

Abstract
Objective
Maintenance therapy with anti-CD25 antibody has emerged as a potentially useful treatment for multiple sclerosis (MS). Constitutive CD25 expression on CD4+CD25+ regulatory T cells (Treg) suggests that anti-CD25 antibody treatment may potentially target a subset of T cells that exhibit immune suppressive properties. We examined changes to CD4+CD25+ Treg in MS subjects receiving maintenance anti-CD25 monoclonal antibody treatment to determine the impact of treatment on Treg, and consequently on immunological tolerance.
Design
Peripheral blood and cerebrospinal fluid samples obtained from a before-after trial of anti-CD25 antibody monotherapy were examined to compare baseline and treatment differences in CD4+CD25+ Treg
Subjects
15 subjects with MS. One subject withdrawn due to adverse effect.
Results
Sustained reduction in the frequency of CD4+CD25+ Treg was observed during treatment. Anti-CD25 antibody treatment led to evidence of impaired in vivo Treg proliferation and impaired ex vivo Treg suppression. MS inflammatory activity was substantially reduced on treatment despite reduction in circulating Treg, and there was no correlation between changes in the frequency of Treg and changes in brain inflammatory activity. However, new onset inflammatory disease, notably dermatitis, was also observed in a number of subjects during treatment.
Conclusion
The reduction in Treg did not negatively impact maintenance of CNS tolerance during anti-CD25 antibody treatment. The incidence of new onset inflammatory disease outside of the CNS in a subset of patients, however, warrant further studies to examine the possibility of compartmental differences in the capacity to maintain tolerance in the setting of reduced CD4+CD25+ Treg.
Introduction
The anti-CD25 monoclonal antibody daclizumab targets the alpha subunit of the high-affinity interleukin-2 (IL-2) cytokine receptor complex. The up-regulation of CD25 following T cell activation and the subsequent IL-2 signaling constitutes a key event in T cell clonal expansion and differentiation. Abnormalities of IL-2/CD25 cytokine pathway have been reported in a number of immune-mediated diseases including multiple sclerosis (MS), and suggest that CD25 is potentially a target for MS immunotherapy. Increased soluble CD25 levels and abnormally high IL-2 responsiveness of autoreactive T cells in subjects with MS implicate an aberrant IL-2/CD25 circuit in the pathogenesis of MS, and constitute the rationale for anti-CD25 antibody treatment to modulate IL-2 signaling in MS 1, 2. A number of clinical studies are beginning to demonstrate the immunomodulatory effect of the anti-CD25 monoclonal antibody daclizumab in subjects with MS3, 4.
Experimental evidence of the past decade have made increasingly clear that a subset of CD25 expressing CD4+ T cells demonstrate suppressive or regulatory properties and contribute to the maintenance of immunological self-tolerance by their inhibitory influence on autoreactive T cells 5. These CD4+CD25+ regulatory T cells (Treg) are distinguished from conventional activated T cells by constitutive high expression of CD25 and by the expression of Treg lineage specification factor Foxp36, 7. Conventional activated T cells, by contrast, express intermediate levels of CD25 and lack Foxp36, 8. Whereas conventional activated T cells coordinate and amplify immune responses, CD4+CD25+ Treg actively suppress immune responses including those involved in autoimmunity9. The development of multi-organ inflammatory disease following Treg depletion indicates that CD4+CD25+ Treg make critical contribution to the maintenance of immunologic self-tolerance10. The loss or dysfunction of CD4+CD25+ Treg has been implicated in the pathogenesis of a growing number of disorders including systemic lupus erythematosis11, psoriasis12, aplastic anemia13 and MS14, suggesting a potentially broad relevance with respect to human autoimmune diseases.
The shared expression of CD25 on conventional activated T cells and CD4+CD25+ Treg suggest that both are potentially targeted by anti-CD25 antibody. Based on knowledge that CD4+CD25+ Treg contribute to maintenance of tolerance, an inhibitory effect on Treg could potentially exacerbate existing inflammatory disease or unmask underlying predilection for new inflammatory disease. We therefore examined the changes to the CD4+CD25+ T cell subsets in subjects with MS undergoing anti-CD25 antibody treatment. In particular, we asked what effect an anti-human CD25 antibody has on CD4+CD25+ Treg; whether changes to CD4+CD25+ Treg impacted the immunomodulatory effect of treatment; and whether changes to CD4+CD25+ Treg impacted maintenance of overall immunological tolerance.
