weeks, both IgG-AMA and IgM-AMA were increased significantly in the sera of mice immunized with 2OA-BSA (n = 8) compared with sera from mice immunized with BSA (n = 7): 0·608 ± 0·174 versus 0·005 ± 0·003, P = 0·0099 and 0·062 ± 0·018 versus 0·008 ± 0·003, P = 0·0205 respectively. At 4 weeks, AMAs of the IgG, IgA and IgM isotypes were all increased significantly in the sera of NOD.1101 mice immunized with 2OA-BSA (n = 8) compared with BSA-immunized mice (n = 7); 2·629 ± 0·098 versus 0·009 ± 0·005, P < 0·0001, 1·373 ± 0·153 versus 0·002 ± 0·001, P < 0·0001 and 0·113 ± 0·034 versus 0·002 ± 0·002, P = 0·0129 respectively (Fig. 2). AMA-specific IgG and IgA were elevated throughout an observation period of 12 weeks, whereas IgM declined after 8 weeks (Fig. 2).
Fig. 2.
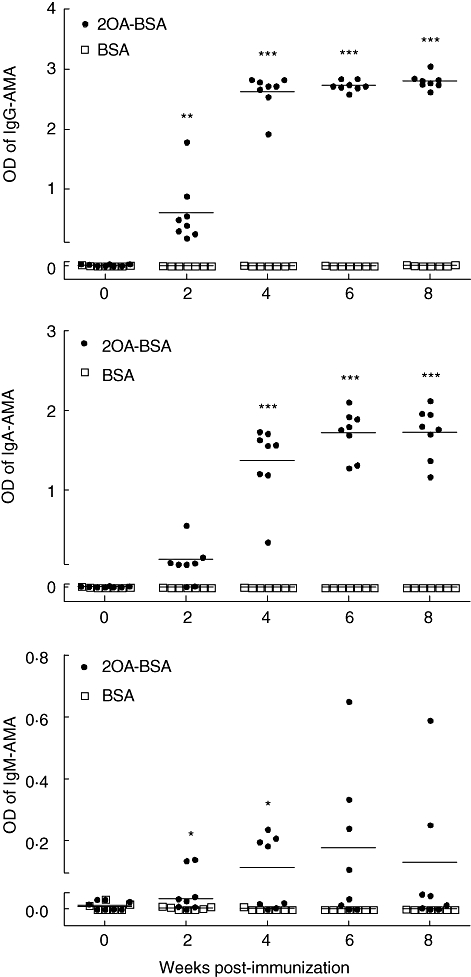
Quantification of serological anti-mitochondrial antibodies by enzyme-linked immunosorbent assay (ELISA) after immunization with 2-octynoic acid (2OA)-bovine serum albumin (BSA) compared with BSA. *P < 0·05; **P < 0·01; ***P < 0·001.
The PBC-like liver histopathology in 2OA-BSA-immunized mice
At 12 weeks after immunization the portal tracts of mice immunized with 2OA-BSA had significant cellular infiltrates comprised of neutrophils and lymphocytes around the bile ducts (Fig. 3a). Some of the portal tract areas showed bile duct cell injury (Fig. 3a, black arrows). No significant change was observed within the central vein areas. These findings were not found in liver tissue sections from BSA-immunized control mice (Fig. 3b). No evidence of either steatosis, eosinophilia or cholestasis was noted. Liver sections from mice at 24 weeks post-immunization showed similar histopathology compared with liver sections at 12 weeks post-immunization. There were no gender-based differences in the pathological findings in the mice. Immunohistological analysis revealed the presence of portal cellular infiltrates comprised of numerous CD4+ and CD8+ lymphocytes (Fig. 3c and d respectively), which were absent in tissue sections from the control mice. The synthesis of TNF-α- and IFN-γ-producing cells was also evaluated and TNF-α- or IFN-γ-producing cells were visualized readily and localized to the portal areas (Fig. 3e and f). No such TNF-α- or IFN-γ-producing cells were found in control mice. Sections other than liver were also evaluated and no cellular infiltrates were found in tissue sections prepared from the thyroid, lung and colon (data not shown). Of note, in 2OA-BSA immunized NOD 1101 mice, numerous granulocytes were observed in portal inflammation whereas those were rarely found in 2OA-BSA immunized B6 mice (Fig. 3g and h) [18]. Immunohistochemical staining for Gr-1 demonstrated qualitatively more Gr-1+ cells in portal cellular infiltrates in 2OA-BSA immunized NOD 1101 mice than those in 2OA-BSA immunized B6 mice (Fig. 3i and j).
