

Abstract
Background
Atopic dermatitis (AD) is a chronic inflammatory skin disease that is characterized by a defective skin barrier function. Recent studies have reported mutations of the skin barrier gene encoding filaggrin in a subset of patients with AD.
Objective
We investigated whether reduced filaggrin expression was found in AD patients who were not carriers of known filaggrin mutations and if filaggrin expression was modulated by the atopic inflammatory response.
Methods
Filaggrin expression was measured in skin biopsies and cultured keratinocytes using real-time RT-PCR and immunohistochemistry. Filaggrin loss-of-function mutations were screened in a total of 69 subjects.
Results
As compared with normal skin, filaggrin expression was significantly reduced (p<0.05) in acute AD skin, with further reduction seen in acute lesions from three European American AD subjects who were heterozygous for the 2282del4 mutation. This was confirmed using immunohistochemistry. AD skin is characterized by the over-expression of IL-4 and IL-13. Keratinocytes differentiated in the presence of IL-4 and IL-13 exhibited significantly reduced filaggrin gene expression (0.04±0.01 ng filaggrin/ng GAPDH, p<0.05) compared to media alone (0.16±0.03).
Conclusion
Patients with AD have an acquired defect in filaggrin expression which can be modulated by the atopic inflammatory response.
Clinical Implications
The atopic immune response contributes to the skin barrier defect in AD; therefore neutralization of IL-4 and IL-13 could improve skin barrier integrity.
Keywords: Atopic Dermatitis, skin barrier, filaggrin
Introduction
Atopic dermatitis (AD) is a chronic inflammatory skin disorder that affects approximately 17% of children and leads to significant disruption in quality of life.1,2 There has been considerable interest in the disease mechanism which drives the so-called “atopic march” reflecting a progression from AD to asthma.3 Recent studies have focused on the impaired epidermal barrier identified in AD patients.4 Using a murine model of AD, it has been shown that skin barrier dysfunction not only enhances allergen sensitization, but also leads to systemic allergic responses such as increased IgE levels and airway hyper-reactivity.5 These observations support the concept that absorption of allergens through the skin of AD patients may predispose to the atopic march.
Filament aggregating protein (filaggrin) is a key protein that plays an important role in formation of the cornified cell envelope (CCE), which is critical for an effective skin barrier. Filaggrin binds to and is responsible for the aggregation of keratins (K1/10) which induce the cytoskeleton to collapse and result in formation of corneocytes. These corneocytes are then heavily cross-linked by the action of transglutaminases and comprise the CCE.6 Recent genetic studies have shown that loss-of-function (null) mutations in the gene encoding filaggrin (FLG) result in impaired skin barrier function and are strongly associated with the development of asthma in AD patients.7-9 Of note, the filaggrin null mutation is observed in less than one-third of the general AD population.7-9 Since most AD patients have a defective skin barrier, this suggests there must be additional mechanisms which modulate filaggrin expression and barrier integrity.
AD skin is characterized by the over-expression of interleukin (IL)-4 and IL-13, Th2 cytokines known to induce atopic responses and down-regulate skin innate immune response genes.10-12 Therefore, we investigated whether filaggrin deficiency in AD may be acquired as the result of Th2 cytokine modulation, and to what extent the two well-characterized FLG mutations (R501X and 2282del4) contribute to altered filaggrin expression in a North American population of AD.
Methods
Study Subjects
Subjects included 39 healthy persons (27 European American, 11 African American, 1 Asian American) with no history of skin disease (mean age: 36.8 ± 2.1 years), 30 patients (17 European American, 12 African American, 1 Hispanic) with moderate AD (mean age: 36.2 ± 1.8 years; 20%-60% skin involvement), and six subjects with lichen planus (mean age: 51.3 ± 7.3 years). Subjects were recruited as part of the NIAID-funded Atopic Dermatitis and Vaccinia Network (ADVN). None of the subjects had received systemic corticosteroids or cyclosporine previously, and none had received topical corticosteroid or calcineurin inhibitors for a period of at least one week before enrollment. The study was approved by the institutional review board at National Jewish Medical and Research Center in Denver, and University of Rochester Medical Center. All subjects gave written informed consent prior to participation in these studies.
Two millimeter punch biopsies were collected from acute erythematous AD lesions (defined as less than three days after onset according to Hamid et al.13 and uninvolved skin of the same AD patients (n=16), lichen planus lesions (n=6), and normal healthy skin (n=15). The skin samples were submerged immediately in either Tri-Reagent (Molecular Research Center, Inc, Cincinnati, OH) or 10% buffered formalin for real-time RT-PCR and immunohistochemical studies, respectively.
Genotypic Analysis of Filaggrin
Blood samples were collected from all 69 subjects for extraction of DNA for genotyping using PaxGene Blood DNA tubes (PreAnalytix GmbH, Hombrechtikon, CH) and genomic DNA was extracted from peripheral blood samples using M48 Biorobot (Qiagen, Inc, Valencia, Calif).
