

Abstract
In addition to cancer endpoints, arsenic exposures can also lead to non-cancerous chronic lung disease. Exposures during sensitive developmental time points can contribute to the adult disease. Using a mouse model, in utero and early postnatal exposures to arsenic (100 ppb or less in drinking water) were found to alter airway reactivity to methacholine challenge in 28 day old pups. Removal of mice from arsenic exposure 28 days after birth did not reverse the alterations in sensitivity to methacholine. In addition, adult mice exposed to similar levels of arsenic in drinking water did not show alterations. Therefore, alterations in airway reactivity were irreversible and specific to exposures during lung development. These functional changes correlated with protein and gene expression changes as well as morphological structural changes around the airways. Arsenic increased the whole lung levels of smooth muscle actin in a dose dependent manner. The level of smooth muscle mass around airways was increased with arsenic exposure, especially around airways smaller than 100 μm in diameter. This increase in smooth muscle was associated with alterations in extracellular matrix (collagen, elastin) expression. This model system demonstrates that in utero and postnatal exposure to environmentally relevant levels of arsenic can irreversibly alter pulmonary structure and function in the adults.
Keywords: arsenic, lung development, pulmonary function, airway smooth muscle, extracellular matrix
Introduction
Growth and development requires the temporal and spatial coordinated expression of genes and gene products. During this critical time, in utero and early postnatal exposure to toxicants has the potential to affect gene expression, altering organ structure and physiological function which can be manifested as adult disease (Merkus et al, 2003; Wei et al, 2007). While the potential adverse health outcomes that result from exposures during these sensitive developmental times are recognized, only limited attention has been paid to the effects of environmentally relevant exposures to toxicants during these critical periods of development (Mazumder, 2007, Vahter, 2008) Inorganic arsenic is a ubiquitous environmental toxicant, found in high concentrations throughout the world. Chronic environmental arsenic exposure through consumption of geologically contaminated drinking water has been correlated with increased incidence of and mortality due to internal cancers of the lung, skin, kidney, urinary bladder and liver (Chen et al., 1988; Chiou et al., 1995; Wu et al., 1989; Hopenhayn-Rich et al., 1998). In addition, reports from human studies in Chile, Bangladesh and the West Bengal region of India show that chronic exposure to arsenic via drinking water is correlated with increased incidence of chronic cough, chronic bronchitis, shortness of breath and obstructive or restrictive lung disease (von Ehrenstein, et al., 2005; Mazumder et al., 2000; Smith et al., 1998). Taken together, these studies argue unequivocally that the lung is targeted by arsenic, producing both carcinogenic and non-carcinogenic endpoints.
That high exposures to arsenic in drinking water (800 ppb) during sensitive developmental times can lead to adverse health outcomes and increased mortality has been reported (Smith et al, 2006). Drinking water exposures to high levels of arsenic either in utero or during early childhood development led to an increased risk of dying from lung cancers and chronic lung disease in young adults. Exposures in early childhood led to a standardized mortality ratio (SMR) for lung cancer of 7.0 and a SMR for bronchiectasis of 12.4. For those exposed both during in utero and early childhood, the SMRs were 6.1 for lung cancer and 46.2 for bronchiectasis. These findings suggest that exposure to arsenic in drinking water during early childhood or in utero has pronounced pulmonary effects, greatly increasing subsequent mortality in young adults from both malignant and nonmalignant lung disease. The effects and the molecular targets for alterations after exposure to environmentally relevant levels (0 to 100 ppb) of arsenic, levels that would be seen in some regions of the United States, are not known.
In addition to the effects reported on the lung, early developmental exposures have also been associated with other adverse outcomes in humans. Arsenic is able to cross the placenta (Concha et al, 1998). In Chilean populations with well defined arsenic exposures, an association between arsenic exposure in the drinking water and adverse reproductive outcomes (increase infant mortality (Hopenhayn-Rich et al, 2000) and decreased birth weight (Hopenhayn et al, 2003)) were suggested. Autopsy tissues from five children living in the Antofagasta area of Chile (high arsenic exposure region) revealed increased arterial intimal thickening (Rosenberg, 1974). No reports exist concerning the relationship of arsenic exposures and lung function in children.