Methods
Samples
Subjects with MS15 were enrolled in an open-label trial of anti-CD25 antibody (daclizumab). Subjects were free of immunomodulatory therapy for 24 weeks prior to enrollment, and received intravenous infusion of daclizumab monotherapy (1mg/kg) every 4 weeks for 54 weeks. Peripheral blood was obtained at baseline and during treatment. CSF was obtained at baseline and during treatment. Whole blood was processed immediately for fluorescence-activated cell sorting (FACS) analysis. Peripheral blood mononuclear cells (PBMC), available from 12 subjects, were processed by Ficoll-Hypaque density centrifugation and cryopreserved for later use. All treatment samples were obtained just prior to dosing (“trough sample”). Unless otherwise stated, baseline PBMC samples were compared to treatment PBMC samples obtained at “month 2.5” (trough sample following third dose). Brain MRI scans were obtained monthly as previously described3. Informed consent was obtained from each subject. The study was reviewed and approved by the National Institute of Neurological Disorders and Stroke Institutional Review Board.
Flow Cytometry
The following antibodies were used according to manufacturer's instructions. CD3, CD4, CD8, CD25 (M-A251), CD56, CD127, IL-2, Ki67, and pSTAT5 antibodies were obtained from BD Biosciences (San Jose, CA). Foxp3 PE or APC antibodies were obtained from eBioscience (San Diego, CA). CD25 (Anti-Tac) FITC was from Immunotech (Westbrook, ME). CD25 (7G7) PE was from (Ancell; Bayport, MN). FACS analysis of surface markers were performed on erythrocyte-lysed and washed whole blood samples. PBMC were used for all other analyses. Green Dead Stain (Invitrogen; Carlsbad, CA) was used for live/dead cell discrimination with Foxp3 staining. Flow cytometric data was acquired on FACSCalibur (BD Biosciences; San Jose, CA), and analyzed on FlowJo (TreeStar, Ashland, OR).
Treg suppression assay
To compare Treg suppression between baseline MS and healthy donors, CD4+ T cells were purified from PBMC by negative selection magnetic beads (Miltenyi, Auburn, CA). Purity was above 95%. CD25-PE labeled CD4+ T cells were then sorted (FACS diva, BD Biosciences; San Jose, CA) into CD4+CD25high Treg (highest 3% CD25 expression) and CD4+CD25- responder cells. Treg coculture suppression assay was performed as previously described8 with minor modifications. Soluble anti-CD3 (HIT3a, 0.1μg/ml, BD Pharmingen) was used for stimulation. Irradiated (3000 rad) T cell depleted (MACS cell separation / anti-CD3 microbeads, Miltenyi) autologous PBMC was used as accessory cells. Cells were plated in triplicate into 96-well plates, incubated at 37°C for 5 days, and pulsed with [3H] thymidine for final 18 hrs of incubation. Percent suppression was calculated as: %suppression = (1- (cpm of coculture well / cpm of responder well)) x100, where coculture well ratio was 1:1 (responder: Treg). To compare baseline and treatment Treg suppression, CD4+ T cells were purified from viably cryopreserved PBMC by negative selection magnetic beads. Purified CD4+ T cells were labeled with CD127-PE and CD25-PE/Cy5 antibodies. CD4+CD25+CD127low/neg (Treg) and CD4+CD25-CD127+ (responder) cells were FACS sorted and used for Treg coculture suppression assay as described above.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections were prepared on poly-L-lysine coated slides. Immunohistochemistry for CD3 (DAKO) and Foxp3 (Abcam) was carried out on consecutive tissue sections and developed with 3,3'-diaminobenzidine, tetrahydrochloride chromogen. A semi-quantitative assessment of CD3 and Foxp3 expression was carried out by counting the number of positively stained lymphocytes under 40x objective microscopy lens.
Statistical Analysis
Statistical significance was determined by unpaired t-test to compare subjects with MS and healthy donors, and by paired t-test to compare baseline and treatment values. Where appropriate, comparisons were made using general linear model repeated measures analysis of variance. Pearson correlation coefficients were used to analyze relationships between parameters.
Results
Baseline CD4+CD25+ Treg characteristics in MS subjects
To establish pre-treatment characteristics of CD4+CD25+ Treg in this cohort of MS subjects, Treg suppression was measured by an in vitro Treg coculture assay8 (Figure 1A). The majority of subjects in this cohort demonstrated baseline Treg suppression within the range for healthy donors (p = 0.266; Figure 1B). Likewise, the frequency of circulating CD4+Foxp3+ Treg in this cohort of MS subjects did not differ significantly at baseline compared to that of age, gender and race matched healthy donors (p = 0.372; Figure 1C). Furthermore, as in healthy donors, Foxp3+ cells from subjects with MS were characterized by negative CD69 and low or negative CD127 expression16, 17, and demonstrated attenuation of IL-2 production6, 18 (Figure 1D).