R501X mutation analysis
Genotyping for R501X was performed with the ABI 3730 Detect System (Applied Biosystems, Foster City, CA). Briefly, a 178-base pair (bp) fragment was amplified by PCR with a pair of primers: R051X-F, 5′-GACCAGCACTGGAGGAAGAC and R501X-R, 5′-ATGGGAACCTGAGTGTCCAG. PCR was performed in a total volume of 20 μl containing 1 × PCR buffer (including 1.5 mM MgCI2), 12 pmol primers, 2mM dNTPs, 20 ng genomic DNA, and 1.0 U DNA polymerase (Qiagen). Amplification was performed as follows: 50°C for two minutes, 95°C for ten minutes, then 40 cycles at 95°C for 15 seconds, 60°C for one minute; and 72°C for five minutes. All PCR products were purified with QIAquick PCR purification Kit (Qiagen) and bi-directionally sequenced on an ABI 3730 automated sequencer with Big-Dye terminator cycle sequencing reagents (Applied Biosystems).
2282del4 mutation analysis
Genotyping for the 2282del4 was performed with an Applied Biosystems 3700 DNA Sequencer with the addition of a size standard and data analysis using Genotyper (Applied Biosystems), an allele calling software program. In brief, a PCR fragment amplifying 152 bp of genomic DNA was amplified with primers: 5′-6-FAM-CCAGTGGTAGTCAGGCCACT and 5′-AAAGACCCTGAACGTCGAGA. PCR was performed in a total volume of 20 μl containing 1 × PCR buffer (including 1.5 mM MgCI2), 12 pmol primers, 2mM dNTPs, 20 ng genomic DNA, and 1.0 U DNA polymerase (Qiagen). Amplification was performed as follows: 50°C for two minutes, 95°C for 10 minutes, then 40 cycles at 95°C for 15 seconds, 60°C for one minute; and 72°C for five minutes. Fragments were diluted 1:20 and sized according to the manufacturer's recommended protocol (Applied Biosystems). The wild-type allele was 152 bp, and the 2282del4 allele was 148 bp.
Quantitative Real-Time RT-PCR
Total RNA was isolated from 2-mm skin biopsy samples by chloroform:phenol extraction and isopropanol precipitation according to manufacturer's guidelines (Molecular Research Center, Inc.). RNeasy Mini Kits (Qiagen, Valencia, CA) were used according to the manufacturer's protocol to isolate RNA from cell cultures and to further purify RNA from skin biopsies. One microgram of RNA was reverse-transcribed in a 20-μl reaction containing Random Primers (500 μg/ml, Invitrogen, Carlsbad, CA), dNTP (10 mM, Invitrogen), 5X First Strand Buffer (Invitrogen), DTT (0.1M, Invitrogen), Superscript III enzyme (200 U/μl, Invitrogen) and RNase inhibitor (10 U/μl, Invitrogen). Real time PCR was performed and analyzed by the dual-labeled fluorigenic probe method using an ABI Prism 7300 sequence detector (Applied Biosystems). Primers and probes for human GAPDH and filaggrin were purchased from Applied Biosystems. Amplification reactions were performed in MicroAmp optical tubes (Applied Biosystems) in a 25 μl volume as previously described.14 Relative expression levels were calculated by the relative standard curve method as outlined in the manufacturer's technical bulletin. A standard curve was generated using the fluorescent data from the ten-fold serial dilutions of total RNA of the highest expression sample. This was then used to calculate the relative amounts of target mRNA in test samples. Quantities of all targets in test samples were normalized to the corresponding GAPDH levels in cultured keratinocytes and skin biopsies.
Filaggrin Immunohistochemical Staining
Paraffin-embedded tissues were cut at 5 μm and placed on frosted microscope slides. Slides were deparaffinized using a series of xylene and ethanol washes and then stained using the Cell and Tissue Staining Kit (R&D Systems). Endogenous peroxidase was blocked by incubating slides with 3% H2O2 for five minutes and then tissue sections were then blocked with serum blocking reagent G (R&D Systems) for 15 minutes. Slides were then stained overnight with a monoclonal mouse anti-human filaggrin (Abcam, Cambridge, MA) at a 1:500 dilution. The secondary antibody and DAB stain were added according to the manufacturer's protocol (R&D Systems). The skin sections were counterstained with hematoxylin. Antibody specificity was confirmed using purified non-immune mouse IgG (Southern Biotechnology, Birmingham, AL). All slides were coded prior to analysis and read blindly, to ensure patient anonymity. The intensity of the immunostaining was graded with the use of microscopy on a scale from 0 to 5, with 0 indicating no staining and 5 the most intense staining.