Fig. 3.
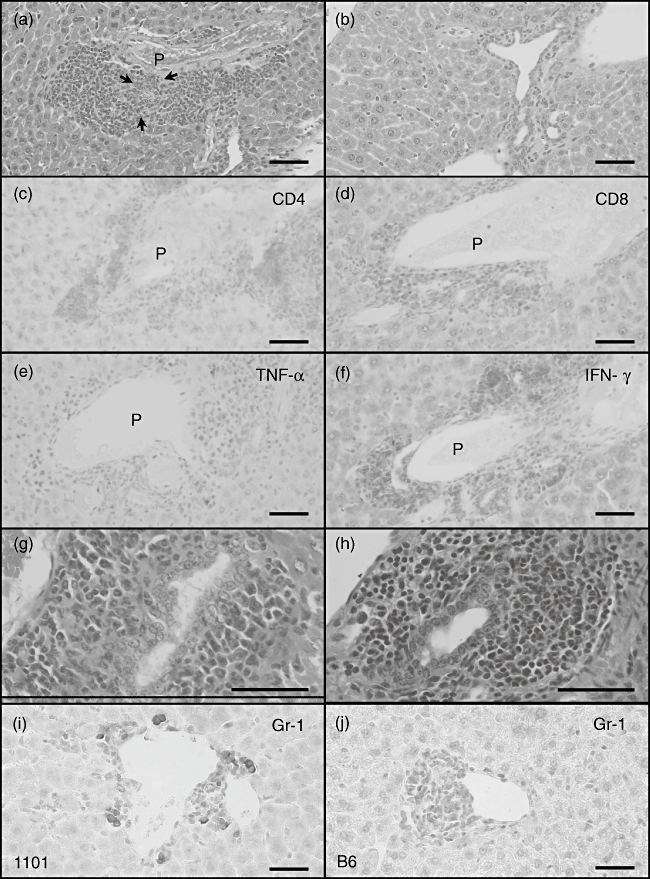
(a–c) Light microscopy of haematoxylin and eosin (H&E)-stained liver sections of representative 2-octynoic acid (2OA)-bovine serum albumin (BSA)-immunized non-obese diabetic (NOD).1101 mice (a, n = 11) demonstrates marked infiltration of neutrophils and lymphocytes in portal tracts and particularly surrounding intralobular bile ducts. Some portal tract shows bile duct injury (a, black arrows). This finding was not found in BSA-immunized mice liver (b, n = 7). (c–f) Immunohistochemical analysis of liver of 2OA-BSA-immunized mice. Livers from three representative mice were stained with monoclonal antibodies CD4 (c), CD8 (d), tumour necrosis factor (TNF)-α (e) and interferon (IFN)-γ (f). (g–j) Compared with previous experiments in B6 mice, 2OA-immunized NOD.1101 mice demonstrated a much higher degree of granulocyte infiltration in portal inflammation in both H&E and immunohistochemical Gr-1 staining (scale bar: 50 µm).
Phenotype of infiltrating cells
The lymphoid cell populations within livers and spleens of 2OA-BSA-immunized mice (n = 5) and control BSA-immunized mice (n = 3) were analysed by flow cytometry. The frequency of CD4+ T cells in the liver and spleen of the 2-OA-BSA immunized mice were decreased with a concomitant increase in the frequency of CD8+T cells compared with control mice (Fig. 4a). The CD4/CD8 ratio in the liver and spleen were decreased significantly: 2·38 ± 0·22 versus 3·58 ± 0·24, P = 0·02 for liver; 1·94 ± 0·09 versus 2·92 ± 0·15, P = 0·002 for spleen (Fig. 4b). These changes in the spleen and liver lymphoid cells were not due to changes in the frequency of naive versus effector memory CD4+ or CD8+ T cells as measured by CD44 expression (Fig. 5a). However, the total cell numbers of liver or spleen lymphoid cells were increased significantly, therefore the absolute numbers of CD4+CD44high or CD8+CD44high memory T cells were increased significantly compared with control mice: 1·18 ± 0·24 versus 0·23 ± 0·03 (×106), P = 0·04 for CD4 and 0·26 ± 0·05 versus 0·04 ± 0·01 (×106), P = 0·02 for CD8 in the liver; 6·19 ± 0·41 versus 1·60 ± 0·61 (×106), P = 0·001 for CD4 and 1·20 ± 0·06 versus 0·37 ± 00·14 (×106), P = 0·002 for CD8 in the spleen (Fig. 5b).