Figure 1. Baseline Treg characteristics in subjects with multiple sclerosis.

A Representative Treg coculture assay [3H] thymidine incorporation data (mean cpm ± standard deviation) showing dose dependent Treg suppression for a subject with MS (86% suppression). Up to 5×103 FACS sorted CD4+CD25high (Treg) cells were titrated in coculture with 5×103 CD4+CD25- (responder) cells for 5-day stimulation with anti-CD3 antibody as described in Methods. B Baseline Treg suppression in subjects with multiple sclerosis “MS” (mean 69.7±18%) compared to healthy donors “HD” (mean 79.7 ± 13%; p = 0.266). C Frequency of peripheral blood CD4+Foxp3+ cells from subjects with MS (mean 2.7 ± 1.6% of lymphocytes) compared to age, race and gender-matched healthy donors (mean 3.2 ± 1.0% of lymphocytes; p = 0.372). D Representative CD4+ gated FACS plots from a subject with MS showing negative CD69 and low or negative CD127 expression in Foxp3+ cells (data representative of 6 MS and 3 healthy donors), and attenuation of IL-2 production following 6 hr PMA/ionomycin stimulation (data representative of 4 MS and 3 healthy donors).
Foxp3+ regulatory T cells were reduced during anti-CD25 antibody treatment
Antibody saturation was monitored during the course of anti-CD25 antibody treatment by flow cytometry using two fluorochrome-labeled antibodies (anti-Tac and 7G7) that bind non-competing epitopes on CD25. Complete antibody saturation of CD25, demonstrated by the absence of fluorochrome-labeled anti-Tac binding, was maintained during the course of treatment (Figure 2A, anti-Tac), whereas reduction in the mean total CD25 expression on lymphocytes was less pronounced (13%) but nevertheless statistically significant (p<0.0001; Figure 2A, 7G7). Examination of CSF demonstrated complete antibody saturation of CD25 on CSF lymphocytes and a 20% decline in mean total CD25 expression during treatment (p=0.0384; Figure 2B).
Figure 2. Reduction in Treg during anti-CD25 antibody treatment.

A Anti-CD25 antibody saturation and total CD25 expression on lymphocytes over time. Antibody saturation and CD25 expression were evaluated by FACS analysis of whole blood using two fluorochrome-labeled antibodies (anti-Tac and 7G7) directed at non-competing epitopes on CD25. Complete antibody saturation of CD25 was demonstrated by lack of fluorochrome-labeled anti-Tac binding. Total CD25 expression (7G7) declined by 13% (p<0.0001 repeated measures ANOVA; n = 12) Months 0 and 0.5 represent the first two infusions. Monthly administration followed thereafter with month13.5 representing the final infusion. B Antibody saturation (anti-Tac) and CD25 expression (7G7) in CSF lymphocytes over time (p = 0.0384; n = 10). C Histograms show representative STAT5 phosphorylation in PBMC obtained during anti-CD25 antibody treatment (bold line) compared to baseline (thin line) and control (shaded) following 15 minute ex vivo IL-2 exposure. Box plot compares STAT5 phosphorylation during treatment (shaded box) compared to baseline (empty box) (p = 0.0050; n = 7). D Representative CD4+ gated FACS analysis of CD4+CD25+Foxp3- conventional activated T cells (upper left quadrants) and CD4+CD25+Foxp3+ Treg (upper right quadrants) at baseline and during treatment. E Reduced Foxp3 MFI (mean fluorescence intensity) during treatment (shaded box) compared to baseline (empty box; p = 0.0007; n = 12). F Reduction in frequency of CD4+Foxp3+ cells (% lymphocyte) over time during the course of anti-CD25 antibody treatment (p<0.0001 repeated measures ANOVA; n = 12). Dashed line indicates end of treatment
IL-2 signaling was inhibited by anti-CD25 antibody treatment. STAT5 (signal transduction and activator of transcription 5) phosphorylation, which mediates downstream IL-2 signaling, was used as a marker of IL-2 signaling. Lymphocytes obtained during treatment demonstrated nearly complete absence of STAT5 phosphorylation in response to low level (10 U/ml) IL-2. Significant reductions of STAT5 phosphorylation were also observed at higher levels (50 and 100 U/ml) of IL-2 (p=0.0050; Figure 2C).