Immuno-Dot-Blot for Filaggrin
Immuno-blot was performed with a Bio-Dot Microfiltration Apparatus from Bio-Rad (Hercules, CA). Protein was isolated from primary keratinocytes by acetone precipitation and washed using 0.3 mol/L guanidine hydrochloride in 95% ethanol with 2.5% glycerol according to the manufacturer's guidelines (Molecular Research Center, Inc). Twenty five micrograms of total protein was loaded in a final volume of 100 μL TRIS-buffered saline with 0.5% Tween 20 (TBST) and allowed to bind to the nitrocellulose membrane (Bio-Rad Laboratories) by gravity filtration. The membrane was then blocked in 5% milk in TBST for 30 minutes at room temperature. After blocking, the membrane was washed once with TBST to remove residual blocking solution and then incubated overnight at 4°C with anti-filaggrin (Abcam) at 4 μg/mL in TBST. The blot was washed 3 times with TBST for 5 minutes and then incubated with anti-mouse horseradish peroxidase conjugated secondary antibody (Amersham Biosciences, Buckinghamshire, UK) at a dilution of 1:5,000 in TBST for 1 hour at room temperature. Blots were washed 3 times for 5 minutes each with TBST and then developed with ECL Western Blotting Detection Reagents (Amersham Biosciences) according to the manufacturer's protocol. The intensity of filaggrin protein was determined by digital imaged analysis using the National Institutes of Health image 1.61 software programs. Specificity of the antibody was determined by replacing the primary antibody with a mouse IgG1 isotype control (Southern Biotechnology, Birmingham, Ala). A standard curve was generated for filaggrin protein by using recombinant human filaggrin protein (Abcam).
Keratinocyte Cell Culture
Primary human keratinocytes (Cascade Biologics, Portland, OR) were grown in serum-free keratinocyte growth medium (EpiLife®, Cascade Biologics), supplemented with 1% human keratinocytes growth supplement V2 (Cascade Biologics), 0.06 mM CaCl2 and 1% of penicillin and streptomycin.
To investigate the effects of cytokines on filaggrin expression, keratinocytes were differentiated for five days in 1.3 mM CaCl2 and cytokines were added for the entire differentiation period or the final 24 hours. IL-4 (50ng/ml) and IL-13 (50ng/ml, R&D Systems) were purchased from R&D Systems (Minneapolis, MN). Total RNA was then isolated for analysis of filaggrin gene expression by real-time RT-PCR.
Statistical Analysis
Statistical analysis of gene expression and immunohistochemical staining was conducted using Graph Pad Prism, version 4.03 (San Diego, California). Statistical differences between groups were determined using an unpaired T test with significant differences conferred when p<0.05. In cases where multiple groups were compared with a control, data were analyzed by a one-way analysis of variance (ANOVA), and significant differences were determined by a Tukey-Kramer test.15 For associations of genetic mutations with disease status, data was analyzed in STATA 8.2 (StataCorp LP., College Station, TX) using Fisher's exact test. Analyses of the European American and African American subjects were conducted separately; subjects from the other ethnic groups were considered for allele frequency only. Departures from Hardy-Weinberg equilibrium (HWE) proportions were tested among cases and controls separately, using SIB-PAIR 1.00 (David Duffy).
Results
Characterization of Filaggrin Expression
Recent studies have shown that the loss-of-function mutations, R501X and 2282del4, are associated with AD in European Americans.7-9 Therefore we genotyped these mutations in 39 healthy subjects (27 European American, 11 African American, 1 Asian American) and 30 subjects with mild to moderate AD (17 European American, 12 African American, 1 Hispanic). None of the subjects genotyped had the R501X mutation. The frequency of the 2282del4 loss-of function mutation was similar to recent reports of frequencies among subjects with and without AD;7-9 however, all five subjects with the 2282del4 in our study were heterozygotes. Counts of European Caucasian AD and healthy subjects by genotype are presented in Table 1, and given the heterozygote status of all subjects with the variant, allele frequencies for 2282del4 among the European American AD and healthy subjects were 8.8%, and 3.7%, respectively. The marker 2282del4 was in Hardy-Weinberg equilibrium at a level of p>0.1 for both cases and controls. Using Fisher's exact test, we found no evidence for association between presence of 2282del4 allele and AD among European Americans (p=0.36). All of the 23 African American subjects genotyped had wild type genotypes for both mutations, providing further evidence that these mutations may be less common in this racial group.7
Table 1. The number of filaggrin mutation genotypes in European American AD subjects and healthy controls.
| Mutation | Genotype | AD | Control |
|---|---|---|---|
| 2284del4 | AA | 14 | 25 |
| Aa | 3 (0.18) | 2 (0.07) | |
| R501X | RR | 17 | 27 |
| RX | 0 | 0 | |
| No. of cases | 17 | 27 | |
To determine whether the 2282del4 mutation affected FLG expression, we compared filaggrin gene expression in a subset of European Americans subjects using real-time RT-PCR. As illustrated in Figure 1, FLG expression was decreased in lesional and uninvolved AD skin as compared to the skin of normal healthy subjects without the mutation (40.77 ± 6.75 ng FLG/ng GAPDH; p<0.05). Whereas similar levels of FLG were observed in the uninvolved skin of AD patients with (13.4 ± 11.4) and without (13.36 ± 8.06) the 2282del4 mutation; FLG expression was higher in lesional skin from AD patients without the mutation (13.54 ± 2.41) as compared to the two patients who were heterozygous for the 2282del4 mutation (2.59 ± 0.07). This difference was determined to be significant (p<0.01) using an un-paired t-test with Welch's correction due to differences in variance. Further investigation on a subject by subject basis revealed higher levels of filaggrin expression in the uninvolved, as compared to lesional, skin of 8 out of 14 AD patients without the mutation. The presence of a single 2282del4 mutation in a normal healthy subject did not significantly affect FLG expression (46.56 ng FLG/ng GAPDH). One normal healthy control and one AD patient with the 2282del4 mutation were excluded from analysis due to a failure to amplify their GAPDH gene.