Fig. 4.
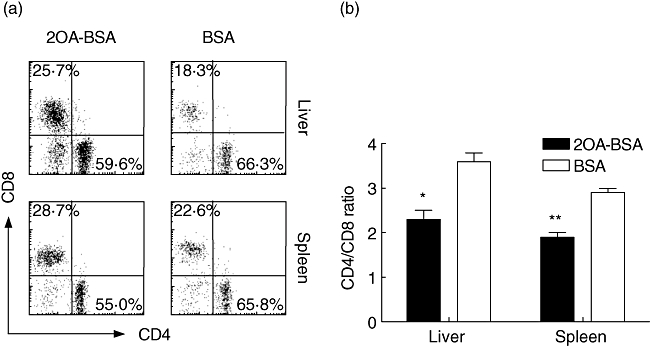
Flow cytometric analysis of liver and spleen cells from 2-octynoic acid (2OA)-bovine serum albumin (BSA) (n = 5) or BSA-immunized mice (n = 3) at 12 weeks post-immunization. Cells were stained with antibodies to T cell receptor (TCR)β, CD49b (DX5), CD4 and CD8a. TCRβ+DX5– cells were gated (a) and the ratio of CD4+ and CD8a+ T cells in the liver and spleen are indicated (b). CD4+ T cells were decreased and CD8a+ cells were increased, therefore CD4/CD8 ratios in both liver and spleen were decreased compared with BSA-immunized mice. *P < 0·05; **P < 0·01.
Fig. 5.
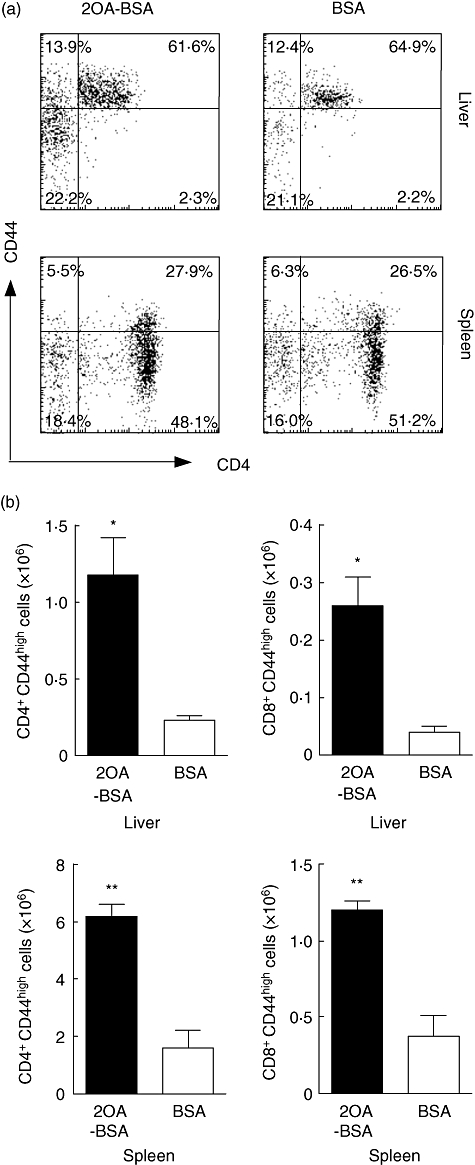
Flow cytometric analysis of liver and spleen cell populations at 12 weeks post-immunization [n = 5 for 2-octynoic acid (2OA)-bovine serum albumin (BSA) and n = 3 for BSA]. Analysis of CD4 and CD44 was performed with gating of T cell receptor (TCR)β+cells (a). Absolute numbers of CD4+CD44+ memory T cells and CD8+CD44+ memory T cells were increased in the liver and spleen from 2OA-BSA-immunized mice compared with BSA-immunized mice (b). *P < 0·05; **P < 0·01.