Animal and in vitro models have been used in attempts to determine the sites and the mechanisms of developmental toxicity of inorganic arsenicals. In a mouse model of transplacental carcinogenesis, arsenic exposure (42.5 and 85 ppm) during gestation days 8 through 18 lead to significant increases in tumor incidence and multiplicity in the lung and several other organs in adult offspring (Waalkes et al, 2003, Shen et al, 2007). While the doses used in the previous mouse studies are high compared to environmental exposure levels, they do show that tumor formation can occur in an animal model of in utero arsenic exposure. There are also a few studies detailing alterations in the developing fetus induced by maternal arsenic exposure. Acute, high dose ip injections of arsenic (30-45 mg/kg) during gestation have been associated with neural tube defects and corresponding aberrant gene expression of developmentally important transcription factors in the neural tube, including Hox 3.1 and Pax 3 (Liu et al, 2006). These doses of arsenic also triggered upregulation of bcl-2 and p53 gene expression in the neural tube, indicative of inhibition of cellular proliferation (Wlodarczyk et al, 1996). In our previous research we have exposed pregnant female rats to 500 ppb arsenite in drinking water from conception until embryonic day 18. Analysis of arsenic-induced alterations in genes and proteins indicated that targets of in utero arsenic exposure in the developing lung appear to be the developing extracellular matrix and the processes of cellular differentiation and branching morphogenesis (Petrick et al, 2008). The most likely affected pathway was alteration in integrin signaling through the β-catenin pathway, altering c-myc.
Since our earlier research had identified extracellular matrix as a potential target for arsenic, we hypothesized that arsenic-induced changes in matrix during lung development would lead to structural and functional alterations in the adult. During fetal and early postnatal lung development, extracellular matrix gene expression is necessary for proper development of lung and blood vessels (Mariani et al, 2002). Agents that can alter this expression during these critical times are known to cause long term morphological alterations in the lung and blood vessels. (Examples are inhibition of elastin expression following maternal cigarette smoking (Collins et al, 1985) or postnatal exposure to hyperoxia (Bruce et al, 1996), viral infections (Castleman et al, 1988) or dexamethasone (Blanco and Frank, 1993)). These exposures lead to permanent alveolar enlargement. We purpose that arsenic exposure during critical times of development will result in similar irreversible and morphological changes in the lung. In this report we present data that show that in utero and early postnatal exposure to environmentally relevant levels of arsenic in drinking water can lead to irreversible alterations in lung function. These are accompanied by changes in matrix gene expression and alterations in airway smooth muscle.
Methods and Materials
Animals and exposure
C57Bl/6 mice were used. For mouse breeding we used non-sterile micro-isolator pan with water from a sterile double deionized source (used to make arsenic water). We used Teklad Global 19% protein diet which is good for breeding mice and sani-chip bedding (Harlan Teklad, Los Angeles, CA). The chow was assessed for total arsenic levels and was found to contain 40 ng/g (40 ppb) of total arsenic. Control animals for this study received distilled water throughout. Treated animals received sodium arsenite in their drinking water at doses of 5, 10, 50 or 100 μg/l (ppb). These are environmental levels found in drinking water throughout the world. All solutions were prepared with distilled water and, if necessary, titrated to a pH of 7.0, a pH similar to that of control water. Each water bottle was placed in a specially made metal casing to reduce light exposure and subsequent chemical breakdown. The amount of water consumed by each pen of animals (4 maximum) was monitored and recorded every 24 hours. Bottles were cleaned and solutions prepared daily. Arsenic speciation was determined and concentrations verified using ICP-MS, available through the Arizona Superfund Core Facility. Females began exposure to arsenic in their drinking water two weeks prior to mating. Females used for multiple breeding cycles were continuously kept on the same arsenic water throughout. Results from pups born as first litters to breeding females were not different when compared to subsequent litters. After birth of the pups, mothers and weaned animals were exposed to the arsenic in the drinking water. Pups were sacrificed using CO2, on day 1, 7, 12 and 28 after birth. During that time, animals were continually exposed to arsenic in the drinking water. Only one pup from a litter was used for each time point and measurement (If, for example, N=5, then each of the five animals used came from a different litter). All protocols were approved by the University of Arizona, Institutional Animal Care and Use Committee.
Quantitative RT-PCR
All lung tissue was harvested and placed in RNAlater solution from Qiagen Cat. No. 76104 in ratio of 1ml/100mg of tissue, stored in 4°C overnight. The solution was then removed and samples were stored in -80°C until used. All RNA isolation was done following procedure using Qiagen RNeasy Mini Kit Cat. No. 74104. We also performed in-column DNAse treatment (DNAse Kit Cat No. 79254) to remove DNA contamination during the RNA isolation. 30 mg of tissue per reaction was homogenized with disposable plastic mortar and pestle.