Figure 1.
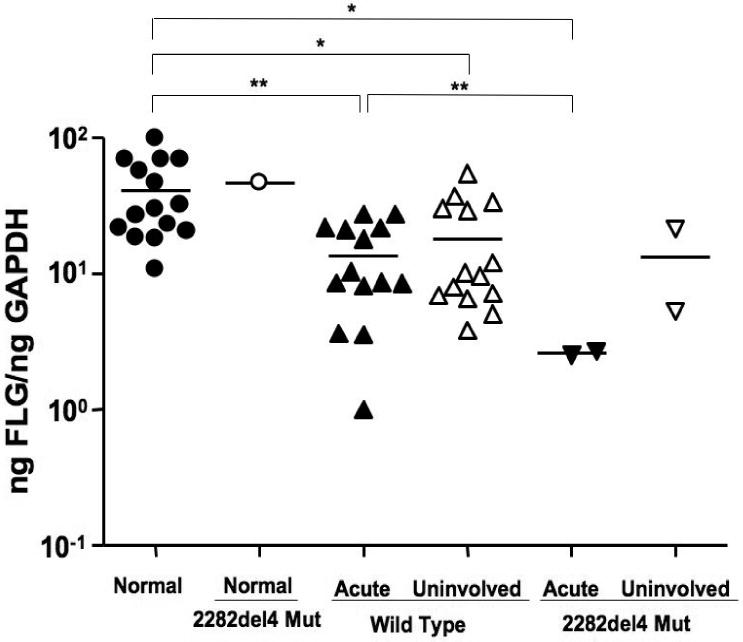
Filaggrin deficiency in AD skin. RNA was isolated from the skin of normal subjects (n=15) and AD patients (n=16) with or without the 2282del4 mutation. Filaggrin gene expression was evaluated using real-time RT-PCR. * and ** indicate significant differences of p<0.05 and p<0.01, respectively.
Immunohistochemical Expression of Filaggrin
Based on our observations, we further evaluated the epidermal expression of filaggrin in healthy skin from normal subjects, uninvolved and lesional skin from AD patients, and lesional skin from patients with lichen planus. Lichen planus was chosen as a positive control since, like AD, it is a T cell-mediated skin disease, however its primary response is Th1 rather than the Th2 observed in AD.16 Additionally, there currently is no known link between lichen planus and a null filaggrin mutation. Differences in filaggrin staining intensity are illustrated in figure 2A. Filaggrin staining was more intense in skin from normal healthy subjects and patients with lichen planus as compared to lesional and uninvolved skin from AD patients. Filaggrin staining was also significantly greater in uninvolved AD skin as compared to lesional AD skin from the same subjects (p<0.05). This suggests that filaggrin mutations don't account for the decrease in filaggrin expression. Additionally, filaggrin protein expression was higher in the uninvolved, as compared to lesional, skin of 8 out of 12 AD patients without the mutation. Lesional skin from AD patients with the 2282del4 mutation exhibited the least intense staining for filaggrin. The composite data for filaggrin immunostaining in all samples are shown in Figure 2B.
Figure 2.
