

SYNOPSIS
Vertebrate gap junctions, composed of proteins from the connexin gene family, play critical roles in embryonic development, coordinated contraction of excitable cells, tissue homeostasis, normal cell growth and differentiation. Phosphorylation of connexin43, the most abundant and ubiquitously expressed connexin, has been implicated in the regulation of gap junctional communication at several stages of the connexin “life cycle” including hemichannel oligomerization, export of the protein to the plasma membrane, hemichannel activity, gap junction assembly, gap junction channel gating and connexin degradation. Consistent with a short (1−5 h) protein half-life, connexin43 phosphorylation is dynamic and changes in response to activation of many different kinases. This review assesses our current understanding of the effects of phosphorylation on connexin43 structure and function that in turn regulate gap junction biology with an emphasis on events occurring in heart and skin.
Keywords: Connexin, Gap junction, Phosphorylation, Kinase, Phosphatase, Cell Signaling, Post translational modifications
INTRODUCTION
Vertebrate gap junctions are composed of integral membrane proteins from the connexin gene family (abbreviated as Cx followed by the mass of the protein in kilodaltons, e.g., Cx43). Gap junction-mediated intercellular communication facilitates direct communication among adjacent cells by allowing passage of ions and small metabolites [1-4]. Embryonic development, coordinated contraction of excitable cells, tissue homeostasis, controlled cell growth and differentiation are all regulated by connexins [2, 3, 5]. Connexin gene mutations have been linked to several diseases [5-8] including oculodentodigital dysplasia, a disease due to connexin43 (Cx43) mutations that can result in small eyes, underdeveloped teeth, syndactyly, atrioseptal defects and arrhythmias [9]. Extensive evidence also indicates that gap junctions play important roles in the control of cell growth and inhibit tumor formation [10].
Connexins are highly regulated integral membrane proteins that contain 4 transmembrane domains, two extracellular loops containing 6 conserved cysteine residues, a cytoplasmic loop and cytoplasmic N- and C-termini [2, 3, 5]. The C-terminal domain varies widely in length and is thought to play key regulatory roles and provide for sites of protein-protein interaction. Connexin export to the plasma membrane, assembly into gap junctions, gating and degradation all appear to be regulated via post-translational modification and interaction with other cellular proteins. Many connexins (i.e., Cx31, Cx32, Cx36, Cx37, Cx40, Cx43, Cx45, Cx46, Cx50, and Cx56) are phosphoproteins as shown by either a phosphatase-sensitive shift in their electrophoretic mobility, direct incorporation of [32P]-phosphate or mass spectrometry [11-13]. Cx43 is the most widely expressed connexin being present in at least 34 tissues and 46 cell types [5], and it is the predominant connexin expressed in most cell lines, so more is known about phosphorylation of Cx43 than for the other connexins. In Cx43, the C-terminal domain appears to be the primary region that becomes phosphorylated, but Cx36 and Cx56 can also be phosphorylated within the cytoplasmic loop [14]. Cx43 does not contain serine residues in its cytoplasmic loop, and we are not aware of any reports of phosphorylation of the N-terminal domain of any connexin. Activation of several kinases can lead to increased Cx43 phosphorylation including protein kinase A (PKA) [15-18], protein kinase C (PKC) [19-23], p34cdc2/cyclin B kinase (p34cdc2) [24-26], casein kinase 1 (CK1) [27], mitogen-activated protein kinase (MAPK) [28-33] and pp60src kinase (src) [34-37].
CONNEXIN BIOCHEMISTRY
Formation and degradation of gap junctions are dynamic processes with reports of connexin half-lives being less than 5 hours in cultured cells and in tissues [22, 37-40]. Therefore, regulation of gap junction assembly and turnover is likely to be critical in the control of intercellular communication [5]. Musil and Goodenough, Lau and collaborators, and several other investigators have shown that Cx43 is differentially phosphorylated [19, 21, 37, 38, 40-43]. Cx43 has multiple electrophoretic isoforms when analyzed by SDS-PAGE, including a faster migrating form that includes non-phosphorylated (P0 or NP) Cx43, and at least two slower migrating forms, commonly termed P1 and P2 (Figure 1A, CON lane). In different cell types, the profile of band migration can vary, in some cases due to differences in gap junction assembly [42]. Both P1 and P2 co-migrate with P0 following alkaline phosphatase treatment, suggesting that phosphorylation is the primary covalent modification reflected in their differences in electrophoretic mobility in SDS-PAGE analysis [42]. Similarly, HeLa cells expressing Cx45 show a 46kDa and 48kDa band by Western blot, that like Cx43, comigrate at 46kDa after alkaline phosphatase treatment [44]. Pulse chase experiments indicate that newly synthesized Cx43 migrates at P0 and then matures to P1 followed by P2 [42]. No direct evidence indicates that P2 is more phosphorylated than P1 [42], and though this has known for many years, only recently have specific phosphorylation sites been linked to specific migration changes, in addition to the finding that some phosphorylated species migrate with the P0 band in SDS-PAGE [45].
Figure 1. Phosphorylation and assembly of Cx43 containing gap junctions.

(A) Association of the SDS-PAGE migration of Cx43 in homeostatic (CON) and TPA treated cells with the sites of phosphorylation and a schematic diagram representing channel activity. The cylindrical channels are green to represent promoting communication and red to represent inhibition or closure. The green cross hatch represents an assembling channel and the red a channel with reduced conductance. (B) Gap junction assembly is denoted by cross hatch green channels assembling into solid green communicating channels and red channels represent those that will be internalized and degraded.
It is evident that these apparent 2−4 kDa or more shifts in connexin migration in SDS-PAGE are not simply due to the addition of the mass of phosphate (80 Da). Instead, there are specific phosphorylation events that induce conformational changes that are being detected. Indeed, recent reports utilizing phosphospecific antibodies have shown that phosphorylation at S365 is necessary for the shift to the P1 isoform [46]. This P1 isoform was found primarily in the plasma membrane and in a subset of gap junction plaques. Similarly, phosphorylation at S325/328/330 is involved in the shift to P2 and this isoform was found exclusively in gap junctions [47]. Additional phosphorylation events could be involved in these and other migration shifts, some of which will be discussed below.
KINASES IN THE CONNEXIN LIFECYCLE
Like most integral membrane proteins, connexins are synthesized in association with the endoplasmic reticulum and traffic through the Golgi apparatus. However, Cx43 atypically delays oligomerization into a hexameric hemi-channel or “connexon” until reaching the trans-Golgi network [48]. Activation of PKA can increase Cx43 movement to the plasma membrane in a process termed enhanced assembly [15, 17, 22, 49-51]. Although directed delivery of connexons to the plasma membrane at sites of cell-cell contact has been reported [52], most evidence indicates assembly from the bulk plasma membrane (Figure 1B). CK1 activity has been shown to be involved in the assembly of Cx43 hemichannnels into the gap junction plaque [27]. Inhibition of CK1 led to an increase in non-junctional Cx43 and a decrease in Triton X-100-insoluble gap junctions, and CK1 has been shown to phosphorylate Cx43 at some combination of S325, S328 and/or S330. Expression of site directed mutants, where these serines are converted to alanines, results in cells that have little to no ability to form gap junctions [27], indicating that the conformational change which occurs upon phosphorylation of these residues may be important in gap junction formation.
Treatment of cells with growth factors and phorbol esters such as 12-O-Tetradecanoylphorbol-13-acetate (TPA) have overlapping consequences leading to downregulation of gap junctions [19-23, 53-57] and shifts in Cx43 SDS-PAGE mobility to slower migrating forms (Figure 1A, TPA lane). Treatment with these effectors is accompanied by increased Cx43 phosphorylation on serines in a wide variety of cell types [19-23, 56, 57]. The kinetics of TPA action can be complex as it can have pleiotropic effects on gap junction assembly, channel gating, and connexin half-life [22, 58, 59]. TPA leads to a dramatic decrease in gap junction assembly [22, 60]. Time course, pulse chase and cell surface biotinylation experiments indicate that TPA acts primarily by destabilizing newly forming gap junctions [22]. However, these effects on assembly have not as yet been clearly linked to phosphorylation at a specific site in Cx43 [61].
Classically, TPA is used to activate PKC [62] and it has been shown that phosphorylation of Cx43 on S262 and S368 occurs directly or indirectly upon PKC activation, and PKC can phosphorylate Cx43 directly in vitro [63, 64]. Phosphorylation at S368 does not apparently affect Cx43 migration [45], whereas Cx43 phosphorylated at S262 always shows reduced migration mostly at the P2 position (Figure 1A). Phosphorylation on S262 appears to be an event that can induce a conformational change resulting in a migration shift seen in many cell types [65]. Both of these sites have been shown to have functional relevance. In cardiac myocytes, overexpression of wild-type Cx43 or a S262A mutant resulted in decreased DNA synthesis, whereas a S262D mutant did not, indicating that this PKC-sensitive site may play a role in cell cycle progression [66]. Phosphorylation at S368 results in a reduction in unitary channel conductance with 50pS channels favored over 100pS channels [58, 64].
TPA can also lead to increased phosphorylation at S255 and S279/282 – sites known to be MAP kinase family substrates [29, 30, 33]. In fact, TPA can activate MAPK pathways in many cell types [67]. Phosphorylation on S279/282 is important in downregulation of gap junction communication as these events are able to decrease gap junction channel “open time” [68] (shown by red or closed channel in Figure 1A). Phosphorylation by MAPK also appears to be targeted to a specific subpopulation of connexins as these phosphorylation events are apparently never found in the P0 form of Cx43, even though evidence indicates that these phosphorylation events themselves do not lead to a migration shift [65].
To add to this complexity, kinase activators can modulate Cx43 cellular localization. As addressed above, activation of PKA leads to increased Cx43 in gap junction plaques. TPA can affect Cx43 half-life [22] and cause internalization of Cx43 [69, 70], and epidermal growth factor has been reported to lead to accumulation into gap junctions followed by internalization [71, 72]. Gap junctional channels are likely internalized by multiple methods including endocytosis and formation of double membrane “annular junctions” [73-76] or “connexosomes” [5, 77] (Figure 1B). The regulation of these processes or why one method might predominate over the other is not known, but it appears to be at least partially cell type specific. Ubiquitination of Cx43 has been invoked to be involved in Cx43 internalization and degradation, and a poly-ubiquitin ladder has been shown in some cell types [70, 72]. Mono-ubiquitinylation has been proposed based on ubiquitin antibody specificity [70], but distinct sites of ubiquitination have not been demonstrated.
All these data indicate that coordinated regulation of gap junctions is occurring through multiple signaling pathways leading to phosphorylation on multiple sites. This complex interplay has made it difficult to assign specific aspects of gap junction regulation to specific kinases or phosphorylation events. It seems clear that future dissection of the roles that different kinases play in the regulation of Cx43 will require many phosphospecific antibodies to identify the sites involved, complemented by Cx43 mutagenized at these different sites and techniques/compounds that can specifically modify kinase activity.
SITE SPECIFIC PHOSPHORYLATION OF Cx43
Several phosphospecific antibodies to Cx43 have been developed recently and are beginning to reveal some specific roles of Cx43 phosphorylation in gap junction function. For example, src-mediated downregulation of gap junctions has been well described, and Cx43 has been shown to be directly phosphorylated by src at Y247 and Y265 [36, 65, 78-80]. However, data generated in different model systems, using inhibitors of various kinase pathways have led to some controversy as to how src activation actually downregulates gap junction communication. In a study utilizing several phosphospecific antibodies it was shown that activation of v-src leads not only to phosphorylation on Y247 and Y265, but also S262, S279/282, and S368 and leads to a decrease in phosphorylation at S364/365 [65]. This implies activation of at least the src, MAPK and PKC pathways and their recruitment to Cx43 upon src activation. Knowing the complexity involved is necessary for the design of future experiments studying the mechanisms behind src–mediated downregulation of gap junctions.