The effect of anti-CD25 antibody treatment on CD4+CD25+ Treg was analyzed by examining Foxp3 as a marker of Treg. Flow cytometric analysis (Figure 2D) demonstrated a reduction in mean fluorescence intensity of Foxp3 expression during treatment compared to baseline (p=0.0007; Figure 2E). Reduction in Foxp3 expression at the single-cell level during treatment is consistent with previous studies implicating STAT5 as a regulator of Foxp3 gene transcription19. Furthermore, the frequency of total Foxp3 expressing CD4+ cells were reduced, with approximately 30% reduction in mean frequency of CD4+Foxp3+ cells observed by month 2.5, and 44% reduction by month 7.5 (p<0.0001; Figure 2F). Similar reductions were observed in the frequency of CD4+CD25+Foxp3+ cells (45% reduction; p<0.0001). Post-treatment samples available from a limited number of subjects demonstrated recovery of Treg frequencies to near baseline levels.
Anti-CD25 antibody treatment led to reduced Treg proliferative capacity and impaired Treg suppression
Based on the known role of IL-2 in promoting cell cycle progression in conventional T cells20, we asked whether altered homeostatic proliferation of Treg could account for the reduction in frequency of Treg during treatment. The effect of anti-CD25 antibody treatment on Treg proliferation was examined using Ki67 expression to estimate the in vivo proliferating fraction21, determined as the proportion of CD4+Foxp3+ cells expressing Ki67. PBMC from baseline and treatment were stained ex vivo for intracellular expression of Ki67. Consistent with a previous report demonstrating high in vivo proliferative kinetics of human Treg22, CD4+Foxp3+ cells demonstrated high Ki67 expression at baseline compared to total CD4+ cells (mean 11.7 +/- 2.2% vs. 2.13 +/- 0.8%). Analysis of treatment samples showed a reduction in the Ki67 expressing proliferating Treg fraction (p=0.0009; Figure 3A), suggesting impaired homeostatic proliferation of Treg during anti-CD25 antibody treatment.
Figure 3. Treg in vivo proliferation and ex vivo suppression are impaired during anti-CD25 antibody treatment.

A Representative FACS analysis comparing Ki67 expression in Foxp3+ cells at baseline and during anti-CD25 antibody treatment (% Ki67 expressing fraction in parentheses). Bar graph compares proportion of CD4+Foxp3+ cells expressing Ki67 (mean +/- standard deviation) at baseline and during treatment (p = 0.0010; n = 12). B Representative CD4+ gated FACS analysis showing that majority of CD4+Foxp3+ cells (blue) are CD25+CD127low/neg (polygonal gate) at baseline and remain CD25+CD127low/neg during treatment. Numbers indicate the frequency of cells that are Foxp3+ within the polygonal gate and, in parentheses, the frequency of total CD4+Foxp3+ cells (% lymphocytes). C Representative [3H] Thymidine incorporation data (mean cpm ± standard deviation) shown for up to 5×103 FACS sorted Treg (CD4+CD25+CD127low/neg) titrated in coculture with 5×103 responder (CD4+CD25-CD127+) cells. Closed circles represent cells from baseline and open circles represent cells obtained during anti-CD25 antibody treatment. D Reduced suppressive capacity (mean % suppression +/- standard deviation) of circulating Treg during anti-CD25 antibody treatment (p = 0.0284; n = 4).
To assess Treg function, coculture suppression assays were performed to compare Treg suppression at baseline and during treatment. Because of altered CD25 expression during treatment, an additional surface marker CD127 was utilized to sort Treg, which express low or no CD127 (CD127low/neg)17. FACS analysis demonstrated that majority of CD4+Foxp3+ cells were contained within the CD25+CD127low/neg subset at baseline and during treatment (Figure 3B). To determine the effect of anti-CD25 antibody treatment on Treg suppressive capacity, CD4+CD25+CD127low/neg cells were FACS sorted from PBMC obtained at baseline and during treatment, and cocultured with autologous CD4+CD25-CD127+ (responder) cells. Treatment samples demonstrated impaired Treg suppression compared to baseline samples (p=0.0284, Figure 3C&D), indicating a functional impairment of ex vivo Treg suppression during anti-CD25 antibody treatment.
Reduction in circulating CD4+CD25+ Treg did not negatively impact acute CNS inflammation
In addition to changes in Treg, anti-CD25 antibody treatment led to significant alteration in conventional activated CD4+CD25+ T cells and CD56bright natural killer (NK) cells. The frequency of conventional activated T cells (CD4+CD25+Foxp3-) was reduced during anti-CD25 antibody treatment (p<0.0001), which corresponded to the contraction of Ki67+ proliferating fraction of conventional activated T cells. In contrast, the proportion of proliferating CD56+ natural killer (NK) cells was increased during anti-CD25 antibody treatment, and corresponded to the expansion of CD56bright NK cells observed during the course of therapy (p=0.0006).