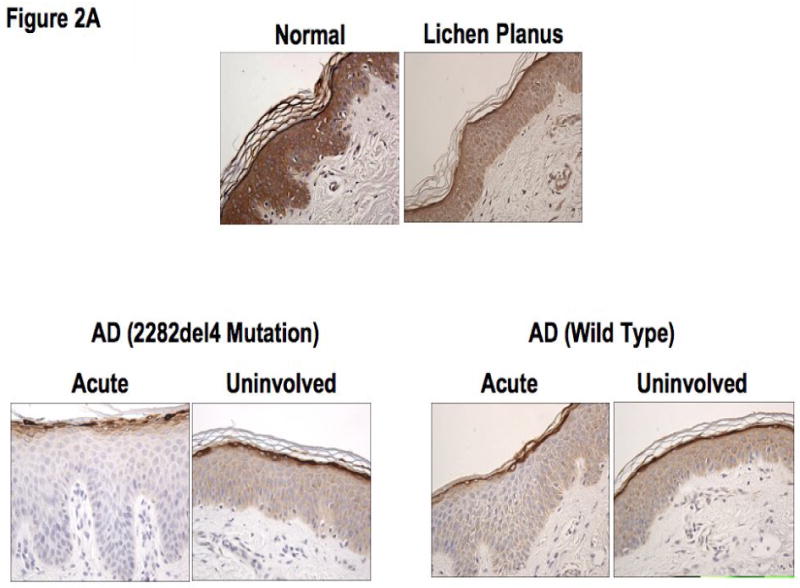
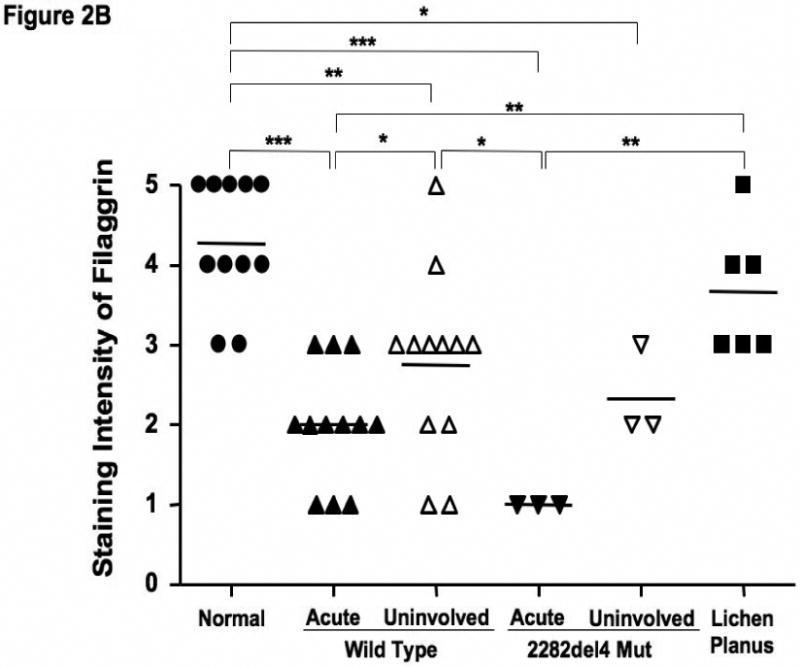
Decreased filaggrin staining in AD skin. A. Representative paraffin embedded skin biopsies from normal subjects (n=11), AD patients (n=12), AD patients with the 2282del4 mutation (n=3), and patients with lichen planus (n=6) stained for filaggrin are shown. Images were collected at 40× magnification and the scale bar represents 50 μm. B. The intensity of the staining was graded visually on a scale from 0 (no staining) to 5 (the most intense staining). *, **, and *** indicate significant differences of p<0.05, p<0.01, and p<0.001, respectively.
Modulation of Filaggrin Expression
Filaggrin is involved in the terminal differentiation of keratinocytes to form the CCE.6 Therefore we initially investigated whether differentiation of keratinocytes induced filaggrin expression. Keratinocytes were differentiated in 1.3 mM CaCl2 as previously described.17 Figure 3 demonstrates that differentiation in increased calcium (1.3 mM) significantly induces filaggrin gene expression (mean: 0.86 ± 0.12) as compared to 0.06 mM (mean: 0.27 ± 0.04; p<0.001). Since acute AD skin is characterized by the over-expression of IL-4 and IL-13,13 and filaggrin expression is increased in uninvolved AD skin as compared to acute AD skin; we investigated whether IL-4 and IL-13 modulates filaggrin expression during keratinocyte differentiation. IFN-γ, which is minimally expressed in acute AD, was used as a Th1 cytokine control. Keratinocytes were differentiated in the presence and absence of IL-4 and IL-13 or IFN-γ for five days. Differentiation of keratinocytes in the presence of IL-4 and IL-13 significantly (p<0.001) reduced filaggrin expression (0.18 ± 0.02) (Figure 3A). In contrast, differentiation in the presence of IFN-γ augmented filaggrin expression (3.31 ± 0.63, p<0.001). Upon further investigation, we also found that incubation of differentiated keratinocytes with IL-4 and IL-13 for 24 hours was able to down-regulate filaggrin expression (0.137 ± 0.023; p<0.05) as compared to media alone (0.606 ± 0.028) (data not shown).
Figure 3.