In addition to antibodies recognizing specific phosphorylation events, antibodies specific for non-phosphorylated residues have generated important information. For example, a monoclonal antibody has been reported to bind primarily to non-phosphorylated Cx43 (Zymed/Invitrogen, 13−8300) and in unstimulated cells recognizes only the P0 form of Cx43 [76]. The epitope of this antibody is not clearly defined but the immunizing peptide was residues 360−376. Since Cx43 is known to be phosphorylated at many sites upstream of these residues and this antibody can recognize other minor bands in homeostatic cells and unmistakably recognizes multiple bands upon TPA treatment [81], it is clear that it could not represent fully dephosphorylated Cx43. Another “non-phosphospecific” monoclonal antibody, termed CT1, which has both overlapping and distinct properties, was epitope mapped to non-phosphorylated S364/S365 (Figure 2A, light green) [82]. Various lines of evidence indicate that the epitopes of these antibodies are key regulators of gap junctions and their life cycle [83-87]. Substitution of S365 in Cx43 with aspartic acid had dramatic effects on the 15N-HSQC solution structure of the C-terminal region of Cx43 [46], consistent with the CT1 antibodies ability to detect a conformational change visible in SDS-PAGE. Functionally, both S364 and S365 have been reported to be phosphorylated in response to increased cAMP levels leading to increased trafficking of Cx43 to gap junction plaques [18, 51, 88]. Thus, it seems likely that phosphorylation of S364 and/or S365 is the modification that eliminates the CT1 epitope [46]. Immunofluorescence analysis with this antibody indicates that this event is correlated with gap junction formation as CT1 recognizes predominantly Golgi apparatus-associated cytoplasmic Cx43 with little to no recognition of Cx43 in the plasma membrane or gap junction plaque [82]. Regulation of this epitope appears to occur after or upon Golgi exit as treatment of cells with Brefeldin A, which results in the loss of P1and P2 and the appearance of a phosphatase-sensitive band migrating just above the P0 form [89], showed that both P0 and P1/2 were CT1 reactive, indicating that the CT1 epitope is present prior to Golgi entry [82].
Figure 2. Model of the Cx43 C-terminus based on NMR structural studies.

(A) Cartoon of a possible structure for the Cx43 C-terminus (amino acids 252−382) with known regulatory sites/binding sequences marked in different colors produced using the PyMOL Molecular Graphics System (http://www.pymol.org). (B and C) Space filled models of amino acids 260−292 and 360−382, respectively. Circles mark residues which show resonance peak shifts in NMR spectra in response to SH3 binding (B) or S365D substitution (C). Blue circles mark residues that shift in response binding of the src SH3 domain (B and C). Green circles mark residues that are shifted in a S365D mutant as compared to wild type (B). In (C) dashed lines represent the only residues shown that do not shift due to ZO-1 binding (pink) or the S365D mutation (yellow).
In addition to the data from these “de-phosphospecific” antibodies indicating a role for S364/365 in the conformation of Cx43 upon gap junction assembly, a phosphospecific antibody for phosphorylation at S365 has shown an important role for this site at the gap junction plaque, in that S365 phosphorylation at this residue prevents phosphorylation at S368 [46]. Since phosphorylation at S368 increases during ischemia/hypoxia, wound healing and phorbol ester treatment [45, 58, 90, 91] phosphorylation at S365 was proposed to serve a “gatekeeper” function that may represent a mechanism to protect cells from ischemia and phorbol ester-induced downregulation of channel conductance [46].
CONNEXIN STRUCTURE
The C-terminal regions of the connexins are thought to contain most of the regulatory and protein-protein interaction domains. Mice that are engineered to lack the C-terminus of Cx43 die shortly after birth [92] and removal of the C-terminal domain leads to prevention of PDGF-induced cell growth [93]. According to models, the 4th transmembrane domain of Cx43 ends at approximately residue 230 leaving a C-terminal domain of about 150 residues. Structure prediction modeling and 15N-heteronuclear single-quantum correlation (HSQC) Nuclear Magnetic Resonance (NMR) experiments on a fusion protein containing residues 252−382 have indicated that the C-terminal region is not highly ordered but does show one alpha helix at A311-S325 and possibly another at D339-K345 or slightly shifted C-terminal [94, 95]. One possible arrangement for the C-terminal domain is shown in Figure 2A. Several known functional domains and phosphorylation sites exist in the Cx43 C-terminal region and these are depicted as colored lines in Figure 2A, where red lines indicate phosphorylation events associated with downregulation of gap junction communication and green lines show sites associated with gap junction assembly. Moving from N- to C-terminus, the juxtamembrane region contains a tubulin-binding domain (region not present in NMR structural model) [96]. Residues 253−256 of Cx43 constitute a SH3 binding domain that promotes binding of connexin43 interacting protein 85 (CIP85), a protein involved in connexin turnover [97] (Figure 2A and B, depicted in light pink). This domain also contains S255, a site associated with downregulated communication, which becomes phosphorylated during mitosis in a manner dependent on p34cdc2 kinase [25] and can also be directly phosphorylated by Big Mitogen-activated Protein Kinase 1/ERK5 [33]. Phosphorylation on S262 has been implicated in controlling cell cycle progression in cardiomyocytes [66]. Y265 and Y247 are phosphorylated by src [79]. Phosphorylation by src occurs through binding of its SH3 domain to proline-rich residues P274-P280 (Figure 2A, blue) with subsequent phosphorylation on Y265 creating a potential SH2 binding site that can lead to phosphorylation on Y247, culminating in channel closure [79]. Residues S279 and S282 are phosphorylated by MAPK and are implicated in channel gating [30, 68]. S282 also overlaps with a proline-rich PY-motif protein interaction domain (xPPxY) (Figure 2A, yellow) and a tyrosine-based sorting signal (Yxxϕ, where ϕ is hydrophobic) (Figure 2A, orange). Elimination of the tyrosine-based signal tripled the Cx43 half-life [98], whereas the PY-motif was shown to bind WW domains of the ubiquitin ligase Nedd4 in a process that may be modulated by phosphorylation at S279 and S282 [99]. WW domains are protein modules that mediate protein-protein interactions through recognition of proline-rich motifs and phosphorylated serine/threonine-proline sites. Phosphorylation sites implicated in gap junction assembly are found between the putative alpha helices at S325/328/330 [27](Figure 2A, green). Epitope mapping using monoclonal antibodies which distinguish Cx43 in specific subcellular locales have shown that phosphorylation on S364/S365 is important for gap junction assembly, as discussed above, (Figure 2A and B, CT1, light green line) and that prolines 375 and 377 are critical for ZO-1 binding and important in generating a conformation of Cx43 found in gap junction plaques (Figure 2A, IF1, light blue) [82]. Finally, the C-terminal LEI residues allow for PDZ domain containing proteins to bind to Cx43, including ZO-1 (Figure 2A, dark pink).
A striking aspect in this structure is that there seem to be 2 regions that contain many of the known regulatory sites; amino acids between 260−290 and the last 20 residues. Possible space filling/surface models of these sequences derived from the NMR structure are shown in Figure 2B and C (color coding from Figure 2A is maintained). In Figure 2B it is clear that several binding domains and phosphorylation sites are clustered, making it easy to imagine a complex interplay between proteins in this region. Notably, in this model the pocket between the src-binding SH3 domain (blue) and SH2 domain (dark red) is quite striking. SH3 recruitment of src could bring it into the pocket where it can phosphorylate Y265 or perhaps SH3 binding opens up the structure around Y265 making it accessible. In Figure 2C, S368 is found towards the interior of the structure (red) and is on the opposite side of S365 (light green). This potential arrangement may be part of the mechanism by which S365 can regulate S368 phosphorylation.
Interestingly, NMR analyses have shown that changes in either of these regions result in conformational effects on the other region [46, 100]. Examples of these are depicted in Figure 2B and C by circles. The circles surround residues that have been reported to shift by NMR in response to either binding by src (blue circles), ZO-1 (pink circle) [100] or by an S to D amino acid substitution at S365 (green circles) [46]. In Figure 2C, though mutagenesis studies show that src binding requires the SH3 (blue) and SH2 (dark red) domains, its binding effects are transduced to the downstream interaction domains as well (blue circle). In addition, this binding affects residues around S368 which can be phosphorylated by PKC in response to src activation [65]. The terminal 20 amino acids of Cx43 appear to have some sort of coordinated behavior as both ZO-1 binding [100] and a S365D mutation [46] altered nearly all of these residues (pink dashed line shows D360 the single residue shown that does not shift in response to ZO-1 binding and yellow dashed line shows S369, the single residue not shifted by S365D). In addition, binding of the cytoplasmic loop of Cx43 could affect a subset of these residues [101]. Thus, it seems possible that S365 may play an important role in regulating interactions at the C-terminus consistent with the distinct epitopes found with the Zymed 13−8300 and CT1 antibodies discussed above. Like src binding, the S365D mutation led to long-range changes by affecting residues in the SH3 domain (Figure 2B green circle, blue domain) and at the overlapping residues of the proline-rich PY and tyrosine-based sorting domains (Figure 2B, green circle, orange domain).
It has been shown that these types of changes have biological consequences as binding of the src SH3 domain could displace ZO-1 [100, 102]. These structural studies have shed some light on studies in cells showing src-mediated regulation of ZO-1 binding to Cx43. In cardiac myocytes, expression of constitutively active c-src inhibited the interaction between ZO-1 and wild type Cx43 but not the Cx43 mutant, Y265F [103]. Additionally, Cx43 from cultured astrocytes exposed to chemical ischemia had an increased association with c-src, extracellular signal-regulated kinase 1/2 (ERK1/2) and MAPK phosphatase-1 [104] and a decreased association with ZO-1 [102].
CONNEXIN LOCALIZATION AND CELL GROWTH
Connexin expression has been shown to affect the growth of cells under numerous conditions [10, 105-108]. While some of these effects may be linked to changes in gap junction communication, there is increasing evidence that at least some of these changes may be independent of molecular conduction through gap junctions and, rather, a consequence of the impact of connexin expression on the expression and localization of other scaffold and signaling molecules [109-114]. Though few cases have looked specifically at the role of phosphorylation in these interactions, it seems likely to be a means of regulating these interactions. Some examples of this type are discussed below.
Nov/CCN3
In the communication deficient Jeg3 trophoblast cell line, exogenous expression of Cx43 but not Cx40 nor a C-terminal truncation mutant of Cx43, led to decreased cell growth in culture and tumor growth in nude mice [115]. Expression of Cx43 was also accompanied by an increase in expression and interaction with NOV/CCN3, a protein which appears regulate growth and can be found in the nucleus and cytoplasm [114, 115]. Cx43 expression led to a shift in NOV/CCN3 from diffuse staining to the plasma membrane, where it co-localized with apparent gap junctions [114, 115].
p27 and Skp2
Cx43 expression has been shown to suppress the G1-S phase transition in U2OS cells by increasing p27 expression [116]. This increase in p27 resulted from decreased degradation via ubiquitin-dependent proteolysis by the SCFSkp2 complex. Specifically, Cx43 expression led to increased degradation of Skp2, a component of the ubiquitinating complex [116]. This effect on Skp2 appeared to be gap junction independent as expression of the C-terminus alone could decrease Skp2 levels [117].
Neuronal Tissue and PDZ-domain containing scaffold proteins
Neuronal tissues contain a high density of gap junctions that allow different types of cells to engage in both homotypic and heterotypic gap junctional communication. These junctions are composed of various connexins which can exhibit both redundant and specific functions in these tissues [118-121]. Several PDZ domain containing scaffold proteins have been shown to localize to these gap junctions including ZO-1 and ZONAB, a transcription factor that can regulate cell proliferation [122]. A role for connexins in maintaining these gap junction complexes has been shown by a loss of gap junctions or their associated scaffold proteins in various connexin knockout animals [123, 124].
Myoblast differentiation and the cytoskeleton
In a cell culture model, myoblast differentiation induced by sphingosine 1-phosphate was shown to depend on both gap junctional communication and a MAPK dependent interaction between Cx43, F-actin and cortactin [125]. Interestingly, in this case, it was the cytoplasmic loop of Cx43 that was required for this interaction and effect.
While none of these studies looked at connexin phosphorylation, per se, evidence indicates that post-translational modifications, such as phosphorylation, are a result of and lead to changes in binding partners and can lead to changes in subcellular localization (e.g., 27, 72, 114, 126). It has become increasingly clear that signaling is highly dependent on subcellular localization and, in particular, that signaling through integral membrane proteins is not confined merely to the plasma membrane but that effects can be transduced throughout the endosomal-lysosomal system [127].