Lung total RNA was quantified and checked for RNA integrity by Agilent 2100 Bioanalyzer. cDNA was synthesized using 2 μg of total RNA following procedure from Taqman Reverse Transcription kit (Applied Biosystems, N808-0234.) Amplification was performed by PCR using Taq Gold polymerase master mix (Applied Biosystems, N808-0241) and 1× SYBR Green (Invitrogen, S7563) performed by Roter-gene 3000 (Corbett Research, Australia.). The PCR conditions were: for collagen 1A1, 1A2, 3A1, smooth muscle actin and beta actin PCR at 95°C for 30 sec; annealing at 50°C for 30 sec, and extension at 72°C for 30 sec and for elastin PCR at 95°C for 30 sec; annealing at 58°C for 30sec, and extension at 72°C 30 sec. The PCR primer sequences are shown in Table 1. Levels of expression were normalized to GAPDH. After birth, lung GAPDH expression levels do not change ((http://lungtranscriptome.bwh.harvard.edu/)).
Table 1. Real Time PCR Primers.
| Elastin: | |
| Forward: | 5′-TGG AGG CAA GGG AGC AAG AAA C-3′ |
| Reverse: | 5′-TAA CAG AAC AGA GAG TGC TGT GGG-3′ |
| Collagen 1A1: | |
| Forward: | 5′-GAG CGG AGA GTA CTG GAT CG-3′ |
| Reverse: | 3′-GTT CGG GCT GAT GTA CCA GT-3′ |
| Collagen 1A2: | |
| Forward: | 5′-GGA GGG AAC GGT CCA CGA T-3′ |
| Reverse: | 5′-GAG TCC GCG TAT CCA CAA-3′ |
| Collagen 3a1: | |
| Forward: | 5′-AAG TTC ACC AGC AAC AGC AG-3′ |
| Reverse: | 5′-TTG GTT AGC CAT GTA GAG CG-3′ |
| Smooth muscle actin: | |
| Forward: | 5′-CTT CCA GCC ATC TTT CAT TGG-3′ |
| Reverse: | 5′-ATA TCA CAC TTC ATG ATG CTG TTA TAG GT-3′ |
| Beta-Actin: | |
| Forward: | 5′-AGA GGG AAA TCG TGC GTG AC-3′ |
| Reverse: | 5′-CAA TAG TGA TGA CCT GGC CGT-3′ |
Relative quantization of expression levels was obtained by developing a standard curve using 2 μg total RNA from control animals. cDNA was serially diluted and PCR was performed on these serially diluted cDNAs to obtain the STD curve. For quantifying, the samples from the control group and treatment groups cDNA were diluted 20 times prior to real-time PCR to obtain a threshold cycle. The standard curve was then used to obtain fold change in expression.
Immunohistochemistry and lung morphometry
Immunohistochemistry was utilized to localize alpha smooth muscle actin. (Rabbit IgG, ab5694, Abcam, MA). Lungs were fixed by intratracheal instillation of buffered formalin at a constant pressure of 20 cm H20. Paraffin embedded sections (5 μm) were baked for 1 hr at 65°C then subjected to a series of de-paraffinization (3 times of 5 min in xylene) and dehydration. Antigen retrieval was performed by microwave treatment. The slides were place in 10 mM citrate buffer at pH 6.0.
All procedures are followed according to Vectorstain ABC kit for Rabbit IgG (PK6101). Primary antibody staining was with a 1:100 dilution of antibody overnight in a humidity chamber. After washing, sections were incubated for 30 min with a biotinylated secondary antibody followed by 30 min incubation with an avidin-HRP complex. Staining was developed (2 min) using the Vector VIP staining solution (SK4600, Vector Laboratories).
The amount of smooth muscle and collagen around airways was quantitated by analyzing digital images collected using PCI software (Pittsburgh, PA). Sections of lung tissue were scanned and all airways cut in cross section (the ratio of maximum to minimum diameter was less than 2) were analyzed. Diameters were determined by filling of the area inside of the airway epithelium. PCI is able to obtain the minimum and maximum diameters of a region of interest. Basement membrane perimeter was determined by tracing. The area of smooth muscle was determined by thresholding the images to detect only the antibody staining and measuring the number of pixels detected (Camateros et al, 2007). Area of collagen staining was obtained using polarized light microscopy of sirius red stained sections (Last et al, 2004). Area of smooth muscle and collagen staining were then normalized to the square of the basement membrane perimeter (Camateros et al, 2007). Data were analyzed for all airways and also were subdivided by airway diameter (small airways, diameter < 100 μm versus large airways, diameter > than 100 μm). The minimum diameter was used a measure of the airway diameter (Weibel, 1979).