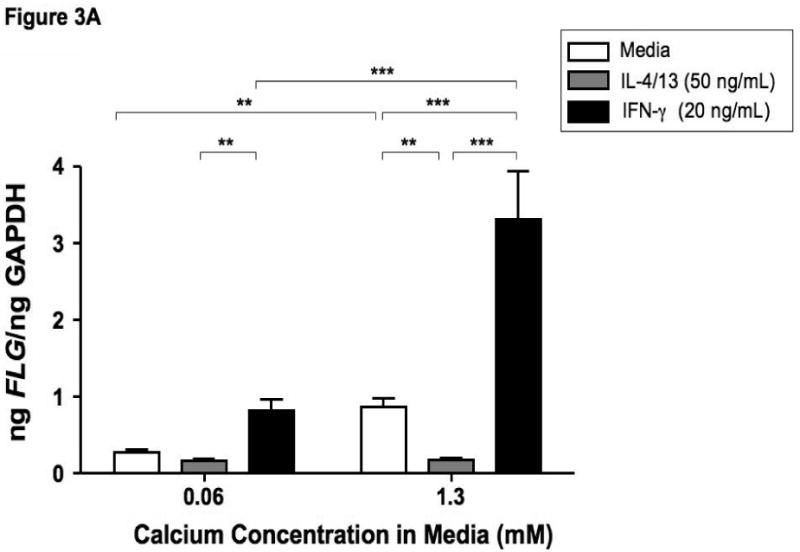
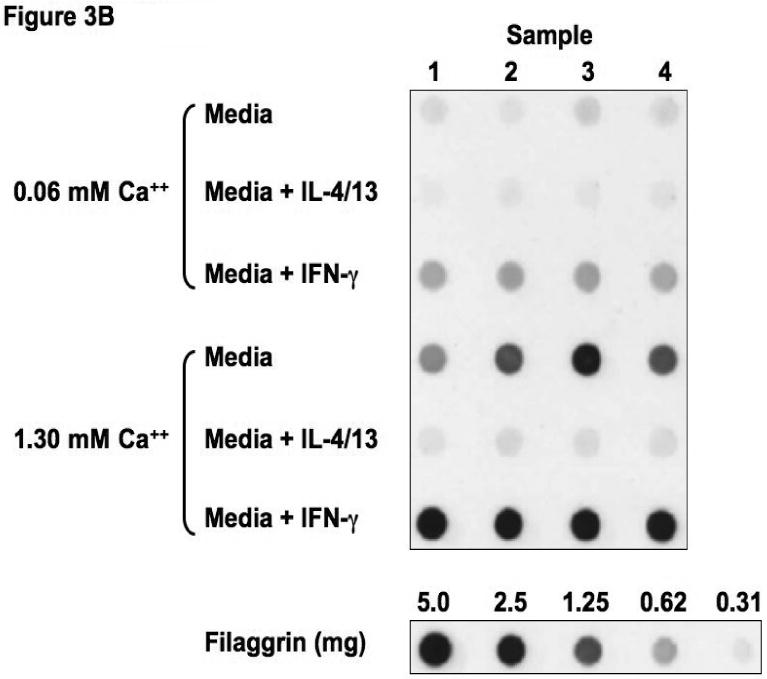
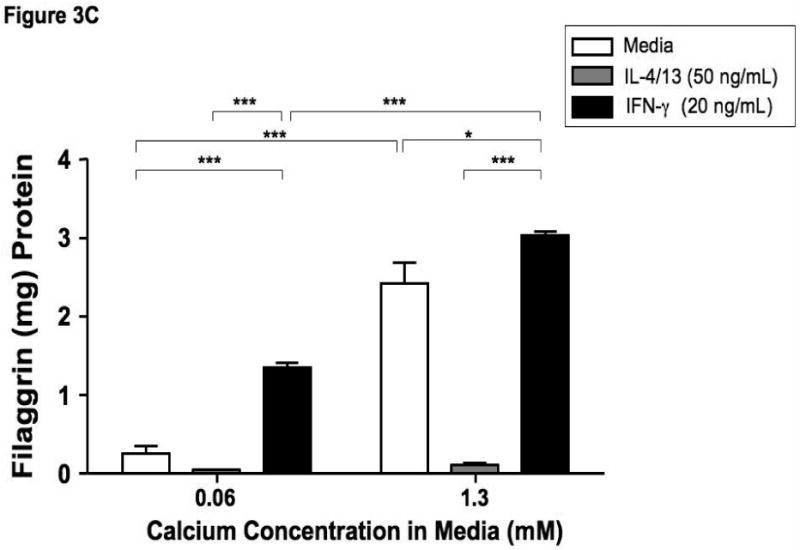
Th2 cytokines down-regulate filaggrin gene expression. Primary human keratinocytes were cultured in 0.06 or 1.3 mM of CaCl2 in the presence and absence of IL-4 and IL-13 or IFN-γ for five days. RNA was collected from the cells, and the levels of filaggrin were evaluated by real-time RT-PCR (A) or Immuno-dot Blot (B & C). *, **, and *** indicate significant differences of p<0.05, p<0.01, and p<0.001, respectively, between exposure groups.
Immuno-dot-blot analysis was used to confirm our observations on the protein level (Figure 3B). We confirmed that IL-4 and IL-13 also down-regulates filaggrin protein expression (0.11 ± 0.03 μg filaggrin protein; p<0.001) as compared to media alone (2.42 ± 0.27) (Figure 3C). As was seen in the gene expression, IFN-γ augmented filaggrin protein expression (3.04 ± 0.04; p<0.001; Figure 3C).