Intriguingly, Cx43 dramatically changes phosphorylation and subcellular localization throughout its life cycle both in homeostatic cells and in response to both acute and chronic stimuli [11, 13, 128, 129]. While changes in gap junction communication have been well documented under these conditions, the impact on signaling is less well understood. One biologically interesting and potentially tractable model to study these types of changes is during the cell cycle, where there are dramatic changes in Cx43 phosphorylation and localization in addition to gap junction function.
CX43 DURING THE CELL CYCLE
Connexin expression and gap junction communication are both regulated during the cell cycle and can impact each other. Cx43 expression can lead to increased duration of G1 and S phase and can affect expression levels of cell cycle regulatory proteins [116, 117, 130]. Cx37 expression has also been shown to profoundly slow all phases of the cell cycle [131] and uncoupling of gap junctions in the mouse neocortex can delay S phase entry [132]. Typically, asynchronous cells exhibit multiple phospho-isoforms of Cx43, readily engage in gap junction communication, as measured by dye transfer, and express Cx43 both in plaques and in the cytoplasm to varying degrees depending on the type of cell. Studies of cells in specific stages of the cell cycle indicate that the level of phosphorylation of Cx43 increases as cells progress from G0/G1 through S phase and into mitosis [24]. During G0/G1, Cx43 endogenously expressed in Normal Rat Kidney cells, is found predominantly at the plasma membrane where it is efficiently assembled into “typical” gap junction plaques at the plasma membrane which are Triton X-100 insoluble and made up largely of the P2 isoform of Cx43 [13]. At this stage of the cell cycle Cx43 has been shown to interact with scaffold proteins, ZO-1 and ZO-2, at the plasma membrane [126] (Figure 3).
Figure 3. Connexin localization and phosphorylation change dramatically during the cell cycle.
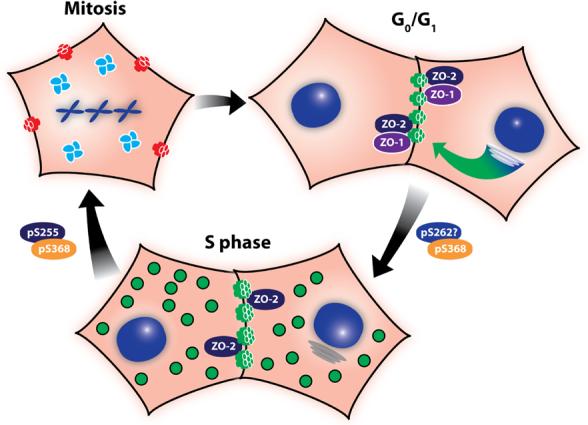
Confluent cells in G0/G1 are very efficient at trafficking and assembling (green arrow) most of the Cx43 into Triton X-100 insoluble gap junctions, shown in green. ZO-1 and ZO-2 are colocalized at these plaques. As cells progress into S phase assembly of gap junctions becomes less efficient and Cx43 is found both in gap junction plaques and in cytoplasmic vesicles (green). Association with ZO-1 is decreased while ZO-2 interaction is maintained. PKC-mediated phosphorylation of Cx43 on S368 and S262 (transient?) begins occurring as cells approach S phase. As cells enter mitosis, gap junction communication ceases (red) and Cx43 is found predominantly in clusters of vesicles in the cytoplasm (green). Cx43 becomes increasingly phosphorylated on S368 and S255 and exhibits a migration shift by SDS-PAGE. Cells are able to quickly resume communication upon cytokinesis, likely due to plasma membrane connexon pools.
As cells progress into and through the cell cycle the assembly of Cx43 into plaques becomes less efficient, with only about 50% of S phase cell:cell interfaces showing newly assembled gap junctions as compared to G0/G1 [45]. This decreased assembly accompanies a shift in subcellular localization where Cx43 is found both in the plasma membrane and in the cytoplasm and an increase in phosphorylation on S368, a known PKC site (Figure 3) [64]. It is not presently clear if the cytoplasmic Cx43 is accumulated through inhibition of gap junction assembly or results from gap junction breakdown. In either case, there appears to be negative regulation of gap junction assembly during S phase and it is likely that PKC is playing a role in these effects, as PKC activation is clearly correlated with decreases in gap junctional communication (described in previous sections). Interestingly, though there is an accumulation of Cx43 in the cytoplasm, many plaques still remain in the plasma membrane and in fact these cells show increased dye transfer as compared to cells in G0/G1. These plaques are also distinct from those in G0/G1 as they show decreased co-localization with ZO-1 while the ZO-2 interaction is maintained [126] (Figure 3). This is consistent with the observation that gap junction communication is highest during S and G2 phase in the mouse neocortex [133]. Though the exact functional consequences of this enhanced gap junctional communication are not clear, it illustrates that protein:protein interactions and gap junction functions are distinctly regulated during the cell cycle. Other PKC-mediated effects during cell cycle progression have been reported including a study in clone 9 cells which showed a PKC-mediated increase in phosphorylation as cells progressed from G0 to S phase [134]. However, in this case there was an accompanying downregulation of gap junctional communication. Another study, utilizing cardiomyocytes and Cx43 containing a serine to alanine mutation at S262 showed that overexpression of the S262A mutant resulted in decreased progression into S phase compared to wild type Cx43 [66]. A S262D mutant, which mimics the phosphorylated charge, showed no effect on cell cycle progression. Notably, this effect appeared to be independent of gap junctional communication. Taken together, these data indicate that Cx43 interactions are differentially regulated during S phase and may in turn, influence S phase progression.
Finally, one of the most striking examples of gap junction reorganization occurs during mitosis, where total phosphorylation reaches its highest levels [24] including specific phosphorylation at S255 [25] and a conformational change evident as the slow migrating Pm isoform by SDS-PAGE, whose formation is dependent on p34cdc2 kinase [24, 25]. At this time gap junction communication ceases [26, 135] and Cx43 is found predominantly in the cytoplasm in what appear to be large clusters of vesicles [25, 26]. As in S phase, it is not clear whether these clusters are comprised of Cx43 trafficking towards the plasma membrane or internalized material. As cells exit mitosis, these structures disperse and gap junctional communication quickly resumes upon cytokinesis, even in the absence of new connexin synthesis or anterograde trafficking [26, 135].
SKIN AND WOUNDING
Expression of connexin genes is tissue specific and Cx26, Cx31, and Cx31.1 and Cx43 are present in skin epidermis. Although Cx43 is the predominant connexin in human epidermis and in cultures of human keratinocytes [136], mutations in the other connexins can lead to keratitis-ichthyosis-deafness syndrome, hystrix-like ichthyosis with deafness, Vohwinkel's syndrome and erythrokeratodermia variabilis [137-141]. Human epidermis is a stratified tissue with a proliferative basal cell layer and multiple layers of terminally differentiating suprabasal and granular cells. Connexin proteins are differentially expressed in human skin with lower expression in the proliferative regions and more expression upon differentiation [25, 142-145].
Epidermal wounding activates cell migration across the wound bed, increases proliferation, and promotes changes in cell-to-cell communication [146-149]. Connexin proteins are temporally and spatially regulated during wounding and distinct communication compartments are formed that likely regulate processes such as proliferation, migration and adhesion. Gap junctional intercellular communication may regulate certain aspects of the wound healing process including synchronization of cell migration [25, 150]. In unwounded human skin, basal keratinocytes show low expression of total Cx43, whereas suprabasal cells show high levels with low phosphorylation at S368 [90]. Upon wounding, Cx43 expression is decreased at the wound edge (Figure 4) but expression is enhanced in basal cells at unwounded adjacent areas [25, 150, 151]. Remarkably, 24 hours after wounding, phosphorylation at S368 was dramatically increased but strictly limited to the basal cells [90]. Since S368 phosphorylation changes the communication properties of Cx43 channels by affecting their conductance levels [58, 64], a distinct communication compartment is formed that regulates exchange within and between this compartment (orange basal cells in Figure 4). The levels and distribution of phosphorylated S368 returned to normal levels at 72 hours [64]. Modulation of Cx43 expression directly affects wound repair. Cx43 antisense application to wounds accelerated migration and the rate of wound repair resulting in less scarring [152]. Also wound closure is delayed in diabetic skin when Cx43 expression remains high [153] or upon Cx43 overexpression [154]. Mice with reduced levels of epidermal Cx43 show more rapid healing [155]. These results clearly indicate that Cx43 regulation plays an important role in wound repair.
Figure 4. Schematic diagram of connexin expression in normal and wounded human skin.

Cx43 expression (denoted in green) in unwounded skin is mainly in the upper more differentiated layers. Upon wounding Cx43 expression drops very near the wound and redistributes to lower layers in the cells several cells distant from the wound. The basal cells near the wound express Cx43 that is highly phosphorylated at S368 (indicated by orange) forming a distinct communication compartment.
Other connexins also show differential regulation as the normally low levels of Cx26 and Cx30 are increased near the wound bed [150, 151, 156-158]. Skin diseases in humans with mutations in these connexins point to their importance in epidermal biology but their assignment of a specific role in human wound healing is less clear as their expression level is low until 24−48 hours after wounding [157]. Furthermore, some real differences exist between rodent and human skin making it difficult to clearly correlate the human and mouse models. For example, in rodent the epidermis is only a few cell layers thick, and Cx43 expression is high in the basal cells, while human epidermis is much more thick and stratified with Cx43 expression low in basal cells and higher in the more differentiated layers. Therefore, a clear understanding of the contribution of connexins to the wound healing response and the differences in these processes between human and mouse models remain to be elucidated.
CARDIAC GAP JUNCTIONS AND ISCHEMIA
Coordinated contraction of the heart requires myocytes to be mechanically and electrically coupled. This coupling is maintained at a specialized structure at the ends of myocytes referred to as the intercalated disc [159, 160], which contains a high concentration of intercellular junctional proteins. Desmosomes and adherens junctions provide mechanical stability [161, 162] and are closely juxtaposed with large gap junction plaques that provide electrical and chemical coupling. This is schematized in Figure 5 where green represents the permeable gap junctions in the intercalated disc. In the ventricle, Cx43 is the main gap junction component [163, 164] and is the best studied; thus it will be the focus of this discussion.
Figure 5. Schematic diagram of changes in connexin expression in heart under ischemic conditions alone or with preconditioning.
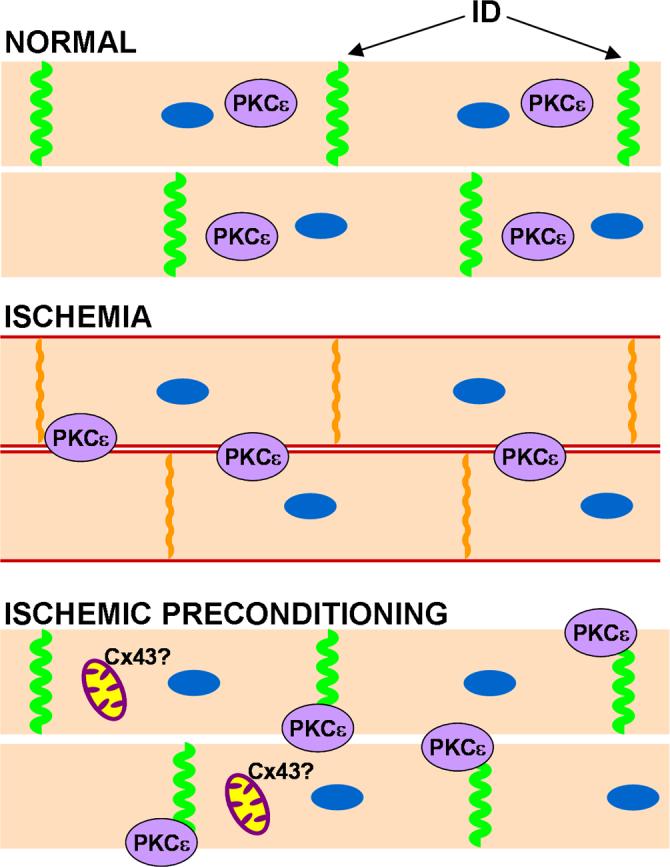
Normally cardiac myocytes express high levels of Cx43 at the intercalated disc (ID) and gap junctions are in their open state, shown in green. Under ischemic conditions there is a loss of gap junctions at the intercalated disc as Cx43 moves to the lateral edges of the myocyte, shown in red. Some Cx43 is retained at the intercalated disc, but it becomes phosphorylated on S368 (indicated by orange). In addition, PKCε translocates from the cytoplasm to the plasma membrane and may be involved in Cx43 phosphorylation on S368. With ischemic preconditioning, gap junctions are retained at the intercalated disc with no subsequent S368 phosphorylation. PKCε is necessary for this retention and is found at the plasma membrane. In addition, increased Cx43 may be found in the mitochondria.