Cytokines response of cultured spleen cells
The levels were increased significantly in the supernatant of the CD4+ and CD8+ T cells cultured from the 2OA-BSA immunized mice compared with control mice (Fig. 6). The levels of the prototypical T helper type 2 (Th2) cytokines IL-6 and IL-10 were also elevated in the supernatant of CD4+ and CD8+ cell cultures from the 2OA-BSA immunized mice compared with the control mice; 6·5 ± 0·4 pg/ml versus 2·9 ± 0·1 pg/ml, P < 0·0001 for IL-6 and 134·2 ± 14·9 pg/ml versus 19·5 ± 11·9 pg/ml, P < 0·0001 for IL-10. The levels of IL-6 and IL-10 from the cultures of CD8+ cells from the control BSA-immunized mice were below the limit of detection.
Fig. 6.

Concentration of tumour necrosis factor (TNF)-α, interferon (IFN)-γ, interleukin (IL)-6 and IL-10 in the supernatant of cultured spleen cells from 2-octynoic acid (2OA)-bovine serum albumin (BSA) or BSA-immunized mice were measured using cytometric bead array. CD4+ or CD8+ T cells were cultured for 3 days with anti-CD3 and CD28 stimulation. Levels of TNF-α, IFN-γ and IL-6 in the supernatant of CD4+ or CD8+ cells and IL-10 in the supernatant of CD4+ cells from 2OA-BSA-immunized mice were all elevated compared with that of BSA-immunized mice. *P < 0·05; **P < 0·01; ***P < 0·001.
Discussion
The long asymptomatic latency period preceding overt PBC in humans calls for the use of animal models to define early events that elucidate the mechanisms involved in loss of self-tolerance to the 2-OADC autoantigens [25]. Several studies suggest a role for environmental factors in initiating loss of tolerance superimposed on a genetic predisposition [8,21,26–30]. To explore these issues, there are several models of autoimmune cholangitis including spontaneous disease in NOD.c3c4 mice [20,31], and genetically manipulated models such as TGF-β receptor II dominant-negative mice [32], IL-2 receptor α knock-out mice [33] and Cl−/HCO3− anion exchanger 2 (Ae2a,b) knock-out mice [34]. As for many other autoimmune diseases, there appear to be important roles for both epigenetic influences and environmental agents [35–37]. Our laboratory has demonstrated that autoimmune cholangitis can be induced by breaking tolerance to PDC in inbred mice [18] and in outbred models [10,13], whether by use of xenobiotics or experimental infection of mice with the bacterium N. aromaticivorans[21].
The NOD mouse is genetically susceptible to T1D. At least 20 genetic susceptibility loci, known as insulin-dependent diabetes (Idd) loci, have been identified in the NOD mouse and localized to different chromosomes [38,39]. NOD.1101 mice contain the B6-derived diabetes resistance alleles at the Idd10 and Idd18 regions on chromosome 3 (Fig. 1a). Results of previous studies suggest that NOD.1101 mice are 50% less likely to develop T1D compared with the parental NOD strain [22]. NOD.c3c4 mice develop severe biliary disease with AMA production [20,31], whereas NOD.1101 mice do not develop biliary disease spontaneously. However, NOD.1101 mice have a portion of the B6-derived chromosome 3 region present in NOD.c3c4 mice (Fig. 1a). Infection of NOD.1101 mice with N. aromaticivorans induces antibodies against PDC-E2 and triggered chronic T cell-mediated autoimmunity against small bile ducts [21]. Therefore, we hypothesized that NOD.1101 mice should be at least as susceptible to induction of autoimmune biliary disease by immunization with 2OA as C57BL/6 mice, which we have reported previously [18].
A feature of early-stage PBC is an intrahepatic transcriptional up-regulation in the liver of genes encoding TNF-α and IFN-γ with a contrasting down-regulation of IL-4 [40–43]. Similarly, as reported in our previous study, B6 mice immunized with 2OA-BSA with CFA and IFA developed a PBC-like autoimmune cholangitis with AMA positivity, and such mice demonstrated a Th1 cytokine bias as judged by increased intrahepatic levels of TNF-α and IFN-γ[18], in keeping with their genetically determined Th1-dominant cytokine responsiveness [44]. Supernatants from CD4+ or CD8+ T cell cultures activated in vitro by anti-CD3 or anti-CD28 in 2OA-BSA-immunized NOD.1101 mice also demonstrated a Th1 cytokine response, but these supernatants also showed increased levels of IL-6 and IL-10. The use of CFA or IFA appears to prevent the development of autoimmune diabetes in NOD mice [45,46], and this prevention is attributed to a decrease in the ratio of IFN-γ to IL-4 mRNA synthesized by the pancreatic islet infiltrating T cells [47]. Because the 2OA-BSA-immunized NOD.1101 mice were shown in the present study to retain much higher Th1 cytokine responsiveness compared with controls immunized with BSA alone (see Fig. 6), we reasoned that this Th1 type response after immunization with 2OA-BSA was unlikely to be due to the use of CFA or IFA.