Discussion
The epidermal barrier serves as the first line of defense against invading pathogens and allergens. In AD skin, this barrier is significantly compromised and is associated with increased allergen sensitization, trans-epidermal water loss, viral and bacterial skin infections.18,19 This has been shown to augment the atopic inflammatory response,5,20 thereby further exacerbating the disease state and potentially modulating the effectiveness of the epidermal barrier. The epidermal differentiation complex (EDC) contains a number of proteins involved in barrier function including filaggrin. Our current study evaluated filaggrin expression in the skin of healthy subjects and AD patients to determine whether there is a relationship between filaggrin expression and the atopic immune response.
Several recent studies have now demonstrated an association between null mutations in the filaggrin gene and AD.7-9 This observation is important in our understanding of the significance of skin barrier function in AD, however, this mutation has been observed in less than one-third of general populations of AD patients of European decent.7-9 Additionally, this mutation was heterozygous in most cases. Genetic variants of filaggrin were absent in AD patients with African American and Asian descent, suggesting that the evolutionary pressure that led to the development of these mutations in subjects of European American descent differs from those observed in other racial groups. 7
In our current study, we observed a heterozygous 2282del4 mutation in 8.8% of European American AD patients and 3.7% of our normal healthy subjects. Interestingly, the acute lesional skin of AD patients with the mutation exhibited lower levels of filaggrin expression as compared to the uninvolved skin in the same patient. This suggests that the heterozygous mutation does not completely abolish filaggrin expression. In fact, a homozygous mutation is necessary to completely eliminate filaggrin.7 Combined with the knowledge that skin barrier disruption is seen in all AD patients, we investigated other factors that may contribute to the deficiency in filaggrin.
Studies by Gustafsson et al.21 and Illi et al.22 demonstrate that most AD patients “outgrow” their skin disease. This suggests that the filaggrin deficiency may be due to the atopic skin immune response in many AD patients. Additionally, our laboratory has previously demonstrated that deficiencies in anti-microbial peptides (AMPs), important components of the innate immune response, are acquired rather than constitutive.14,23 Further studies demonstrated that this acquired defect was due to the Th2 cytokine milieu within AD skin. Additionally, AD patients respond well to topical treatments such as tacrolimus and pimecrolimus which suppress the inflammatory reaction in AD skin.1,19 This further suggests that the defect in AD skin barrier function can be acquired.24
Acute AD skin is characterized by the over-expression of the Th2 cytokines, IL-4 and IL-13. Combined with the observation that filaggrin expression is increased in uninvolved AD skin as compared to acute AD skin, we therefore investigated whether these cytokines altered the expression of filaggrin. During the differentiation process, profilaggrin is cleaved into multiple 37 kDa filaggrin subunits which are critical for the formation of the intact CCE. Therefore we differentiated normal primary keratinocytes in the presence and absence of IL-4 and IL-13 for 5 days to determine if filaggrin gene expression would be modulated. Differentiation alone induced filaggrin, however this was significantly inhibited in the presence of IL-4 and IL-13. In additional experiments, we demonstrate that this suppression could also occur in keratinocytes that have been differentiated first and then treated with IL-4 and IL-13 for the final 24 hours. This strongly suggests that the filaggrin deficiency in many AD patients is acquired due to the Th2 cytokine milieu.
Our current study not only demonstrates that filaggrin gene expression and protein are decreased in the skin of AD patients, but also indicates that this deficiency is due, in part, to the over expression of Th2 cytokines which down-regulate filaggrin expression during the differentiation process. Our data suggests that many AD patients acquire the filaggrin deficiency and subsequent barrier disruption as the result of the local inflammatory immune response. This may contribute to decreased filaggrin gene expression in patients with wild type, as well as heterozygous null mutations in FLG. Further studies are needed to determine whether early, effective treatment of AD may impede the development of asthma in such patients by repairing their skin barrier defect.
Acknowledgments
The authors are additionally indebted to the nursing staff in the General Clinical Research Center for the recruitment of patients and collection of specimens. The authors also thank Maureen Sandoval for her help in preparing this manuscript.
This work was supported in part by the Inje University Research grant (B.E.K); National Institutes of Health (NIH) grants HHSN266200400029C, Mary Beryl Patch Turnbull Scholar Program (K.C.B); AI 50024, AAAAI Interest Section Award (L.A.B.); NIH grants NIH/NIAID contracts N01 AI40029 and N01 AI40030, AR41256, R21 AR051634-01, General Clinical Research Center grant MO1 RR00051 from the Division of Research Resources, the Edelstein Family Chair in Pediatric Allergy and Immunology, and the University of Colorado Cancer Center (D.Y.M.L).