In the setting of simultaneous changes to activated conventional T cell and NK cell compartments, Treg were not necessarily the major determinants of acute CNS inflammation. Contrast (Gd-DTPA) enhancement of MS lesions on brain MRI, a marker of MS inflammatory activity, was assessed on a monthly basis. The number of MS lesions demonstrating Gd-DTPA enhancement (Gd-DTPA+ lesions) was significantly reduced during treatment with anti-CD25 antibody (p=0.0006; Figure 4A). To assess whether the reduction in Treg had any negative impact on the immunomodulatory effect of anti-CD25 antibody, we analyzed the relationship between changes in the frequency of Treg and brain inflammatory activity. No significant correlations were observed between changes in the frequency of Treg and changes in brain inflammatory activity measured as the total number of Gd-DTPA+ lesions per month (r2 = 0.0171; p=0.685; Figure 4B) or the number of new Gd-DTPA+ lesions per month (r2 = 0.0157; p=0.713) when assessed at month 7.5. Analysis of earlier and later time points (months 2.5 & 12.5) yielded similar results (r2 = 0.068 and r2 = 0.0391 respectively for correlation between change in total number of Gd-DTPA+ and Treg).
Figure 4. Lack of correlation between reduction in the frequency of Treg and changes in acute CNS inflammatory activity.

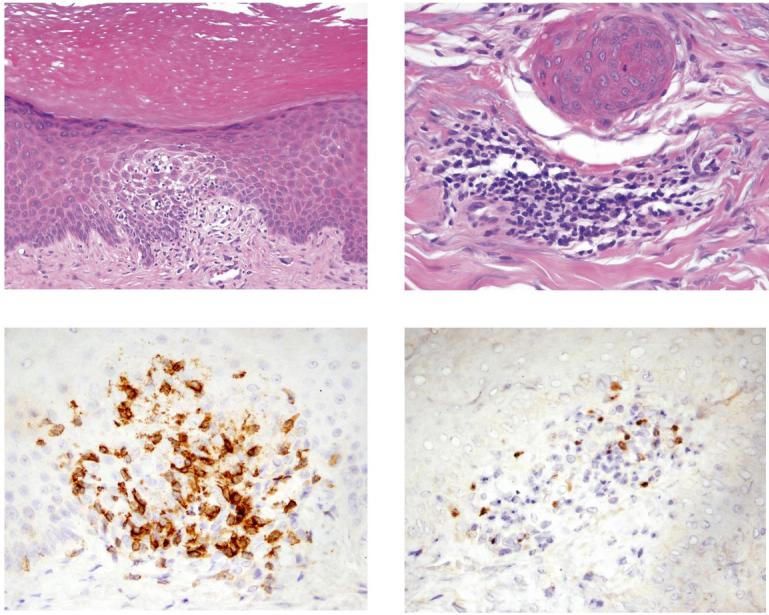
A Reduction in brain inflammatory activity indicated by reduction in the total number of contrast enhancing MS lesions per month (total # of Gd-DTPA lesions) on serial brain magnetic resonance images (p = 0.0006; n = 13). B Regression analysis of correlation between change in total number of contrast enhancing lesions per month (% reduction in Gd-DTPA+ lesions) and change in the frequency of circulating Treg (% reduction in # of Treg) at month 7.5 during anti-CD25 antibody treatment (r2 = 0.0171; p=0.685). Pearson correlation coefficient. Analysis of Treg in lesional skin. C Hematoxylin and Eosin (H&E) stained tissue section (subject MS9) showing epidermal changes of the skin lesion, characterized by compact hyperkeratosis, acanthosis and focal spongiosis with exocytosis of lymphocytes (20x). D H&E stained section showing histologic changes in the superficial dermis, characterized by a perivascular chronic inflammatory infiltrate comprised predominantly of lymphocytes. E Immunohistochemical staining for CD3. F Immunohistochemical staining for Foxp3.
Adverse events during anti-CD25 antibody treatment included new onset inflammatory dermatitis
Dermatitis occurred in 3 out of 15 individuals on anti-CD25 antibody treatment (Table). Onset of dermatitis occurred during anti-CD25 antibody treatment in two subjects, and at the end of treatment in one subject who nevertheless still demonstrated >80% saturation of CD25 at the onset of dermatitis. An additional subject with a family history of rheumatoid arthritis developed palindromic rheumatism during treatment. Reductions in the frequencies of Foxp3+ cells for subjects who developed dermatitis are shown in Table. Histologic examination of lesional skin from two subjects who developed dermatitis during treatment showed spongiotic to psoriasiform epidermal changes with perivascular lymphocytic inflammatory infiltrate in the subjacent superficial dermis (Figure 4C&D). In situ quantitative detection of Foxp3 in the lesional skin showed that approximately 13% of infiltrating CD3+ cells were Treg (Figure 4E&F).