Looking at cellular events in the liver, 2OA-BSA immunized NOD.1011 (and B6) mice had a decrease in the CD4/CD8 ratio that was associated with an increase in the frequency of CD4+CD44high or CD8+CD44high memory T cells [18]. However, comparing the two strains, the characteristics of the infiltrating cells were distinct. Thus, while the portal areas were the site of the cellular infiltrates in liver tissue from both strains, the infiltrates in the 2OA-BSA immunized NOD.1101 mice also included granulocytes, which is rarely seen in haematoxylin and eosin-stained liver sections of 2OA-immunized B6 mice, evaluated by a liver pathologist (K. T.) (see Fig. 3, [18]). These findings are significant, because our laboratory has reported that myeloperoxidase-positive inflammatory cells participate in bile duct damage in PBC [48]. Myeloperoxidase-positive infiltrating cells are associated closely with the expression of 3-nitrotyrosine, the enzyme responsible for nitric oxide production, in bile duct epithelial cells (BECs) with apoptotic changes. Also its expression was elevated at early stage (stages I–II according to Ludwig et al. [49]) compared with late stage (stages III–IV) in BECs of human PBC [48]. Because 2OA-BSA immunized NOD.1101 mice did not demonstrate liver fibrosis, granulocyte infiltration in livers of this strain is compatible with that in early-stage livers of human PBC. Human BEC express and produce IL-8 and monocyte chemotactic protein (MCP)-1, which promote transendothelial migration of neutrophils when stimulated by proinflammatory cytokines such as IL-1 and TNF-α[50]. Also, TNF-α were expressed in the cytoplasm of BECs of damaged small bile duct and bile ductules in PBC [42]. Further, MCP-2 and MCP-3 expressing mononuclear cells infiltrated contiguous to damaged bile ducts of PBC livers [51]. Thus, these reports suggest a possible mechanism that TNF-α-producing cells, demonstrated in Fig. 3g, may promote IL-8, MCP-1 production in BECs, leading to enhanced granulocyte infiltration around portal tracts of 2OA-BSA immunized NOD.1101. Perhaps there is a distinct genetic element responsible for this granulocytic response in the NOD.1101 strain, but not in the B6 strain. Further studies should attempt identification of granulocytes and chemokines such as IL-8 and MCP-1 around portal tracts of this strain.
Our data support further the likelihood that genetic susceptibility determines an earlier occurrence of autoimmune cholangitis in mice after chemical xenobiotic exposure and reinforces the idea that loss of tolerance to mitochondrial antigens will lead inevitably to biliary damage. This is also substantiated by coincidentally ascertained AMA positivity in humans who progress subsequently after some years to overt PBC [52]. However, there is still the need to understand the unique immunobiology that renders the intrahepatic biliary ductular epithelial cells overly susceptible to damage following autoimmune responses to 2-OADC mitochondrial autoantigens, although we have suggested that preferential expression of these antigens on the apoptotic blebs of such cells renders them uniquely susceptible to destruction.
Acknowledgments
This work was supported by National Institute of Health grants DK39588, DK067003 and DK074768. L. S. W. is a Juvenile Diabetes Research Foundation/Wellcome Trust Principal Research Fellow.