Immunoblot analysis of lysates from normal heart show that essentially none of the Cx43 protein migrates in SDS-PAGE as the P0 form and all adopts a conformation that exhibits slower migration that is sensitive to alkaline phosphatase [84, 165-167]. Consistent with this observation, analysis with phosphospecific antibodies show that Cx43 is heavily phosphorylated on S365 and S325/328/330, sites which are known to affect Cx43 SDS-PAGE migration [46, 58]. Other phosphorylation sites have been identified via mass spectrometry [168], however, the abundance and intracellular location of these events these sites have not been addressed.
There is increasing evidence and interest in the idea that regulation of Cx43 containing gap junctions can play an important role in cardioprotection. This is evident from studies aimed at understanding the phenomenon of ischemic preconditioning, where periods of brief ischemia in intact hearts can minimize damage from subsequent longer bouts of ischemia [169]. It is clear that Cx43 plays an important and necessary role in this process as mice that are heterozygous for a null mutation in Cx43 are not protected by preconditioning [170-172]. This involvement of gap junctions in cardiac protection could prove to be medically important as rotigaptide, a compound that has been developed to inhibit cardiac re-entry arrhythmias and is being tested in humans [173], is hypothesized to work through effects on gap junctions and connexin phosphorylation. Cx43 dephosphorylation in response to low-flow ischemia was significantly prevented by rotigaptide [174], and during conditions of acute cardiac ischemia, rotigaptide effectively prevented induction of both ventricular and atrial tachyarrhythmia [173]. Additionally, a peptide termed RXP-E has been developed which has been shown to bind to the C-terminus of Cx43 and can inhibit downregulation of gap junction mediated action potentials in cultured myocytes [175].
Data from several animal models show that during ischemia a majority of the Cx43 moves out of the intercalated disc and is found throughout the plasma membrane of the cardiomyocyte, a redistribution often referred to as lateralization (Fig B, red) (e.g., 165). Connexin found in these lateral membranes of the cardiomyocyte is likely to be in the form of closed hemichannels [171, 176]. A functional consequence of this loss of gap junctions is decreased electrical coupling in the ischemic and presumably damaged cells, though dye transfer of Lucifer Yellow between the cells was not apparently downregulated [166, 177]. These changes are concomitant with changes in Cx43 phosphorylation detectable as a shift to the fast migrating P0 isoform of Cx43. Work with phosphospecific antibodies has shown that this shift in migration results from a loss of phosphorylation at S365 [46] and S325/328/330 [47], sites which are typically seen in gap junction plaques. Dephosphorylation at S364/S365, at least, is quite rapid and can be seen after 5 minutes of hypoxia when examined with the monoclonal antibody, CT1, which specifically recognizes Cx43 where S364/S365 are not phosphorylated [82]. This antibody also very clearly recognizes Cx43 that has translocated to the lateral membranes of cardiomyocytes (unpublished data). The rapid appearance and visibility of CT1-positive Cx43 in the lateral membrane of the myocytes indicates that a phosphatase may be acting on S365 and likely on S325/328/330. Consistent with this idea, a report has shown that an inhibitor of PP1-like phosphatases could inhibit ischemia-induced dephosphorylation of Cx43 [178]. In contrast to these decreases in S365 and S325/328/330 phosphorylation, S368 phosphorylation increases during ischemia including the fraction of Cx43 that remains in the intercalated disc [58]. As noted in previous sections, phosphorylation on S365 inhibits S368 phosphorylation, indicating that dephosphorylation of S365 is occurring on Cx43 both in the lateral membranes and at the intercalated disc. This S368 phosphorylation at the intercalated disc likely indicates a change in the permselectivity of the channels remaining under ischemic conditions [58]. In addition a S364P mutation, which had been reported in viseral atrial heterotaxia [179] though not subsequently confirmed [180], can cause alteration of gating or expression in experimental model systems [181, 182]. It seems plausible that this mutation could perturb proper regulation between S365 and S368.
Preconditioning likely maintains phosphorylation at S325/328/330 as the slow migrating phosphoforms are preserved. Interestingly, phosphorylation on S368 is increased to an equal [91] or greater degree [177] under ischemic preconditioning, when analyzed by immunoblotting, and has been invoked to be the event leading to inhibition of dye transfer [177]. However, it is not clear how much of the phosphorylation on S368 is occurring at the plaque as immunohistochemistry on preconditioned hearts showed decreased pS368 at the intercalated disc [91]. Although the kinases and phosphatases affecting S365 and S325/328/330 are not known, S368 is a known PKC site [63, 64]. Indeed, there is a clear role for PKC in ischemia as inhibition of PKC activity can block the ability of preconditioning to delay uncoupling and inhibit lateralization of Cx43 [84]. PKCε has been shown to translocate from the cytosol to the membrane during ischemia regardless of preconditioning [91] (Fig B and C) and has been shown to interact with Cx43 in heart [183].
Very clear evidence that PKCε, in particular, plays a critical role in ischemic preconditioning comes from the PKCε knock-out mice which do not respond to preconditioning [184]. Studies of Cx43 in these mice showed that PKCε was necessary for maintenance of gap junctions at the intercalated disc during ischemic preconditioning as there was a loss of gap junctions from the intercalated disc during ischemia regardless of preconditioning. Interestingly, PKCε seemed to play a role in plaque maintenance under normal conditions, as evidenced by an increase in gap junctions in the knockout animal. Somewhat surprisingly, S368 phosphorylation was maintained in the PKCε knock-out animal, indicating that other PKCs can phosphorylate Cx43 under these conditions [184]. In fact, the complex interplay between PKC isoforms and Cx43 was illustrated in this system through the observation that PKCδ translocated to the plasma membrane specifically in the preconditioned KO mouse and not in the wild type and hence, may have been the kinase responsible for phosphorylation on S368 [184] under these conditions.
Clearly, preconditioning has dramatic effects on the organization of gap junction plaques during ischemia which will have important consequences on impulse propagation. However, there is some evidence that Cx43 at the intercalated disc may not be the only mediator of protection during ischemia. It has been reported that a small fraction of Cx43 can be found in the mitochondria and that this amount rapidly increases over 2.5 times during ischemic preconditioning [86], though it is not at all clear how Cx43 could be imported into the mitochondria. During ischemia at least some of the damage incurred by cells is due to reactive oxygen species generation in the mitochondria that in combination with other events culminates in damaged mitochondria and eventually cell necrosis and rupture. Preconditioning can dramatically decrease reactive oxygen species production and this is likely one of the endpoint mechanisms resulting in cardioprotection [185]. Mitochondrial Cx43 could somehow decrease reactive oxygen species production and protect the mitochondria from damage [176]. There has been some data utilizing cardiomyocytes from mice which are heterozygous for Cx43 indicating that this decrease in Cx43 results in cells which do not attenuate reactive oxygen species in response to preconditioning stimuli [186]. It will be interesting to see whether this hypothesis will, indeed, yield new functions for Cx43.
CONCLUDING REMARKS
The multiple essential roles that gap junctions play in tissue homeostasis are evident from the diseases that result from connexin mutations including deafness, skin diseases, demyelination, arrhythmia and a variety of developmental defects. Acute function in response to wounding and hypoxia also requires connexin function for proper response. Connexin phosphorylation appears to be the primary mechanism for these acute responses. Phosphorylation affects both the structure and function of Cx43 and leads to changes in localization, interacting protein partners and channel selectivity. Further studies with other connexins will likely show similar roles for phosphorylation in their regulation in a manner specific for the tissue in which they are expressed.
ACKNOWLEDGEMENTS
The work performed in the author's lab reviewed here was supported by NIH GM55632.
Abbreviations used
- Cx
Connexin
- SDS-PAGE
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- PKA
protein kinase A
- PKC
protein kinase C
- p34cdc2
p34cdc2/cyclin B kinase
- CK1
casein kinase 1
- MAPK
mitogen-activated protein kinase
- src
pp60src kinase
- TPA
12-O-Tetradecanoylphorbol-13-acetate
- SH2
src homology 2
- SH3
src homology 3
- ZO-1
zonula occludens 1
- ZO-2
zonula occludens 2
- HSQC
15N-heteronuclear single-quantum correlation
- D1gh1
Disc Large homolog 1
- CIP85
connexin43 interacting protein 85
- PDZ
post synaptic density protein-Drosophila disc large tumor suppressor-zonula occludens-1 protein domain
- NMR
Nuclear Magnetic Resonance
REFERENCES
- 1.White T, Paul D. Genetic diseases and gene knockouts reveal diverse connexin functions. Ann. Rev. Physiology. 1999;61:283–310. doi: 10.1146/annurev.physiol.61.1.283. [DOI] [PubMed] [Google Scholar]
- 2.Sohl G, Willecke K. Gap junctions and the connexin protein family. Cardiovasc Res. 2004;62:228–232. doi: 10.1016/j.cardiores.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 3.Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 2003;83:1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- 4.Harris AL. Connexin channel permeability to cytoplasmic molecules. Prog Biophys Mol Biol. 2007;94:120–143. doi: 10.1016/j.pbiomolbio.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laird DW. Life cycle of connexins in health and disease. Biochem J. 2006;394:527–543. doi: 10.1042/BJ20051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Laing JN, Parry G, Mueller RF, Leigh IM. Connexin 26 mutations in hereditary nonsyndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 7.Gong X, Li E, Klier G, Huang Q, Wu Y, Lei H, Kumar NM, Horwitz J, Gilula NB. Disruption of alpha3 connexin gene leads to proteolysis and cataractogenesis in mice. Cell. 1997;91:833–843. doi: 10.1016/s0092-8674(00)80471-7. [DOI] [PubMed] [Google Scholar]