Western Blots
The following antibodies and dilutions were used for Western blot analysis: Procollagen Type I(M-60) (Santa Cruz, sc30136), 1′: 1:200, 2nd : 1:10,000; Collagen 1A2(M80) (Santa Cruz, sc28654), 1′: 1:200, 2nd : 1:20,000; Collagen Type III(H300) (Santa Cruz, sc28888), 1′: 1:200, 2nd : 1:20,000; Tropoelatsin (Elastin Product Co, Inc, PR385), 1′: 1:500, 2nd : 1:50,000; GAPDH (FL-335) (Santa Cruz, sc25778), 1′: 1:500, 2nd : 1:20,000; and Alpha SMA, (Abcam, ab5694), 1′: 1:100, 2nd : 1:10,000. All primary antibodies were rabbit IgG. The secondary antibodies for all were goat anti-rabbit IgG –HRP cat# 31460 from Pierce and developed with Super signal West Pico chemiluminescent substrate Cat # 34080 from Pierce.
Lung tissue was homogenized with a hand held polytron in buffer contain (20mM Tris-HCl, pH7.5, 1mM DTT, 1mMEDTA 50mM PMSF plus Protease inhibitor cocktail tablet from ROCHE) and spun for 10 min at 13000 g to collect total protein. 45 μg of lung total protein was added 1:2 ratio to Laemmli sample buffer (BioRad-161-0737) containing βME (50 μL βME in 950 μL sample buffer) then boiled for 3 min. The protein was separated by 10% Tris-HCl SDS PAGE gel (Bio Rad, 161-1155), then transferred over night at 4°C to a PVDF Membrane (Perkin Elmer, 370790.) The membrane was blocked with 5% non fat milk in TBS for 1 hr, then was incubated for 1 hr with primary antibody (antibody in TBS with 2% milk) After washing 3 times in TBS with 0.5% Tween 20 for 5 min each the membranes were incubated with secondary goat anti-rabbit IgG –HRP conjugate. After performing the washing step, the membrane was developed for 5 min with the chemiluminescent substrate (SuperSignal West Pico kit, 34080, Pierce) and exposed to clear blue X-ray film (Pierce, 34093). Suitable signal strength was obtained after 2 min exposure.
The same membrane was then stripped for 15 min at room temperature with Restore Western Blot Stripping Buffer (Pierce, 21059) and then re-blotting following the same above procedures for internal standard using GAPDH. Assuming equal loading, arsenic did not affect the level of expression of GAPDH.
Pulmonary Function
Air way responsiveness was measure on live unrestrained mice at various ages, beginning at 28 days after birth. Lung functions were analyzed by comparing the airways responsiveness to methacholine in unrestrained conscious mice using a Biosystem XA whole body plethysmographs from Buxco Electronics Inc. (Wilmington, North Carolina) as described (Hamelmann et al., 1997). The system was calibrated with 1 ml of air for each chamber prior to use. Mice were then placed in the chamber for 5 to 10 min to allow them to become familiar with the box and until all breathing was normal and Penh values (enhance pause) were constant. Enhanced pause (Penh) index of airway hyper-reactivity was used as an indicator of changes in airway resistance. Penh was calculated by Biosystem XA software using the following formula: (peak expiratory pressure/peak inspiratory pressure) × (expiratory time - relaxation time)/relaxation time. Baseline Penh readings were taken and averaged for 5 minutes before the PBS exposure. Mice were then exposed for 2 min to the nebulized PBS or nebulized methacholine (1- 100 mg/ml in PBS) (Sigma, A2251) and Penh was recorded for 5 minutes and averaged. The aerosol delivery system was set to deliver 75 μl of PBS or methacholine per chamber over the period of 2 min.