New onset inflammatory disorders during anti-CD25 antibody treatment
| Subject ID | Onset | Clinical Diagnosis | Treatment | % Reduction in CD4+Foxp3+ cellsA |
|---|---|---|---|---|
| MS 1 | Month 16.5 | Photodermatitis | Topical corticosteroid | 56% |
| MS 8 | Month 2.5 | Palindromic rheumatism | Discontinue daclizumab | 46% |
| MS 9 | Month 5.5 | Sebopsoriasis | Selenium sulfide | 57% |
| MS 10 | Month 11.5 | Sebosporiasis | Topical corticosteroid | N/A |
Based on frequency of Foxp3+ cells (% CD4) at baseline and month 7.5. Mean % reduction = 42.5, median % reduction = 44.1% and range = 20.6 to 61.8% for this cohort. N/A = not available.
Discussion
We demonstrate that long-term maintenance anti-CD25 antibody treatment led to sustained reduction of CD4+CD25+ Treg in subjects with MS. In contrast to the hypoproliferative nature of Treg in culture, we and others22 find evidence that Treg exhibit high replicative capacity in vivo. Reduced proliferating Treg fraction corresponds to decline in Treg numbers in the setting of impaired IL-2 signaling, and suggest that IL-2-supported Treg proliferation accounts for a substantial portion of the human circulating Treg pool. Clinical trials in cancer patients demonstrated up-regulation of Foxp3 and increased frequency of Treg following administration of IL-223, 24. Our data demonstrates, conversely, that negative perturbation of IL-2 signaling reduces Foxp3 expression and reduces the circulating Treg pool. Collectively, these studies indicate that IL-2 plays a major role in controlling the homeostatic set-point for the size of human circulating Treg pool.
We asked whether reduction in circulating Treg during anti-CD25 antibody treatment negatively impacted MS inflammatory activity. Overall brain inflammatory activity was reduced during treatment, suggesting a shift towards tolerance. Lack of a correlation between changes in frequency of Treg and changes in brain inflammatory activity suggests that sustained reduction in circulating Treg did not negatively impact disease activity. One likely explanation is that simultaneous changes in other cell subsets during anti-CD25 antibody treatment countered any negative impact of reduced Treg. Anti-CD25 antibody treatment led to a contraction of the CD4+CD25+ activated conventional T cell fraction and an expansion of CD56bright NK cells. A previous study demonstrated the capacity of CD56bright NK cells to suppress inflammation; expansion of CD56bright NK population is potentially a major determinant of acute brain inflammatory activity during anti-CD25 antibody treatment25. Alternatively, the lack of correlation between changes in circulating Treg and changes in brain inflammatory activity suggests the possibility that Treg are not a major determinant of acute inflammatory activity in MS. Data from experimental autoimmune encephalomyelitis (EAE), an animal model of MS, have not yet reconciled what role CD4+CD25+ Treg play in modulating acute CNS inflammation. Loss of Treg appears to confer susceptibility to EAE in an otherwise resistant strain of mice26, but CNS antigen-specific Treg failed to inhibit CNS effector T cells during the acute phase of EAE, possibly due to the in situ cytokine milieu, particularly IL-6, that renders effector T cells resistant to Treg suppression27.
Dermatitis occurred in 3 out of the 15 subject cohort on treatment. An additional subject with a family history of rheumatoid arthritis developed migratory tenosynovitis during treatment, diagnosed as palindromic rheumatism. The incidence of new onset inflammatory disease during anti-CD25 antibody treatment raised the possibility that there may be compartmental differences in the capacity to maintain tolerance in the setting of reduced circulating Treg. A recent study showed that CD4+CD25+ Treg contribute to routine immune surveillance and inflammatory response in the human skin28. Furthermore, the availability of circulating Treg capable of migrating into the skin was shown to be critical to the maintenance of skin-specific tolerance in an animal model, suggesting that the skin may be particularly vulnerable to reduction in circulating Treg29. Histologic findings from skin biopsies taken from our subjects were relatively non-specific, but not inconsistent with what has been described in the Foxp3 deficiency syndrome IPEX30. In situ quantitative detection of Foxp3 in the lesional skin showed that approximately 13% of infiltrating CD3+ cells were Treg, which represents a lower frequency of Treg at the site of skin inflammation compared to historical controls31. Reduction in the frequency of Treg were relatively high (above the cohort mean/median) in subjects who developed new onset inflammatory disease, but did not reach statistical significance compared to those who did not develop new inflammatory disease on treatment. The relationship between reduction in Treg and new onset inflammatory disease during anti-CD25 antibody treatment, though suggestive, is inconclusive, and further work is required to determine whether there are compartmental differences in requirements for CD4+CD25+ Treg to maintain organ-specific tolerance.