References
- 1.Gershwin ME, Ansari AA, Mackay IR, et al. Primary biliary cirrhosis: an orchestrated immune response against epithelial cells. Immunol Rev. 2000;174:210–25. doi: 10.1034/j.1600-0528.2002.017402.x. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–73. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 3.Van de Water J, Fregeau D, Davis P, et al. Autoantibodies of primary biliary cirrhosis recognize dihydrolipoamide acetyltransferase and inhibit enzyme function. J Immunol. 1988;141:2321–4. [PubMed] [Google Scholar]
- 4.Leung PS, Chuang DT, Wynn RM, et al. Autoantibodies to BCOADC-E2 in patients with primary biliary cirrhosis recognize a conformational epitope. Hepatology. 1995;22:505–13. [PubMed] [Google Scholar]
- 5.Moteki S, Leung PS, Dickson ER, et al. Epitope mapping and reactivity of autoantibodies to the E2 component of 2-oxoglutarate dehydrogenase complex in primary biliary cirrhosis using recombinant 2-oxoglutarate dehydrogenase complex. Hepatology. 1996;23:436–44. doi: 10.1002/hep.510230307. [DOI] [PubMed] [Google Scholar]
- 6.Shimoda S, Van de Water J, Ansari A, et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest. 1998;102:1831–40. doi: 10.1172/JCI4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dubel L, Tanaka A, Leung PS, et al. Autoepitope mapping and reactivity of autoantibodies to the dihydrolipoamide dehydrogenase-binding protein (E3BP) and the glycine cleavage proteins in primary biliary cirrhosis. Hepatology. 1999;29:1013–8. doi: 10.1002/hep.510290403. [DOI] [PubMed] [Google Scholar]
- 8.Gershwin ME, Mackay IR. The causes of primary biliary cirrhosis: convenient and inconvenient truths. Hepatology. 2008;47:737–45. doi: 10.1002/hep.22042. [DOI] [PubMed] [Google Scholar]
- 9.Long SA, Quan C, Van de Water J, et al. Immunoreactivity of organic mimeotopes of the E2 component of pyruvate dehydrogenase: connecting xenobiotics with primary biliary cirrhosis. J Immunol. 2001;167:2956–63. doi: 10.4049/jimmunol.167.5.2956. [DOI] [PubMed] [Google Scholar]
- 10.Leung PS, Quan C, Park O, et al. Immunization with a xenobiotic 6-bromohexanoate bovine serum albumin conjugate induces antimitochondrial antibodies. J Immunol. 2003;170:5326–32. doi: 10.4049/jimmunol.170.10.5326. [DOI] [PubMed] [Google Scholar]
- 11.Leung PS, Matsumura S, Park O, et al. Induction of primary biliary cirrhosis in guinea pigs following immunization with a chemical xenobiotics. Hepatology. 2003;38:203A. doi: 10.4049/jimmunol.179.4.2651. [DOI] [PubMed] [Google Scholar]
- 12.Amano K, Leung PS, Xu Q, et al. Xenobiotic-induced loss of tolerance in rabbits to the mitochondrial autoantigen of primary biliary cirrhosis is reversible. J Immunol. 2004;172:6444–52. doi: 10.4049/jimmunol.172.10.6444. [DOI] [PubMed] [Google Scholar]
- 13.Leung PS, Park O, Tsuneyama K, et al. Induction of primary biliary cirrhosis in guinea pigs following chemical xenobiotic immunization. J Immunol. 2007;179:2651–7. doi: 10.4049/jimmunol.179.4.2651. [DOI] [PubMed] [Google Scholar]
- 14.Amano K, Leung PS, Rieger R, et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J Immunol. 2005;174:5874–83. doi: 10.4049/jimmunol.174.9.5874. [DOI] [PubMed] [Google Scholar]
- 15.Rieger R, Leung PS, Jeddeloh MR, et al. Identification of 2-nonynoic acid, a cosmetic component, as a potential trigger of primary biliary cirrhosis. J Autoimmun. 2006;27:7–16. doi: 10.1016/j.jaut.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Rieger R, Gershwin ME. The X and why of xenobiotics in primary biliary cirrhosis. J Autoimmun. 2007;28:76–84. doi: 10.1016/j.jaut.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Opdyke DL. Monographs on fragrance raw materials. Food Cosmet Toxicol. 1979;17:357–90. doi: 10.1016/0015-6264(79)90330-4. [DOI] [PubMed] [Google Scholar]