- 8.Bergoffen J, Scherer SS, Wang S, Oronzi Scott M, Bone LJ, Paul DL, Chen K, Lensch MW, Chance PF, Fishbeck KH. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 9.Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, Hannibal MC, Jabs EW. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cronier L, Crespin S, Strale PO, Defamie N, Mesnil M. Gap Junctions and Cancer: New Functions for an Old Story. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2153. [DOI] [PubMed] [Google Scholar]
- 11.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int. J. of Biochem. Cell Biol. 2004;36:1171–1186. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saez JC, Martinez AD, Branes MC, Gonzalez HE. Regulation of gap junctions by protein phosphorylation. Brazilian J. Med. Biol. Res. 1998;31:593–600. doi: 10.1590/s0100-879x1998000500001. [DOI] [PubMed] [Google Scholar]
- 13.Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711:154–163. doi: 10.1016/j.bbamem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 14.Urschel S, Hoher T, Schubert T, Alev C, Sohl G, Worsdorfer P, Asahara T, Dermietzel R, Weiler R, Willecke K. Protein kinase A-mediated phosphorylation of connexin36 in mouse retina results in decreased gap junctional communication between AII amacrine cells. J Biol Chem. 2006;281:33163–33171. doi: 10.1074/jbc.M606396200. [DOI] [PubMed] [Google Scholar]
- 15.Atkinson MM, Lampe PD, Lin HH, Kollander R, Li X-R, Kiang DT. Cyclic AMP modifies the cellular distribution of connexin 43 and induces a persistent increase in the junctional permeability of mouse mammary tumor cells. J. Cell Sci. 1995;108:3079–3090. doi: 10.1242/jcs.108.9.3079. [DOI] [PubMed] [Google Scholar]
- 16.Burghardt RC, Barhoumi R, Sewall TC, Bowen JA. Cyclic AMP induces rapid increases in gap junction permeability and changes in the cellular distribution of connexin43. J. Membr. Biol. 1995;148:243–253. doi: 10.1007/BF00235042. [DOI] [PubMed] [Google Scholar]
- 17.Paulson AF, Lampe PD, Meyer RA, Atkinson MA, Walseth TF, Johnson RG. Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J. Cell Sci. 2000;113:3037–3049. doi: 10.1242/jcs.113.17.3037. [DOI] [PubMed] [Google Scholar]
- 18.Yogo K, Ogawa T, Akiyama M, Ishida-Kitagawa N, Sasada H, Sato E, Takeya T. PKA implicated in the phosphorylation of Cx43 induced by stimulation with FSH in rat granulosa cells. J Reprod Dev. 2006;52:321–328. doi: 10.1262/jrd.17107. [DOI] [PubMed] [Google Scholar]
- 19.Berthoud VM, Ledbetter MLS, Hertzberg EL, Saez JC. Connexin43 in MDCK cells: Regulation by a tumor-promoting phorbol ester and calcium. Eur. J. Cell Biol. 1992;57:40–50. [PubMed] [Google Scholar]
- 20.Berthoud VM, Rook M, Hertzberg EL, Saez JC. On the mechanism of cell uncoupling induced by a tumor promoter phorbol ester inclone 9 cells, a rat liver epithelial cell line. Eur. J. Cell Biol. 1993;62:384–396. [PubMed] [Google Scholar]
- 21.Brissette JL, Kumar NM, Gilula NB, Dotto GP. The tumor promoter 12-O-tetradecanoylphorbol-13-acetate and the ras oncogene modulate expression and phosphorylation of gap junction proteins. Mol. Cell. Biol. 1991;11:5364–5371. doi: 10.1128/mcb.11.10.5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lampe PD. Analyzing phorbol ester effects on gap junction communication: A dramatic inhibition of assembly. J. Cell Biol. 1994;127:1895–1905. doi: 10.1083/jcb.127.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reynhout JK, Lampe PD, Johnson RG. An activator of protein kinase C inhibits gap junction communication between cultured bovine lens cells. Exp. Cell Res. 1992;198:337–342. doi: 10.1016/0014-4827(92)90388-o. [DOI] [PubMed] [Google Scholar]
- 24.Kanemitsu MY, Jiang W, Eckhart W. Cdc2-mediated phosphorylation of the gap junction protein, connexin43, during mitosis. Cell Growth Differ. 1998;9:13–21. [PubMed] [Google Scholar]
- 25.Lampe PD, Kurata WE, Warn-Cramer B, Lau AF. Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J. Cell Sci. 1998;111:833–841. doi: 10.1242/jcs.111.6.833. [DOI] [PubMed] [Google Scholar]
- 26.Xie H, Laird DW, Chang T-H, Hu VW. A mitosis-specific phosphorylation of the gap junction protein connexin43 in human vascular cells: biochemical characterization and localization. J. Cell Biol. 1997;137:203–210. doi: 10.1083/jcb.137.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper CD, Lampe PD. Casein kinase 1 regulates connexin43 gap junction assembly. J. Biol. Chem. 2002;277:44962–44968. doi: 10.1074/jbc.M209427200. [DOI] [PubMed] [Google Scholar]
- 28.Lau AF, Kanemitsu MY, Kurata WE, Danesh S, Boynton AL. Epidermal growth factor disrupts gap-junctional communication and induces phosphorylation of connexin43 on serine. Mol. Biol. Cell. 1992;3:865–874. doi: 10.1091/mbc.3.8.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanemitsu MY, Lau AF. Epidermal growth factor stimulates the disruption of gap junctional communication and connexin43 phosphorylation independent of 12-O-tetradecanoyl 13-acetate-sensitive protein kinase C: The possible involvement of mitogen-activated protein kinase. Mol. Biol. Cell. 1993;4:837–848. doi: 10.1091/mbc.4.8.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warn-Cramer BJ, Cottrell GT, Burt JM, Lau AF. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 1998;273:9188–9196. doi: 10.1074/jbc.273.15.9188. [DOI] [PubMed] [Google Scholar]
- 31.Polontchouk L, Ebelt B, Jackels M, Dhein S. Chronic effects of endothelin 1 and angiotensin II on gap junctions and intercellular communication in cardiac cells. Faseb J. 2002;16:87–89. doi: 10.1096/fj.01-0381fje. [DOI] [PubMed] [Google Scholar]
- 32.Petrich BG, Gong X, Lerner DL, Wang X, Brown JH, Saffitz JE, Wang Y. c-Jun N-terminal kinase activation mediates downregulation of connexin43 in cardiomyocytes. Circ Res. 2002;91:640–647. doi: 10.1161/01.res.0000035854.11082.01. [DOI] [PubMed] [Google Scholar]
- 33.Cameron SJ, Malik S, Akaike M, Lerner-Marmarosh N, Yan C, Lee JD, Abe J, Yang J. Regulation of epidermal growth factor-induced connexin 43 gap junction communication by big mitogen-activated protein kinase1/ERK5 but not ERK1/2 kinase activation. J Biol Chem. 2003;278:18682–18688. doi: 10.1074/jbc.M213283200. [DOI] [PubMed] [Google Scholar]
- 34.Loo LWM, Berestecky JM, Kanemitsu MY, Lau AF. pp60src-mediated phosphorylation of connexin 43, a gap junction protein. J. Biol. Chem. 1995;270:12751–12761. doi: 10.1074/jbc.270.21.12751. [DOI] [PubMed] [Google Scholar]
- 35.Crow DS, Kurata WE, Lau AF. Phosphorylation of connexin43 in cells containing mutant src oncogenes. Oncogene. 1992;7:999–1003. [PubMed] [Google Scholar]
- 36.Swenson KI, Piwnica-Worms H, McNamee H, Paul DL. Tyrosine phosphorylation of the gap junction protein connexin43 is required for pp60src-induced inhibition of communication. Cell Regul. 1990;1:989–1002. doi: 10.1091/mbc.1.13.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crow DS, Beyer EC, Paul DL, Kobe SS, Lau AF. Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol. Cell. Biol. 1990;10:1754–1763. doi: 10.1128/mcb.10.4.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laird DW, Puranam KL, Revel JP. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem. J. 1991;273:67–72. doi: 10.1042/bj2730067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beardslee M, Laing J, Beyer E, Saffitz J. Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 1998;83:629–635. doi: 10.1161/01.res.83.6.629. [DOI] [PubMed] [Google Scholar]
- 40.Musil LS, Beyer EC, Goodenough DA. Expression of the gap junction protein connexin43 in embryonic chick lens: Molecular cloning, ultrastructural localization, and post-translational phosphorylation. J. Membr. Biol. 1990;116:163–175. doi: 10.1007/BF01868674. [DOI] [PubMed] [Google Scholar]
- 41.Kadle R, Zhang JT, Nicholson BJ. Tissue-specific distribution of differentially phosphorylated forms of Cx43. Mol. Cell. Biol. 1991;11:363–369. doi: 10.1128/mcb.11.1.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Musil LS, Cunningham BA, Edelman GM, Goodenough DA. Differential phosphorylation of gap junction protein connexin43 in junctional communication-competent and deficient cell lines. J. Cell Biol. 1990;111:2077–2088. doi: 10.1083/jcb.111.5.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation and assembly into gap junctional plaques. J. Cell Biol. 1991;115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Veen TAB, van Rijen H. v. M., Jongsma HJ. Electrical conductance of mouse connexin45 gap junction channels is modulated by phosphorylation. Cardiovascular Res. 2000;46:496–510. doi: 10.1016/s0008-6363(00)00047-x. [DOI] [PubMed] [Google Scholar]
- 45.Solan JL, Fry MD, TenBroek EM, Lampe PD. Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J. Cell Sci. 2003;116:2203–2211. doi: 10.1242/jcs.00428. [DOI] [PubMed] [Google Scholar]
- 46.Solan JL, Marquez-Rosado L, Sorgen PL, Thornton PJ, Gafken PR, Lampe PD. Phosphorylation of Cx43 at S365 is a gatekeeper event that changes the structure of Cx43 and prevents downregulation by PKC. 2007;179:1301–1309. doi: 10.1083/jcb.200707060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lampe PD, Cooper CD, King TJ, Burt JM. Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci. 2006;119:3435–3442. doi: 10.1242/jcs.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Musil LS, Goodenough DA. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell. 1993;74:1065–1077. doi: 10.1016/0092-8674(93)90728-9. [DOI] [PubMed] [Google Scholar]
- 49.Li H, Liu T-F, Lazrak A, Peracchia C, Goldberg GS, Lampe PD, Johnson RG. Properties and regulation of gap junctional hemichannels in the plasma membranes of cultured cells. J. Cell Biol. 1996;134:1019–1030. doi: 10.1083/jcb.134.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lampe PD, Qiu Q, Meyer RA, TenBroek EM, Walseth TF, Starich TA, Grunenwald HL, Johnson RG. Gap junction assembly: PTX-sensitive G proteins regulate the distribution of connexin43 within cells. American Journal of Physiology - Cell Physiology. 2001;281:C1211–1222. doi: 10.1152/ajpcell.2001.281.4.C1211. [DOI] [PubMed] [Google Scholar]
- 51.TenBroek EM, Lampe PD, Solan JL, Reynhout JK, Johnson RG. Ser364 of connexin43 and the upregulation of gap junction assembly by cAMP. J. Cell Biol. 2001;155:1307–1318. doi: 10.1083/jcb.200102017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan YN, Jan LY. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007;128:547–560. doi: 10.1016/j.cell.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fitzgerald DJ, Knowles SE, Ballard FJ, Murray AW. Rapid and Reversible Inhibition of Junctional Communication by Tumor Promoters in a Mouse Cell Line. Cancer Res. 1983;43:3614–3618. [PubMed] [Google Scholar]
- 54.Trosko JE, Chang CC, Madhukar BV, Klaunig JE. Chemical, oncogene, and growth factor inhibition of gap junctional intercellular communication: an integrative hypothesis of carcinogenesis. Pathobiol. 1990;58:265–278. doi: 10.1159/000163596. [DOI] [PubMed] [Google Scholar]
- 55.Rivedal E, Opsahl H. Role of PKC and MAP kinase in EGF- and TPA-induced connexin43 phosphorylation and inhibition of gap junction intercellular communication in rat liver epithelial cells. Carcinogenesis. 2001;22:1543–1550. doi: 10.1093/carcin/22.9.1543. [DOI] [PubMed] [Google Scholar]