A functional defect of CD4+CD25+ Treg cells has been reported in subjects with MS14, 32. We found no significant difference in mean Treg suppression between our cohort of MS subjects and healthy donors, suggesting either that a functional defect of Treg may not be a uniform finding in all subjects with MS or that the differences are of a scale that requires a larger cohort to adequately power such comparisons. Studies comparing Treg suppression in MS subjects and healthy volunteers suggest that the differences may be age-dependent33 or disease stage-dependent34.
The collective clinical experience with anti-CD25 antibody treatment constitutes a large body of data that demonstrates its safety and efficacy as induction therapy in the prevention of allograft rejection35 and suggest its utility in an array of human disorders36. The effect of short-term anti-CD25 antibody induction therapy on Treg are likely transient or modified by concomitant use of immunosuppressive agents37. We now demonstrate that a consequence of long-term maintenance monotherapy with anti-CD25 antibody is a sustained reduction of CD4+CD25+ Treg. Maintenance therapy with anti-CD25 antibody is likely to be a valuable therapeutic option in a number of immune-mediated inflammatory diseases3, 38. Our findings underscore the need to clarify the organ-specific consequences of sustained reduction in human CD4+CD25+ Treg.
Acknowledgement
We thank Joan Ohayon for clinical assistance, and Azita Kashani for technical assistance. This research was supported by the Intramural Research Program of the NIH, NINDS. Unsong Oh had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Disclosure: Dr. McFarland is a coinventor on NIH patents related to the use of daclizumab in multiple sclerosis and as such received patent royalty payments.
References
- 1.Waldmann TA. The IL-2/IL-2 receptor system: a target for rational immune intervention. Immunol Today. 1993;14:264–270. doi: 10.1016/0167-5699(93)90043-K. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Markovic-Plese S, Lacet B, et al. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med. 1994;179:973–984. doi: 10.1084/jem.179.3.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bielekova B, Richert N, Howard T, et al. Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon beta. Proc Natl Acad Sci U S A. 2004;101:8705–8708. doi: 10.1073/pnas.0402653101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rose JW, Burns JB, Bjorklund J, et al. Daclizumab phase II trial in relapsing and remitting multiple sclerosis: MRI and clinical results. Neurology. 2007;69:785–789. doi: 10.1212/01.wnl.0000267662.41734.1f. [DOI] [PubMed] [Google Scholar]
- 5.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 6.Yagi H, Nomura T, Nakamura K, et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:1643–1656. doi: 10.1093/intimm/dxh165. [DOI] [PubMed] [Google Scholar]
- 7.Fontenot JD, Rasmussen JP, Williams LM, et al. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 8.Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–1253. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- 9.Shevach EM. Certified professionals: CD4(+)CD25(+) suppressor T cells. J Exp Med. 2001;193:F41–46. doi: 10.1084/jem.193.11.f41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 11.Valencia X, Yarboro C, Illei G, Lipsky PE. Deficient CD4+CD25high T regulatory cell function in patients with active systemic lupus erythematosus. J Immunol. 2007;178:2579–2588. doi: 10.4049/jimmunol.178.4.2579. [DOI] [PubMed] [Google Scholar]
- 12.Sugiyama H, Gyulai R, Toichi E, et al. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol. 2005;174:164–173. doi: 10.4049/jimmunol.174.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solomou EE, Rezvani K, Mielke S, et al. Deficient CD4+ CD25+ FOXP3+ T regulatory cells in acquired aplastic anemia. Blood. 2007;110:1603–1606. doi: 10.1182/blood-2007-01-066258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50:121–127. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- 16.Seddiki N, Santner-Nanan B, Martinson J, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203:1693–1700. doi: 10.1084/jem.20060468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006;203:1701–1711. doi: 10.1084/jem.20060772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bettelli E, Dastrange M, Oukka M. Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc Natl Acad Sci U S A. 2005;102:5138–5143. doi: 10.1073/pnas.0501675102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao Z, Kanno Y, Kerenyi M, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp3. Blood. 2007;109:4368–4375. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nourse J, Firpo E, Flanagan WM, et al. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature. 1994;372:570–573. doi: 10.1038/372570a0. [DOI] [PubMed] [Google Scholar]