- 18.Wakabayashi K, Lian ZX, Leung PS, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology. 2008;48:531–40. doi: 10.1002/hep.22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wicker LS, Clark J, Fraser HI, et al. Type 1 diabetes genes and pathways shared by humans and NOD mice. J Autoimmun. 2005;25:29–33. doi: 10.1016/j.jaut.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 20.Irie J, Wu Y, Wicker LS, et al. NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. J Exp Med. 2006;203:1209–19. doi: 10.1084/jem.20051911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mattner J, Savage PB, Leung P, et al. Liver autoimmunity triggered by microbial activation of natural killer T cells. Cell Host Microbe. 2008;3:304–15. doi: 10.1016/j.chom.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Podolin PL, Denny P, Armitage N, et al. Localization of two insulin-dependent diabetes (Idd) genes to the Idd10 region on mouse chromosome 3. Mamm Genome. 1998;9:283–6. doi: 10.1007/s003359900749. [DOI] [PubMed] [Google Scholar]
- 23.Moteki S, Leung PS, Coppel RL, et al. Use of a designer triple expression hybrid clone for three different lipoyl domain for the detection of antimitochondrial autoantibodies. Hepatology. 1996;24:97–103. doi: 10.1002/hep.510240117. [DOI] [PubMed] [Google Scholar]
- 24.Kumada T, Tsuneyama K, Hatta H, Ishizawa S, Takano Y. Improved 1-h rapid immunostaining method using intermittent microwave irradiation: practicability based on 5 years application in Toyama Medical and Pharmaceutical University Hospital. Mod Pathol. 2004;17:1141–9. doi: 10.1038/modpathol.3800165. [DOI] [PubMed] [Google Scholar]
- 25.Prieto J, Qian C, Garcia N, Diez J, Medina JF. Abnormal expression of anion exchanger genes in primary biliary cirrhosis. Gastroenterology. 1993;105:572–8. doi: 10.1016/0016-5085(93)90735-u. [DOI] [PubMed] [Google Scholar]
- 26.Lan RY, Mackay IR, Gershwin ME. Regulatory T cells in the prevention of mucosal inflammatory diseases: patrolling the border. J Autoimmun. 2007;29:272–80. doi: 10.1016/j.jaut.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lan RY, Selmi C, Gershwin ME. The regulatory, inflammatory, and T cell programming roles of interleukin-2 (IL-2) J Autoimmun. 2008;31:7–12. doi: 10.1016/j.jaut.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Lleo A, Battezzati PM, Selmi C, Gershwin ME, Podda M. Is autoimmunity a matter of sex? Autoimmun Rev. 2008 doi: 10.1016/j.autrev.2008.06.009. in press. [DOI] [PubMed] [Google Scholar]
- 29.Lleo A, Invernizzi P, Selmi C, et al. Autophagy: highlighting a novel player in the autoimmunity scenario. J Autoimmun. 2007;29:61–8. doi: 10.1016/j.jaut.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lleo A, Selmi C, Invernizzi P, Podda M, Gershwin ME. The consequences of apoptosis in autoimmunity. J Autoimmun. 2008 doi: 10.1016/j.jaut.2008.04.009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koarada S, Wu Y, Fertig N, et al. Genetic control of autoimmunity: protection from diabetes, but spontaneous autoimmune biliary disease in a nonobese diabetic congenic strain. J Immunol. 2004;173:2315–23. doi: 10.4049/jimmunol.173.4.2315. [DOI] [PubMed] [Google Scholar]
- 32.Oertelt S, Lian ZX, Cheng CM, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J Immunol. 2006;177:1655–60. doi: 10.4049/jimmunol.177.3.1655. [DOI] [PubMed] [Google Scholar]
- 33.Wakabayashi K, Lian ZX, Moritoki Y, et al. IL-2 receptor alpha(-/-) mice and the development of primary biliary cirrhosis. Hepatology. 2006;44:1240–9. doi: 10.1002/hep.21385. [DOI] [PubMed] [Google Scholar]
- 34.Salas JT, Banales JM, Sarvide S, et al. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology. 2008;134:1482–93. doi: 10.1053/j.gastro.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 35.Selmi C, Mayo MJ, Bach N, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology. 2004;127:485–92. doi: 10.1053/j.gastro.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 36.Richardson B. Primer: epigenetics of autoimmunity. Nat Clin Pract Rheumatol. 2007;3:521–7. doi: 10.1038/ncprheum0573. [DOI] [PubMed] [Google Scholar]