- 56.Lin D, Zhou J, Zelenka PS, Takemoto DJ. Protein kinase Cgamma regulation of gap junction activity through caveolin-1-containing lipid rafts. Invest Ophthalmol Vis Sci. 2003;44:5259–5268. doi: 10.1167/iovs.03-0296. [DOI] [PubMed] [Google Scholar]
- 57.Lin D, Boyle DL, Takemoto DJ. IGF-I-induced phosphorylation of connexin 43 by PKCgamma: regulation of gap junctions in rabbit lens epithelial cells. Invest Ophthalmol Vis Sci. 2003;44:1160–1168. doi: 10.1167/iovs.02-0737. [DOI] [PubMed] [Google Scholar]
- 58.Ek-Vitorin JF, King TJ, Heyman NS, Lampe PD, Burt JM. Selectivity of Connexin 43 Channels Is Regulated Through Protein Kinase C-Dependent Phosphorylation. Circ Res. 2006;98:1498–1505. doi: 10.1161/01.RES.0000227572.45891.2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moreno AP, Lau AF. Gap junction channel gating modulated through protein phosphorylation. Prog Biophys Mol Biol. 2007;94:107–119. doi: 10.1016/j.pbiomolbio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pitts JD, Burk RR. Mechanism of inhibition of junctional communication between animal cells by phorbol ester. Cell Tissue Kinet. 1987;20:145–151. doi: 10.1111/j.1365-2184.1987.tb01093.x. [DOI] [PubMed] [Google Scholar]
- 61.Akoyev V, Takemoto DJ. ZO-1 is required for protein kinase C gamma-driven disassembly of connexin 43. Cell Signal. 2007;19:958–967. doi: 10.1016/j.cellsig.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishizuka Y. Studies and perspectives of protein kinase C. Science. 1986;233:305–312. doi: 10.1126/science.3014651. [DOI] [PubMed] [Google Scholar]
- 63.Saez JC, Nairn AC, Czernik AJ, Fishman GI, Spray DC, Hertzberg EL. Phosphorylation of connexin43 and the regulation of neonatal rat cardiac myocyte gap junctions. J. Mol. Cell. Cardiol. 1997;29:2131–2145. doi: 10.1006/jmcc.1997.0447. [DOI] [PubMed] [Google Scholar]
- 64.Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 2000;126:1503–1512. doi: 10.1083/jcb.149.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Solan JL, Lampe PD. Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun Adhes. 2008;15:75–84. doi: 10.1080/15419060802014016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doble BW, Dang X, Ping P, Fandrich RR, Nickel BE, Jin Y, Cattini PA, Kardami E. Phosphorylation of serine 262 in the gap junction protein connexin-43 regulates DNA synthesis in cell-cell contact forming cardiomyocytes. J. Cell Sci. 2004;117:507–514. doi: 10.1242/jcs.00889. [DOI] [PubMed] [Google Scholar]
- 67.Johnson G, Vaillancourt R. Sequential protein kinase reactions controlling cell growth and differentiation. Curr Opin Cell Biol. 1994;6:230–238. doi: 10.1016/0955-0674(94)90141-4. [DOI] [PubMed] [Google Scholar]
- 68.Cottrell GT, Lin R, Warn-Cramer BJ, Lau AF, Burt JM. Mechanism of v-Src- and mitogen-activated protein kinase-induced reduction of gap junction communication. Am J Physiol Cell Physiol. 2003;284:C511–520. doi: 10.1152/ajpcell.00214.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ruch RJ, Trosko JE, Madhukar BV. Inhibition of connexin43 gap junctional intercellular communication by TPA requires ERK activation. J Cell Biochem. 2001;83:163–169. doi: 10.1002/jcb.1227. [DOI] [PubMed] [Google Scholar]
- 70.Leithe E, Rivedal E. Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J Biol Chem. 2004;279:50089–50096. doi: 10.1074/jbc.M402006200. [DOI] [PubMed] [Google Scholar]
- 71.Leykauf K, Durst M, Alonso A. Phosphorylation and subcellular distribution of connexin43 in normal and stressed cells. Cell Tissue Res. 2003;311:23–30. doi: 10.1007/s00441-002-0645-5. [DOI] [PubMed] [Google Scholar]
- 72.Leithe E, Rivedal E. Epidermal growth factor regulates ubiquitination, internalization and proteasome-dependent degradation of connexin43. J Cell Sci. 2004;117:1211–1220. doi: 10.1242/jcs.00951. [DOI] [PubMed] [Google Scholar]
- 73.Archard HO, Denys FR. Development of annular gap junctions in guinea pig epithelia. J Oral Pathol. 1979;8:187–197. doi: 10.1111/j.1600-0714.1979.tb01885.x. [DOI] [PubMed] [Google Scholar]
- 74.Severs NJ, Shovel KS, Slade AM, Powell T, Twist VW, Green CR. Fate of gap junctions in isolated adult mammalian cardiomyocytes. Circ Res. 1989;65:22–42. doi: 10.1161/01.res.65.1.22. [DOI] [PubMed] [Google Scholar]
- 75.Naus CC, Hearn S, Zhu D, Nicholson BJ, Shivers RR. Ultrastructural analysis of gap junctions in C6 glioma cells transfected with connexin43 cDNA. Exp Cell Res. 1993;206:72–84. doi: 10.1006/excr.1993.1122. [DOI] [PubMed] [Google Scholar]
- 76.Nagy JI, Li WEI, Roy C, Doble BW, Gilchrist JS, Kardami E, Hertzberg EL. Selective monoclonal antibody recognition and cellular localization of an unphosporylated form of connexin43. Exper. Cell Res. 1997;236:127–136. doi: 10.1006/excr.1997.3716. [DOI] [PubMed] [Google Scholar]
- 77.Jordan K, Chodock R, Hand AR, Laird DW. The origin of annular junctions: a mechanism of gap junction internalization. J Cell Sci. 2001;114:763–773. doi: 10.1242/jcs.114.4.763. [DOI] [PubMed] [Google Scholar]
- 78.Kanemitsu MY, Loo LW, Simon S, Lau AF, Eckhart W. Tyrosine phosphorylation of connexin 43 by v-Src is mediated by SH2 and SH3 domain interactions. J. Biol. Chem. 1997;272:22824–22831. doi: 10.1074/jbc.272.36.22824. [DOI] [PubMed] [Google Scholar]
- 79.Lin R, Warn-Cramer BJ, Kurata WE, Lau AF. v-Src phosphorylation of connexin 43 on Tyr247 and Tyr265 disrupts gap junctional communication. J. Cell Biol. 2001;154:815–827. doi: 10.1083/jcb.200102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin R, Martyn KD, Guyette CV, Lau AF, Warn-Cramer BJ. v-Src tyrosine phosphorylation of connexin43: regulation of gap junction communication and effects on cell transformation. Cell Commun Adhes. 2006;13:199–216. doi: 10.1080/15419060600848516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cruciani V, Mikalsen SO. Stimulated phosphorylation of intracellular connexin43. Exp. Cell Res. 1999;251:285–298. doi: 10.1006/excr.1999.4574. [DOI] [PubMed] [Google Scholar]
- 82.Sosinsky GE, Solan JL, Gaietta GM, Ngan L, Lee GJ, Mackey MR, Lampe PD. The C-terminus of Connexin43 adopts different conformations in the golgi and gap junction as detected with structure specific antibodies. Biochem J. 2007;408:375–385. doi: 10.1042/BJ20070550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li WE, Ochalski PA, Hertzberg EL, Nagy JI. Immunorecognition, ultrastructure and phosphorylation status of astrocytic gap junctions and connexin43 in rat brain after cerebral focal ischaemia. Eur J Neurosci. 1998;10:2444–2463. doi: 10.1046/j.1460-9568.1998.00253.x. [DOI] [PubMed] [Google Scholar]
- 84.Jain SK, Schuessler RB, Saffitz JE. Mechanisms of delayed electrical uncoupling induced by ischemic preconditioning. Circ Res. 2003;92:1138–1144. doi: 10.1161/01.RES.0000074883.66422.C5. [DOI] [PubMed] [Google Scholar]
- 85.Li J, Levin MD, Xiong Y, Petrenko N, Patel VV, Radice GL. N-cadherin haploinsufficiency affects cardiac gap junctions and arrhythmic susceptibility. J Mol Cell Cardiol. 2008;44:597–606. doi: 10.1016/j.yjmcc.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boengler K, Dodoni G, Rodriguez-Sinovas A, Cabestrero A, Ruiz-Meana M, Gres P, Konietzka I, Lopez-Iglesias C, Garcia-Dorado D, Di Lisa F, Heusch G, Schulz R. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc Res. 2005;67:234–244. doi: 10.1016/j.cardiores.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 87.Sundset R, Ytrehus K, Zhang Y, Saffitz JE, Yamada KA. Repeated simulated ischemia and protection against gap junctional uncoupling. Cell Commun Adhes. 2007;14:239–249. doi: 10.1080/15419060701821149. [DOI] [PubMed] [Google Scholar]
- 88.Yogo K, Ogawa T, Akiyama M, Ishida N, Takeya T. Identification and functional analysis of novel phosphorylation sites in Cx43 in rat primary granulosa cells. FEBS Lett. 2002;531:132–136. doi: 10.1016/s0014-5793(02)03441-5. [DOI] [PubMed] [Google Scholar]
- 89.Laird DL, Castillo M, Kasprzak L. Gap junction turnover, intracellular trafficking, and phosphorylation of connexin43 in Brefeldin A-treated rat mammary tumor cells. J. Cell Biol. 1995;131:1193–1203. doi: 10.1083/jcb.131.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Richards TS, Dunn CA, Carter WG, Usui ML, Olerud JE, Lampe PD. Protein kinase C spatially and temporally regulates gap junctional communication during human wound repair via phosphorylation of connexin43 on serine368. J Cell Biol. 2004;167:555–562. doi: 10.1083/jcb.200404142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hund TJ, Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Protein kinase Cepsilon mediates salutary effects on electrical coupling induced by ischemic preconditioning. Heart Rhythm. 2007;4:1183–1193. doi: 10.1016/j.hrthm.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maass K, Ghanem A, Kim JS, Saathoff M, Urschel S, Kirfel G, Grummer R, Kretz M, Lewalter T, Tiemann K, Winterhager E, Herzog V, Willecke K. Defective epidermal barrier in neonatal mice lacking the C-terminal region of connexin43. Mol Biol Cell. 2004;15:4597–4608. doi: 10.1091/mbc.E04-04-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Moorby C. A connexin 43 mutant lacking the carboxyl cytoplasmic domain inhibits both growth and motility of mouse 3T3 fibroblasts. Molecular Carcinogenesis. 2000;28:23–30. [PubMed] [Google Scholar]
- 94.Sorgen PL, Duffy HS, Cahill SM, Coombs W, Spray DC, Delmar M, Girvin ME. Sequence-specific resonance assignment of the carboxyl terminal domain of Connexin43. J Biomol NMR. 2002;23:245–246. doi: 10.1023/a:1019892719979. [DOI] [PubMed] [Google Scholar]
- 95.Sorgen PL, Duffy HS, Spray DC, Delmar M. pH-dependent dimerization of the carboxyl terminal domain of Cx43. Biophys J. 2004;87:574–581. doi: 10.1529/biophysj.103.039230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Giepmans BN, Verlaan I, Hengeveld T, Janssen H, Calafat J, Falk MM, Moolenaar WH. Gap junction protein connexin-43 interacts directly with microtubules. Curr Biol. 2001;11:1364–1368. doi: 10.1016/s0960-9822(01)00424-9. [DOI] [PubMed] [Google Scholar]
- 97.Lan Z, Kurata WE, Martyn KD, Jin C, Lau AF. Novel rab GAP-like protein, CIP85, interacts with connexin43 and induces its degradation. Biochemistry. 2005;44:2385–2396. doi: 10.1021/bi048306w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thomas MA, Zosso N, Scerri I, Demaurex N, Chanson M, Staub O. A tyrosine-based sorting signal is involved in connexin43 stability and gap junction turnover. J Cell Sci. 2003;116:2213–2222. doi: 10.1242/jcs.00440. [DOI] [PubMed] [Google Scholar]
- 99.Leykauf K, Salek M, Bomke J, Frech M, Lehmann WD, Durst M, Alonso A. Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J Cell Sci. 2006;119:3634–3642. doi: 10.1242/jcs.03149. [DOI] [PubMed] [Google Scholar]
- 100.Sorgen PL, Duffy HS, Sahoo P, Coombs W, Delmar M, Spray DC. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem. 2004;279:54695–54701. doi: 10.1074/jbc.M409552200. [DOI] [PubMed] [Google Scholar]