In addition to in vivo testing of airway reactivity, pulmonary resistance and compliance were also tested in additional mice (Robledo et al, 2000). Mice were anesthetized with an intramuscular injection mixture of ketamine HCL (80 mg/kg), xylaxine (10 mg/kg) and acepromazine maleate (3 mg/kg). Following this a tracheostomy was performed, inserting a Teflon IV catheter (20 gauge, Critikon, Tampa Bay, FL) as an endotracheal tube. The mice were placed on a small animal ventilator (Kent Scientific, Litchfield, CT) under pressure controlled ventilation. Airflow was measured with a pneumotachograph (Fleish #0000, Instrumentation Associates, New York, NY), which was connected to a differential pressure transducer (Validyne, Northridge, CA). Pulmonary function measurements were obtained using a computerized pulmonary function system (PEDS-LAB, Medical Associates Services, Hatfield, PA) which records airflow and pressure signals, and normalizes them to animal body weight.
Data Analysis
Statistical analyses were performed using ANOVA (Winer et al., 1991), requiring p<0.05 for statistical significance.
Results
Twenty eight day old mice exposed to arsenic in utero from conception through birth and weaning and continuously after weaning, were analyzed for their response to methacholine challenge. Figure 1A shows the response as a function of baseline responsiveness. As can be seen, animals that had been exposed to arsenic had increased Penh levels at lower levels of methacholine challenge. Penh levels were significantly increased at 10 mg/ml methacholine exposure in animals that had been exposed to 100 ppb arsenic during in utero and postnatal development. At 33 mg/ml, Penh continued to increase in animals exposed to 100 ppb, while those exposed to 10 and 50 ppb arsenic also demonstrated a dose-dependent increase in Penh.
Figure 1. Response to methacholine challenge in unrestrained conscious mice.
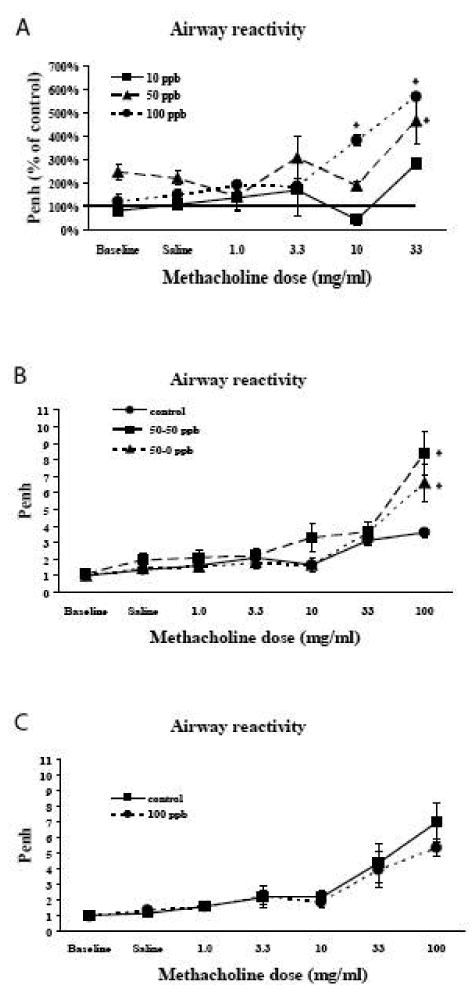
A - Response to methacholine challenge in 28 day old mice that had been continuously exposed to arsenic in utero and during postnatal development. Animals that had been exposed to higher levels of arsenic during development responded with increased Penh levels at similar methacholine challenge levels. Values are means ± sem compared to control animals. N=3 in at each arsenic dose. *=significantly different from control (p<0.05).
B – Response of twelve week old mice to methacholine challenge. All arsenic exposed mice were exposed to 50 ppb arsenic during in utero and early postnatal development. At 28 days of age, animals were split into two groups. One group continued to receive 50 ppb arsenic in their drinking water (50-50 ppb) while arsenic was removed from the water given to the second group (50-0 ppb). Controls received no arsenic throughout. Animals were kept on this regimen until they reached three months of age, when they were tested for their response to methacholine challenge. Removal of arsenic from the water at 28 days of age did not return methacholine response to control levels. The response of the 50-50 and 50-0 animals were not significantly different from each other. Both 50-50 animals and 50-0 animals showed increased response to methacholine compared to controls. (controls, N=5; 50-50, N=4; 50-0, N=3). Values are mean ± sem. *=significantly different from control (p<0.05).