- 21.Gerdes J, Lemke H, Baisch H, et al. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133:1710–1715. [PubMed] [Google Scholar]
- 22.Vukmanovic-Stejic M, Zhang Y, Cook JE, et al. Human CD4+ CD25hi Foxp3+ regulatory T cells are derived by rapid turnover of memory populations in vivo. J Clin Invest. 2006;116:2423–2433. doi: 10.1172/JCI28941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zorn E, Nelson EA, Mohseni M, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571–1579. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmadzadeh M, Rosenberg SA. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood. 2006;107:2409–2414. doi: 10.1182/blood-2005-06-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bielekova B, Catalfamo M, Reichert-Scrivner S, et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A. 2006;103:5941–5946. doi: 10.1073/pnas.0601335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reddy J, Illes Z, Zhang X, et al. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2004;101:15434–15439. doi: 10.1073/pnas.0404444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korn T, Reddy J, Gao W, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirahara K, Liu L, Clark RA, et al. The majority of human peripheral blood CD4+CD25highFoxp3+ regulatory T cells bear functional skin-homing receptors. J Immunol. 2006;177:4488–4494. doi: 10.4049/jimmunol.177.7.4488. [DOI] [PubMed] [Google Scholar]
- 29.Sather BD, Treuting P, Perdue N, et al. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med. 2007;204:1335–1347. doi: 10.1084/jem.20070081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nieves DS, Phipps RP, Pollock SJ, et al. Dermatologic and immunologic findings in the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Arch Dermatol. 2004;140:466–472. doi: 10.1001/archderm.140.4.466. [DOI] [PubMed] [Google Scholar]
- 31.de Boer OJ, van der Loos CM, Teeling P, et al. Immunohistochemical analysis of regulatory T cell markers FOXP3 and GITR on CD4+CD25+ T cells in normal skin and inflammatory dermatoses. J Histochem Cytochem. 2007;55:891–898. doi: 10.1369/jhc.6A7119.2007. [DOI] [PubMed] [Google Scholar]
- 32.Haas J, Hug A, Viehover A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. 2005;35:3343–3352. doi: 10.1002/eji.200526065. [DOI] [PubMed] [Google Scholar]
- 33.Haas J, Fritzsching B, Trubswetter P, et al. Prevalence of newly generated naive regulatory T cells (Treg) is critical for Treg suppressive function and determines Treg dysfunction in multiple sclerosis. J Immunol. 2007;179:1322–1330. doi: 10.4049/jimmunol.179.2.1322. [DOI] [PubMed] [Google Scholar]
- 34.Venken K, Hellings N, Hensen K, et al. Secondary progressive in contrast to relapsing-remitting multiple sclerosis patients show a normal CD4+CD25+ regulatory T-cell function and FOXP3 expression. J Neurosci Res. 2006;83:1432–1446. doi: 10.1002/jnr.20852. [DOI] [PubMed] [Google Scholar]
- 35.Vincenti F, Kirkman R, Light S, et al. Interleukin-2-receptor blockade with daclizumab to prevent acute rejection in renal transplantation. Daclizumab Triple Therapy Study Group. N Engl J Med. 1998;338:161–165. doi: 10.1056/NEJM199801153380304. [DOI] [PubMed] [Google Scholar]
- 36.Waldmann TA. Anti-Tac (daclizumab, Zenapax) in the treatment of leukemia, autoimmune diseases, and in the prevention of allograft rejection: a 25-year personal odyssey. J Clin Immunol. 2007;27:1–18. doi: 10.1007/s10875-006-9060-0. [DOI] [PubMed] [Google Scholar]
- 37.Kreijveld E, Koenen HJ, Klasen IS, et al. Following anti-CD25 treatment, a functional CD4+CD25+ regulatory T-cell pool is present in renal transplant recipients. Am J Transplant. 2007;7:249–255. doi: 10.1111/j.1600-6143.2006.01604.x. [DOI] [PubMed] [Google Scholar]
- 38.Nussenblatt RB, Thompson DJ, Li Z, et al. Humanized anti-interleukin-2 (IL-2) receptor alpha therapy: long-term results in uveitis patients and preliminary safety and activity data for establishing parameters for subcutaneous administration. J Autoimmun. 2003;21:283–293. doi: 10.1016/s0896-8411(03)00113-6. [DOI] [PubMed] [Google Scholar]