- 101.Hirst-Jensen BJ, Sahoo P, Kieken F, Delmar M, Sorgen PL. Characterization of the pH-dependent interaction between the gap junction protein connexin43 carboxyl terminus and cytoplasmic loop domains. J Biol Chem. 2007;282:5801–5813. doi: 10.1074/jbc.M605233200. [DOI] [PubMed] [Google Scholar]
- 102.Duffy HS, Ashton AW, O'Donnell P, Coombs W, Taffet SM, Delmar M, Spray DC. Regulation of connexin43 protein complexes by intracellular acidification. Circ Res. 2004;94:215–222. doi: 10.1161/01.RES.0000113924.06926.11. [DOI] [PubMed] [Google Scholar]
- 103.Toyofuku T, Akamatsu Y, Zhang H, Kuzuya T, Tada M, Hori M. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J Biol Chem. 2001;276:1780–1788. doi: 10.1074/jbc.M005826200. [DOI] [PubMed] [Google Scholar]
- 104.Li W, Nagy JI. Connexin43 phosphorylation state and intercellular communication in cultured astrocytes following hypoxia and protein phosphatase inhibition. Eur. J. Neurosci. 2000;12:2644–2650. doi: 10.1046/j.1460-9568.2000.00162.x. [DOI] [PubMed] [Google Scholar]
- 105.Kardami E, Dang X, Iacobas DA, Nickel BE, Jeyaraman M, Srisakuldee W, Makazan J, Tanguy S, Spray DC. The role of connexins in controlling cell growth and gene expression. Prog Biophys Mol Biol. 2007;94:245–264. doi: 10.1016/j.pbiomolbio.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 106.Pointis G, Fiorini C, Gilleron J, Carette D, Segretain D. Connexins as precocious markers and molecular targets for chemical and pharmacological agents in carcinogenesis. Curr Med Chem. 2007;14:2288–2303. doi: 10.2174/092986707781696564. [DOI] [PubMed] [Google Scholar]
- 107.Omori Y, Li Q, Nishikawa Y, Yoshioka T, Yoshida M, Nishimura T, Enomoto K. Pathological significance of intracytoplasmic connexin proteins: implication in tumor progression. J Membr Biol. 2007;218:73–77. doi: 10.1007/s00232-007-9048-6. [DOI] [PubMed] [Google Scholar]
- 108.King TJ, Bertram JS. Connexins as targets for cancer chemoprevention and chemotherapy. Biochim Biophys Acta. 2005;1719:146–160. doi: 10.1016/j.bbamem.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 109.Duflot-Dancer A, Mesnil M, Yamasaki H. Dominant-negative abrogation of connexin-mediated cell growth control by mutant connexin genes. Oncogene. 1997;15:2151–2158. doi: 10.1038/sj.onc.1201393. [DOI] [PubMed] [Google Scholar]
- 110.Huang R, Lin Y, Wang CC, Gano J, Lin B, Shi Q, Boynton A, Burke J, Huang RP. Connexin 43 suppresses human glioblastoma cell growth by down-regulation of monocyte chemotactic protein 1, as discovered using protein array technology. Cancer Res. 2002;62:2806–2812. [PubMed] [Google Scholar]
- 111.Krutovskikh VA, Troyanovsky SM, Piccoli C, Tsuda H, Asamoto M, Yamasaki H. Differential effect of subcellular localization of communication impairing gap junction protein connexin43 on tumor cell growth in vivo. Oncogene. 2000;19:505–513. doi: 10.1038/sj.onc.1203340. [DOI] [PubMed] [Google Scholar]
- 112.Moorby C, Patel M. Dual functions for connexins: Cx43 regulates growth independently of gap junction formation. Exp Cell Res. 2001;271:238–248. doi: 10.1006/excr.2001.5357. [DOI] [PubMed] [Google Scholar]
- 113.Olbina G, Eckhart W. Mutations in the second extracellular region of connexin 43 prevent localization to the plasma membrane, but do not affect its ability to suppress cell growth. Mol Cancer Res. 2003;1:690–700. [PubMed] [Google Scholar]
- 114.Fu CT, Bechberger JF, Ozog MA, Perbal B, Naus CC. CCN3 (NOV) interacts with connexin43 in C6 glioma cells: possible mechanism of connexin-mediated growth suppression. J Biol Chem. 2004;279:36943–36950. doi: 10.1074/jbc.M403952200. [DOI] [PubMed] [Google Scholar]
- 115.Gellhaus A, Dong X, Propson S, Maass K, Klein-Hitpass L, Kibschull M, Traub O, Willecke K, Perbal B, Lye SJ, Winterhager E. Connexin43 interacts with NOV: A possible mechanism for negative regulation of cell growth in choriocarcinoma cells. J Biol Chem. 2004 doi: 10.1074/jbc.M404073200. [DOI] [PubMed] [Google Scholar]
- 116.Zhang YW, Nakayama K, Morita I. A novel route for connexin 43 to inhibit cell proliferation: negative regulation of S-phase kinase-associated protein (Skp 2). Cancer Res. 2003;63:1623–1630. [PubMed] [Google Scholar]
- 117.Zhang YW, Kaneda M, Morita I. The gap junction-independent tumor-suppressing effect of connexin 43. J Biol Chem. 2003;278:44852–44856. doi: 10.1074/jbc.M305072200. [DOI] [PubMed] [Google Scholar]
- 118.Bruzzone R, Dermietzel R. Structure and function of gap junctions in the developing brain. Cell Tissue Res. 2006;326:239–248. doi: 10.1007/s00441-006-0287-0. [DOI] [PubMed] [Google Scholar]
- 119.Nagy JI, Dudek FE, Rash JE. Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Res Brain Res Rev. 2004;47:191–215. doi: 10.1016/j.brainresrev.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 120.Sohl G, Maxeiner S, Willecke K. Expression and functions of neuronal gap junctions. Nat Rev Neurosci. 2005;6:191–200. doi: 10.1038/nrn1627. [DOI] [PubMed] [Google Scholar]
- 121.Orthmann-Murphy JL, Abrams CK, Scherer SS. Gap junctions couple astrocytes and oligodendrocytes. J Mol Neurosci. 2008;35:101–116. doi: 10.1007/s12031-007-9027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Penes MC, Li X, Nagy JI. Expression of zonula occludens-1 (ZO-1) and the transcription factor ZO-1-associated nucleic acid-binding protein (ZONAB)-MsY3 in glial cells and colocalization at oligodendrocyte and astrocyte gap junctions in mouse brain. Eur J Neurosci. 2005;22:404–418. doi: 10.1111/j.1460-9568.2005.04225.x. [DOI] [PubMed] [Google Scholar]
- 123.Ciolofan C, Li XB, Olson C, Kamasawa N, Gebhardt BR, Yasumura T, Morita M, Rash JE, Nagy JI. Association of connexin36 and zonula occludens-1 with zonula occludens-2 and the transcription factor zonula occludens-1-associated nucleic acid-binding protein at neuronal gap junctions in rodent retina. Neuroscience. 2006;140:433–451. doi: 10.1016/j.neuroscience.2006.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li X, Penes M, Odermatt B, Willecke K, Nagy JI. Ablation of Cx47 in transgenic mice leads to the loss of MUPP1, ZONAB and multiple connexins at oligodendrocyte-astrocyte gap junctions. Eur J Neurosci. 2008;28:1503–1517. doi: 10.1111/j.1460-9568.2008.06431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Squecco R, Sassoli C, Nuti F, Martinesi M, Chellini F, Nosi D, Zecchi-Orlandini S, Francini F, Formigli L, Meacci E. Sphingosine 1-phosphate induces myoblast differentiation through Cx43 protein expression: a role for a gap junction-dependent and -independent function. Mol Biol Cell. 2006;17:4896–4910. doi: 10.1091/mbc.E06-03-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Singh D, Solan JL, Taffet SM, Javier R, Lampe PD. Connexin 43 interacts with zona occludens-1 and -2 proteins in a cell cycle stage-specific manner. J Biol Chem. 2005;280:30416–30421. doi: 10.1074/jbc.M506799200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Polo S, Di Fiore PP. Endocytosis conducts the cell signaling orchestra. Cell. 2006;124:897–900. doi: 10.1016/j.cell.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 128.Pahujaa M, Anikin M, Goldberg GS. Phosphorylation of connexin43 induced by Src: regulation of gap junctional communication between transformed cells. Exp Cell Res. 2007;313:4083–4090. doi: 10.1016/j.yexcr.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 129.Herve JC, Sarrouilhe D. Protein phosphatase modulation of the intercellular junctional communication: importance in cardiac myocytes. Prog Biophys Mol Biol. 2006;90:225–248. doi: 10.1016/j.pbiomolbio.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 130.Chen S-C, Pelletier DB, Ao P, Boynton AL. Connexin43 reverses the phenotype of transformed cells and alters their expression of cyclin/cyclin dependent kinases. Cell Growth Differ. 1995;6:681–690. [PubMed] [Google Scholar]
- 131.Burt JM, Nelson TK, Simon AM, Fang JS. Connexin 37 profoundly slows cell cycle progression in rat insulinoma cells. Am J Physiol Cell Physiol. 2008;295:C1103–1112. doi: 10.1152/ajpcell.299.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bittman K, Owens DF, Kriegstein AR, LoTurco JJ. Cell coupling and uncoupling in the ventricular zone of developing neocortex. J. Neurosci. 1997;17:7037–7044. doi: 10.1523/JNEUROSCI.17-18-07037.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bittman KS, LoTurco JJ. Differential regulation of connexin 26 and 43 in murine neocortical precursors. Cerebral Cortex. 1999;9:188–195. doi: 10.1093/cercor/9.2.188. [DOI] [PubMed] [Google Scholar]
- 134.Koo SK, Kim DY, Park SD, Kang KW, Joe CO. PKC phosphorylation disrupts gap junctional communication at G0/S phase in clone 9 cells. Mol. Cell. Biochem. 1997;167:41–49. doi: 10.1023/a:1006831114120. [DOI] [PubMed] [Google Scholar]
- 135.Stein LS, Boonstra J, Burghardt RC. Reduced cell-cell communication between mitotic and nonmitotic cells. Exper. Cell Res. 1992;198:1–7. doi: 10.1016/0014-4827(92)90141-t. [DOI] [PubMed] [Google Scholar]
- 136.Fitzgerald DJ, Fusenig NE, Boukamp P, Piccoli C, Mesnil M, Yamasaki H. Expression and function of connexin in normal and transformed human keratinocytes in culture. Carcinogenesis. 1994;15:1859–1865. doi: 10.1093/carcin/15.9.1859. [DOI] [PubMed] [Google Scholar]
- 137.Maestrini E, Korge B, Ocaña-Sierra J, Calzolari E, Cambiaghi S, Scudder P, Hovnanian A, Monaco A, Munro C. A missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel's syndrome) in three unrelated families. Hum Mol Genet. 1999;8:1237–1243. doi: 10.1093/hmg/8.7.1237. [DOI] [PubMed] [Google Scholar]
- 138.Common JE, O'Toole EA, Leigh IM, Thomas A, Griffiths WA, Venning V, Grabczynska S, Peris Z, Kansky A, Kelsell DP. Clinical and genetic heterogeneity of erythrokeratoderma variabilis. J Invest Dermatol. 2005;125:920–927. doi: 10.1111/j.0022-202X.2005.23919.x. [DOI] [PubMed] [Google Scholar]
- 139.Richard G, Brown N, Rouan F, Van der Schroeff JG, Bijlsma E, Eichenfield LF, Sybert VP, Greer KE, Hogan P, Campanelli C, Compton JG, Bale SJ, DiGiovanna JJ, Uitto J. Genetic heterogeneity in erythrokeratodermia variabilis: novel mutations in the connexin gene GJB4 (Cx30.3) and genotype-phenotype correlations. J Invest Dermatol. 2003;120:601–609. doi: 10.1046/j.1523-1747.2003.12080.x. [DOI] [PubMed] [Google Scholar]
- 140.Simon AM, Goodenough DA, Paul DL. Mice lacking connexin40 have cardiac conduction abnormalities characteristic of atrioventricular block and bundle branch block. Curr. Biol. 1998;8:295–298. doi: 10.1016/s0960-9822(98)70113-7. [DOI] [PubMed] [Google Scholar]
- 141.van Steensel MA. Gap junction diseases of the skin. Am J Med Genet C Semin Med Genet. 2004;131C:12–19. doi: 10.1002/ajmg.c.30030. [DOI] [PubMed] [Google Scholar]
- 142.Salomon D, Masgrau E, Vischer S, Ullrich S, Dupont E, Sappino P, Saurat J-H, Meda P. Topography of mammalian connexins in human skin. J. Invest. Dermatol. 1994;103:240–247. doi: 10.1111/1523-1747.ep12393218. [DOI] [PubMed] [Google Scholar]