- 37.Strickland FM, Richardson BC. Epigenetics in human autoimmunity. Epigenetics in autoimmunity – DNA methylation in systemic lupus erythematosus and beyond. Autoimmunity. 2008;41:278–86. doi: 10.1080/08916930802024616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waldner H, Sobel RA, Price N, Kuchroo VK. The autoimmune diabetes locus Idd9 regulates development of type 1 diabetes by affecting the homing of islet-specific T cells. J Immunol. 2006;176:5455–62. doi: 10.4049/jimmunol.176.9.5455. [DOI] [PubMed] [Google Scholar]
- 39.Morin J, Boitard C, Vallois D, Avner P, Rogner UC. Mapping of the murine type 1 diabetes locus Idd20 by genetic interaction. Mamm Genome. 2006;17:1105–12. doi: 10.1007/s00335-006-0076-9. [DOI] [PubMed] [Google Scholar]
- 40.Shindo M, Mullin GE, Braun-Elwert L, Bergasa NV, Jones EA, James SP. Cytokine mRNA expression in the liver of patients with primary biliary cirrhosis (PBC) and chronic hepatitis B (CHB) Clin Exp Immunol. 1996;105:254–9. doi: 10.1046/j.1365-2249.1996.d01-759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harada K, Van de Water J, Leung PS, et al. In situ nucleic acid hybridization of cytokines in primary biliary cirrhosis: predominance of the Th1 subset. Hepatology. 1997;25:791–6. doi: 10.1002/hep.510250402. [DOI] [PubMed] [Google Scholar]
- 42.Yasoshima M, Kono N, Sugawara H, Katayanagi K, Harada K, Nakanuma Y. Increased expression of interleukin-6 and tumor necrosis factor-alpha in pathologic biliary epithelial cells: in situ and culture study. Lab Invest. 1998;78:89–100. [PubMed] [Google Scholar]
- 43.Nagano T, Yamamoto K, Matsumoto S, et al. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol. 1999;19:422–7. doi: 10.1023/a:1020511002025. [DOI] [PubMed] [Google Scholar]
- 44.Shi Z, Wakil AE, Rockey DC. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc Natl Acad Sci USA. 1997;94:10663–8. doi: 10.1073/pnas.94.20.10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin HY, Sadelain MW, Hitchon C, Lauzon J, Singh B. Complete Freund's adjuvant-induced T cells prevent the development and adoptive transfer of diabetes in nonobese diabetic mice. J Immunol. 1993;150:2072–80. [PubMed] [Google Scholar]
- 46.Liddi R, Beales PE, Rosignoli G, Pozzilli P. Incomplete Freund's adjuvant reduces diabetes in the non-obese diabetic mouse. Horm Metab Res. 2000;32:201–6. doi: 10.1055/s-2007-978622. [DOI] [PubMed] [Google Scholar]
- 47.Serreze DV, Chapman HD, Post CM, Johnson EA, Suarez-Pinzon WL, Rabinovitch A. Th1 to Th2 cytokine shifts in nonobese diabetic mice: sometimes an outcome, rather than the cause, of diabetes resistance elicited by immunostimulation. J Immunol. 2001;166:1352–9. doi: 10.4049/jimmunol.166.2.1352. [DOI] [PubMed] [Google Scholar]
- 48.Wu CT, Eiserich JP, Ansari AA, et al. Myeloperoxidase-positive inflammatory cells participate in bile duct damage in primary biliary cirrhosis through nitric oxide-mediated reactions. Hepatology. 2003;38:1018–25. doi: 10.1053/jhep.2003.50407. [DOI] [PubMed] [Google Scholar]
- 49.Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis) Virchows Arch A Pathol Anat Histol. 1978;379:103–12. doi: 10.1007/BF00432479. [DOI] [PubMed] [Google Scholar]
- 50.Morland CM, Fear J, McNab G, Joplin R, Adams DH. Promotion of leukocyte transendothelial cell migration by chemokines derived from human biliary epithelial cells in vitro. Proc Assoc Am Physicians. 1997;109:372–82. [PubMed] [Google Scholar]
- 51.Tsuneyama K, Harada K, Yasoshima M, et al. Monocyte chemotactic protein-1, -2, and -3 are distinctively expressed in portal tracts and granulomata in primary biliary cirrhosis: implications for pathogenesis. J Pathol. 2001;193:102–9. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH725>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 52.Lazaridis KN, Juran BD, Boe GM, et al. Increased prevalence of antimitochondrial antibodies in first-degree relatives of patients with primary biliary cirrhosis. Hepatology. 2007;46:785–92. doi: 10.1002/hep.21749. [DOI] [PubMed] [Google Scholar]