Abbreviations
- AD
Atopic dermatitis
- CCE
Cornified cell envelope
- EDC
Epidermal differentiation complex
- FCS
Fetal calf serum
- MEM
Minimum essential media
- FLG
Filaggrin
- IL
Interleukin
- MEM
Minimum essential media
Footnotes
We have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Leung DY, Bieber T. Atopic dermatitis. Lancet. 2003;361:151–60. doi: 10.1016/S0140-6736(03)12193-9. [DOI] [PubMed] [Google Scholar]
- 2.Homey B, Steinhoff M, Ruzicka T, Leung DY. Cytokines and chemokines orchestrate atopic skin inflammation. J Allergy Clin Immunol. 2006;118:178–89. doi: 10.1016/j.jaci.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 3.Warner JO. A double-blinded, randomized, placebo-controlled trial of cetirizine in preventing the onset of asthma in children with atopic dermatitis: 18 months' treatment and 18 months' posttreatment follow-up. J Allergy Clin Immunol. 2001;108:929–37. doi: 10.1067/mai.2001.120015. [DOI] [PubMed] [Google Scholar]
- 4.Bowcock AM, Cookson WO. The genetics of psoriasis, psoriatic arthritis and atopic dermatitis. Hum Mol Genet. 2004;13(Spec No 1):R43–55. doi: 10.1093/hmg/ddh094. [DOI] [PubMed] [Google Scholar]
- 5.Spergel JM, Mizoguchi E, Brewer JP, Martin TR, Bhan AK, Geha RS. Epicutaneous sensitization with protein antigen induces localized allergic dermatitis and hyperresponsiveness to methacholine after single exposure to aerosolized antigen in mice. J Clin Invest. 1998;101:1614–22. doi: 10.1172/JCI1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6:328–40. doi: 10.1038/nrm1619. [DOI] [PubMed] [Google Scholar]
- 7.Palmer CN, Irvine AD, Terron-Kwiatkowski A, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–6. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 8.Weidinger S, Illig T, Baurecht H, et al. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J Allergy Clin Immunol. 2006;118:214–9. doi: 10.1016/j.jaci.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Marenholz I, Nickel R, Ruschendorf F, et al. Filaggrin loss-of-function mutations predispose to phenotypes involved in the atopic march. J Allergy Clin Immunol. 2006;118:866–71. doi: 10.1016/j.jaci.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 10.Ong PY, Ohtake T, Brandt C, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–60. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 11.Fiset PO, Leung DY, Hamid Q. Immunopathology of atopic dermatitis. J Allergy Clin Immunol. 2006;118:287–90. doi: 10.1016/j.jaci.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 12.Howell MD, Gallo RL, Boguniewicz M, et al. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity. 2006;24:341–8. doi: 10.1016/j.immuni.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 13.Hamid Q, Boguniewicz M, Leung DY. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest. 1994;94:870–6. doi: 10.1172/JCI117408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howell MD, Novak N, Bieber T, et al. Interleukin-10 downregulates anti-microbial peptide expression in atopic dermatitis. J Invest Dermatol. 2005;125:738–45. doi: 10.1111/j.0022-202X.2005.23776.x. [DOI] [PubMed] [Google Scholar]
- 15.Tukey J. Exploratory Data Analysis. Reading, NY: Addison Wesley; 1977. [Google Scholar]
- 16.Wenzel J, Scheler M, Proelss J, Bieber T, Tuting T. Type I interferon-associated cytotoxic inflammation in lichen planus. J Cutan Pathol. 2006;33:672–8. doi: 10.1111/j.1600-0560.2006.00527.x. [DOI] [PubMed] [Google Scholar]
- 17.Nomura I, Goleva E, Howell MD, et al. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J Immunol. 2003;171:3262–9. doi: 10.4049/jimmunol.171.6.3262. [DOI] [PubMed] [Google Scholar]
- 18.Boguniewicz M, Leung DY. 10. Atopic dermatitis. J Allergy Clin Immunol. 2006;117:S475–80. doi: 10.1016/j.jaci.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 19.Leung DY, Boguniewicz M, Howell MD, Nomura I, Hamid QA. New insights into atopic dermatitis. J Clin Invest. 2004;113:651–7. doi: 10.1172/JCI21060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laouini D, Kawamoto S, Yalcindag A, et al. Epicutaneous sensitization with superantigen induces allergic skin inflammation. J Allergy Clin Immunol. 2003;112:981–7. doi: 10.1016/j.jaci.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Gustafsson D, Sjoberg O, Foucard T. Development of allergies and asthma in infants and young children with atopic dermatitis--a prospective follow-up to 7 years of age. Allergy. 2000;55:240–5. doi: 10.1034/j.1398-9995.2000.00391.x. [DOI] [PubMed] [Google Scholar]
- 22.Illi S, von Mutius E, Lau S, et al. The natural course of atopic dermatitis from birth to age 7 years and the association with asthma. J Allergy Clin Immunol. 2004;113:925–31. doi: 10.1016/j.jaci.2004.01.778. [DOI] [PubMed] [Google Scholar]
- 23.Howell MD, Boguniewicz M, Pastore S, et al. Mechanism of HBD-3 deficiency in atopic dermatitis. Clin Immunol. 2006;121:332–8. doi: 10.1016/j.clim.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 24.Aalto-Korte K. Improvement of skin barrier function during treatment of atopic dermatitis. J Am Acad Dermatol. 1995;33:969–72. doi: 10.1016/0190-9622(95)90288-0. [DOI] [PubMed] [Google Scholar]