- 143.Wilgenbus KK, Kirkpatrick CJ, Knuechel R, Willecke K, Traub O. Expression of Cx26, Cx32 and Cx43 gap junction proteins in normal and neoplastic human tissues. Int J Cancer. 1992;51:522–529. doi: 10.1002/ijc.2910510404. [DOI] [PubMed] [Google Scholar]
- 144.Guo H, Acevedo P, Parsa FD, Bertram JS. Gap-junctional protein connexin 43 is expressed in dermis and epidermis of human skin: differential modulation by retinoids. J Invest Dermatol. 1992;99:460–467. doi: 10.1111/1523-1747.ep12616154. [DOI] [PubMed] [Google Scholar]
- 145.Arita K, Akiyama M, Tsuji Y, McMillan JR, Eady RA, Shimizu H. Changes in gap junction distribution and connexin expression pattern during human fetal skin development. J Histochem Cytochem. 2002;50:1493–1500. doi: 10.1177/002215540205001109. [DOI] [PubMed] [Google Scholar]
- 146.Clark RAF. Cutaneous tissue repair: basic biological considerations. J. Am. Acad. Derm. 1985;13:701–725. doi: 10.1016/s0190-9622(85)70213-7. [DOI] [PubMed] [Google Scholar]
- 147.Grinnell F. Wound repair, keratinocyte activation and integrin modulation. J. Cell Sci. 1992;101:1–5. doi: 10.1242/jcs.101.1.1. [DOI] [PubMed] [Google Scholar]
- 148.Gailit J, Clark RA. Wound repair in the context of extracellular matrix. Curr. Opin. Cell Biol. 1994;6:717–725. doi: 10.1016/0955-0674(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 149.Martin P. Wound healing--aiming for perfect skin regeneration. Science. 1997;276:75–81. doi: 10.1126/science.276.5309.75. [DOI] [PubMed] [Google Scholar]
- 150.Goliger JA, Paul DL. Wounding alters epidermal connexin expression and gap junction-mediated intercellular communication. Mol. Biol. Cell. 1995;6:1491–1501. doi: 10.1091/mbc.6.11.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Saitoh M, Oyamada M, Oyamada Y, Kaku T, Mori M. Changes in the expression of gap junction proteins (connexins) in hamster tongue epithelium during would healing and carcinogenesis. Carcinogenesis. 1997;18:1319–1328. doi: 10.1093/carcin/18.7.1319. [DOI] [PubMed] [Google Scholar]
- 152.Qiu C, Coutinho P, Frank S, Franke S, Law LY, Martin P, Green CR, Becker DL. Targeting connexin43 expression accelerates the rate of wound repair. Curr. Biol. 2003;13:1697–1703. doi: 10.1016/j.cub.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 153.Wang CM, Lincoln J, Cook JE, Becker DL. Abnormal connexin expression underlies delayed wound healing in diabetic skin. Diabetes. 2007;56:2809–2817. doi: 10.2337/db07-0613. [DOI] [PubMed] [Google Scholar]
- 154.Nakano Y, Oyamada M, Dai P, Nakagami T, Kinoshita S, Takamatsu T. Connexin43 knockdown accelerates wound healing but inhibits mesenchymal transition after corneal endothelial injury in vivo. Invest Ophthalmol Vis Sci. 2008;49:93–104. doi: 10.1167/iovs.07-0255. [DOI] [PubMed] [Google Scholar]
- 155.Kretz M, Euwens C, Hombach S, Eckardt D, Teubner B, Traub O, Willecke K, Ott T. Altered connexin expression and wound healing in the epidermis of connexin-deficient mice. J. Cell Sci. 2003;116:3443–3452. doi: 10.1242/jcs.00638. [DOI] [PubMed] [Google Scholar]
- 156.Brandner JM, Houdek P, Husing B, Kaiser C, Moll I. Connexins 26, 30, and 43: differences among spontaneous, chronic, and accelerated human wound healing. J Invest Dermatol. 2004;122:1310–1320. doi: 10.1111/j.0022-202X.2004.22529.x. [DOI] [PubMed] [Google Scholar]
- 157.Coutinho P, Qiu C, Frank S, Tamber K, Becker D. Dynamic changes in connexin expression correlate with key events in the wound healing process. Cell Biol Int. 2003;27:525–541. doi: 10.1016/s1065-6995(03)00077-5. [DOI] [PubMed] [Google Scholar]
- 158.Tada J, Hashimoto K. Ultrastructural localization of gap junction protein connexin 43 in normal human skin, basal cell carcinoma, and squamous cell carcinoma. J Cutan Pathol. 1997;24:628–635. doi: 10.1111/j.1600-0560.1997.tb01094.x. [DOI] [PubMed] [Google Scholar]
- 159.Revel JP, Karnovsky MJ. Hexagonal array of subunits in intercellular junctions of the mouse heart and liver. J Cell Biol. 1967;33:C7–C12. doi: 10.1083/jcb.33.3.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Severs NJ. The cardiac gap junction and intercalated disc. Int J Cardiol. 1990;26:137–173. doi: 10.1016/0167-5273(90)90030-9. [DOI] [PubMed] [Google Scholar]
- 161.Angst BD, Khan LU, Severs NJ, Whitely K, Rothery S, Thompson RP, Magee AI, Gourdie RG. Dissociated spatial patterning of gap junctions and cell adhesion junctions during postnatal differentiation of ventricular myocardium. Circ Res. 1997;80:88–94. doi: 10.1161/01.res.80.1.88. [DOI] [PubMed] [Google Scholar]
- 162.Peters NS, Severs NJ, Rothery SM, Lincoln C, Yacoub MH, Green CR. Spatiotemporal relation between gap junctions and fascia adherens junctions during postnatal development of human ventricular myocardium. Circulation. 1994;90:713–725. doi: 10.1161/01.cir.90.2.713. [DOI] [PubMed] [Google Scholar]
- 163.Davis LM, Kanter HL, Beyer EC, Saffitz JE. Distinct gap junction protein phenotypes in cardiac tissues with disparate conduction properties. J Am Coll Cardiol. 1994;24:1124–1132. doi: 10.1016/0735-1097(94)90879-6. [DOI] [PubMed] [Google Scholar]
- 164.Davis LM, Rodefeld ME, Green K, Beyer EC, Saffitz JE. Gap junction protein phenotypes of the human heart and conduction system. J Cardiovasc Electrophysiol. 1995;6:813–822. doi: 10.1111/j.1540-8167.1995.tb00357.x. [DOI] [PubMed] [Google Scholar]
- 165.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kleber AG, Schuessler RB, Saffitz JE. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656–662. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- 166.Miura T, Ohnuma Y, Kuno A, Tanno M, Ichikawa Y, Nakamura Y, Yano T, Miki T, Sakamoto J, Shimamoto K. Protective role of gap junctions in preconditioning against myocardial infarction. Am J Physiol Heart Circ Physiol. 2004;286:H214–221. doi: 10.1152/ajpheart.00441.2003. [DOI] [PubMed] [Google Scholar]
- 167.Schulz R, Gres P, Skyschally A, Duschin A, Belosjorow S, Konietzka I, Heusch G. Ischemic preconditioning preserves connexin 43 phosphorylation during sustained ischemia in pig hearts in vivo. Faseb J. 2003;17:1355–1357. doi: 10.1096/fj.02-0975fje. [DOI] [PubMed] [Google Scholar]
- 168.Axelsen L, Stahlhut M, Mohammed S, Larsen B, Nielsen M, Holstein-Rathlou N, Andersen S, Jensen O, Hennan J, Kjølbye A. J Mol Cell Cardiol. . 2006;40:790–798. doi: 10.1016/j.yjmcc.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 169.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 170.Li X, Heinzel FR, Boengler K, Schulz R, Heusch G. Role of connexin 43 in ischemic preconditioning does not involve intercellular communication through gap junctions. J Mol Cell Cardiol. 2004;36:161–163. doi: 10.1016/j.yjmcc.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 171.Schwanke U, Konietzka I, Duschin A, Li X, Schulz R, Heusch G. No ischemic preconditioning in heterozygous connexin43-deficient mice. Am J Physiol Heart Circ Physiol. 2002;283:H1740–1742. doi: 10.1152/ajpheart.00442.2002. [DOI] [PubMed] [Google Scholar]
- 172.Schwanke U, Li X, Schulz R, Heusch G. No ischemic preconditioning in heterozygous connexin 43-deficient mice--a further in vivo study. Basic Res Cardiol. 2003;98:181–182. doi: 10.1007/s003950300002. [DOI] [PubMed] [Google Scholar]
- 173.Kjolbye AL, Haugan K, Hennan JK, Petersen JS. Pharmacological modulation of gap junction function with the novel compound rotigaptide: a promising new principle for prevention of arrhythmias. Basic Clin Pharmacol Toxicol. 2007;101:215–230. doi: 10.1111/j.1742-7843.2007.00123.x. [DOI] [PubMed] [Google Scholar]
- 174.Kjolbye AL, Dikshteyn M, Eloff BC, Deschenes I, Rosenbaum DS. Maintenance of intercellular coupling by the antiarrhythmic peptide rotigaptide suppresses arrhythmogenic discordant alternans. Am J Physiol Heart Circ Physiol. 2008;294:H41–49. doi: 10.1152/ajpheart.01089.2006. [DOI] [PubMed] [Google Scholar]
- 175.Lewandowski R, Procida K, Vaidyanathan R, Coombs W, Jalife J, Nielsen MS, Taffet SM, Delmar M. RXP-E: a connexin43-binding peptide that prevents action potential propagation block. Circ Res. 2008;103:519–526. doi: 10.1161/CIRCRESAHA.108.179069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ruiz-Meana M, Rodriguez-Sinovas A, Cabestrero A, Boengler K, Heusch G, Garcia-Dorado D. Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2008;77:325–333. doi: 10.1093/cvr/cvm062. [DOI] [PubMed] [Google Scholar]
- 177.Miura T, Yano T, Naitoh K, Nishihara M, Miki T, Tanno M, Shimamoto K. Delta-opioid receptor activation before ischemia reduces gap junction permeability in ischemic myocardium by PKC-epsilon-mediated phosphorylation of connexin 43. Am J Physiol Heart Circ Physiol. 2007;293:H1425–1431. doi: 10.1152/ajpheart.01115.2006. [DOI] [PubMed] [Google Scholar]
- 178.Jeyaraman M, Tanguy S, Fandrich RR, Lukas A, Kardami E. Ischemia-induced dephosphorylation of cardiomyocyte connexin-43 is reduced by okadaic acid and calyculin A but not fostriecin. Mol Cell Biochem. 2003;242:129–134. [PubMed] [Google Scholar]
- 179.Britz-Cunningham SH, Shah MM, Zuppan CW, Fletcher WH. Mutations of the connexin43 gap junction gene in patients with heart malformations and defects of laterality. New Engl. J. Med. 1995;332:1323–1329. doi: 10.1056/NEJM199505183322002. [DOI] [PubMed] [Google Scholar]
- 180.Debrus S, Tuffery S, Matsuoka R, Galal O, Sarda P, Sauer U, Bozio A, Tanman B, Toutain A, Claustres M, Le Paslier D, Bouvagnet P. Lack of evidence for connexin 43 gene mutations in human autosomal recessive lateralization defects. J. Mol. Cell. Cardiol. 1997;29:1423–1431. doi: 10.1006/jmcc.1997.0380. [DOI] [PubMed] [Google Scholar]
- 181.Ek-Vitorin JF, Calero G, Morley GE, Coombs W, Taffet SM, Delmar M. pH regulation of connexin43: molecular analysis of the gating particle. Biophys. J. 1996;71:1273–1284. doi: 10.1016/S0006-3495(96)79328-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Levin M, Mercola M. Gap junctions are involved in the early generation of left-right asymmetry. Dev. Biol. 1998;203:90–105. doi: 10.1006/dbio.1998.9024. [DOI] [PubMed] [Google Scholar]
- 183.Bowling N, Huang X, Sandusky GE, Fouts RL, Mintze K, Esterman M, Allen PD, Maddi R, McCall E, Vlahos CJ. Protein kinase C-alpha and -epsilon modulate connexin-43 phosphorylation in human heart. J. Mol. Cell. Cardiol. 2001;33:789–798. doi: 10.1006/jmcc.2000.1349. [DOI] [PubMed] [Google Scholar]
- 184.Saurin AT, Pennington DJ, Raat NJ, Latchman DS, Owen MJ, Marber MS. Targeted disruption of the protein kinase C epsilon gene abolishes the infarct size reduction that follows ischaemic preconditioning of isolated buffer-perfused mouse hearts. Cardiovasc Res. 2002;55:672–680. doi: 10.1016/s0008-6363(02)00325-5. [DOI] [PubMed] [Google Scholar]
- 185.Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. 2007;1767:1007–1031. doi: 10.1016/j.bbabio.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Heinzel FR, Luo Y, Li X, Boengler K, Buechert A, Garcia-Dorado D, Di Lisa F, Schulz R, Heusch G. Impairment of diazoxide-induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ Res. 2005;97:583–586. doi: 10.1161/01.RES.0000181171.65293.65. [DOI] [PubMed] [Google Scholar]