C- Response to methacholine challenge in mice that have only been exposed to arsenic as adults. Adult mice were given 100 ppb arsenic in their drinking water for three months and tested for alterations in Penh after methacholine challenge. Values for animals exposed to 100 ppb arsenic were not different from animals that received no arsenic in their water. (controls, N=4; 100 ppb, N=6). Values are mean ± sem.
To determine if the increases in Penh caused by arsenic exposures was reversible, 28 day old animals that had been continuously exposed to 50 ppb arsenic since conception were split into two groups. One group was continued on 50 ppb in their drinking water (50-50 ppb) while arsenic was eliminated from the drinking water in the second group (50-0 ppb). A parallel control group was also analyzed. Methacholine-induced changes in Penh were again evaluated when the animals reached 3 months of age (Figure 1B). As can be seen, removal of the animals from the arsenic exposure on day 28 did not reduce the increased response to methacholine challenge. Therefore, alterations in the response to methacholine that were caused by in utero and early postnatal exposure to arsenic were not reversible by removal of arsenic after the early developmental time period.
In order to evaluate whether alterations in response to methacholine challenge was specific to exposure during developmental times, adult male mice were exposed to arsenic in their drinking water for three months and evaluated for methacholine-induced changes in Penh. Parallel control animals were also analyzed. Adult only exposure to 100 ppb arsenite in drinking water did not lead to altered pulmonary response to methacholine challenge (Figure 1C). Therefore, the alterations in lung function were specific to in utero and early postnatal exposures.
Results from unanaesthetized animals were validated by determining alterations in pulmonary resistance and compliance in anesthetized animals. As shown in Figure 2, exposure to arsenic resulted in a dose dependent increase in pulmonary resistance in 4 week old animals. (Figure 2A). This increase was no altered by removal of arsenic from the animals at 28 days of age (Figure 2B). There were no changes in lung compliance.
Figure 2. Pulmonary resistance was measured in anesthetized mice.
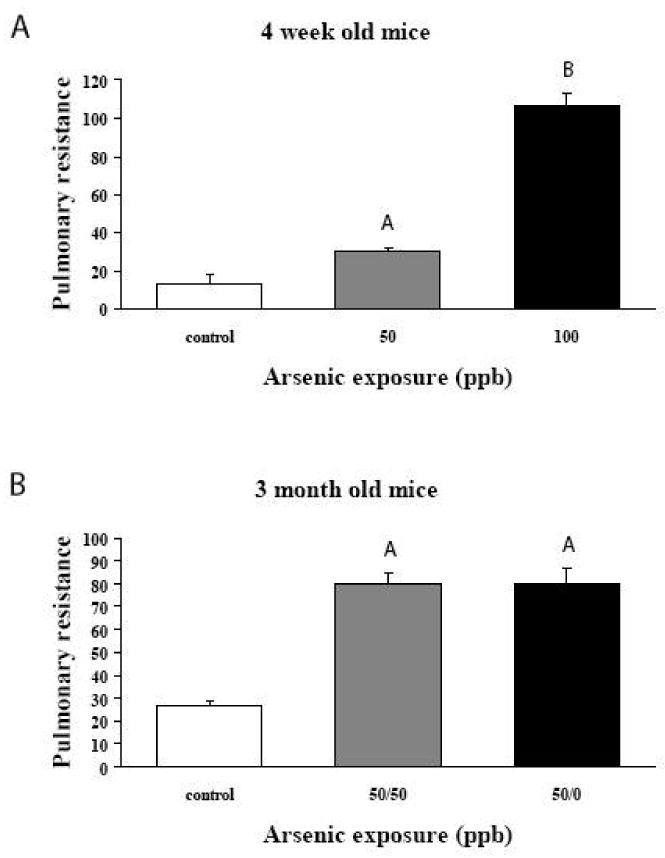
A – Mice were exposed continuously during in utero and early postnatal development to 50 or 100 ppb arsenic. At 28 days of age, mice were anesthetized and pulmonary resistance was measured. Arsenic exposure resulted in increased resistance in a dose dependent manner. (Controls, N=4; 50 ppb, N=4; 100 ppb N=5), Values are mean ± sem. A=significantly different from Control (p<0.05). B=significantly different from Control and 50 ppb arsenic (p<0.05).
B – Pulmonary resistance in twelve week old mice. All arsenic exposed mice were exposed to 50 ppb arsenic during in utero and early postnatal development. At 28 days of age, animals were split into two groups. One group continued to receive 50 ppb arsenic in their drinking water (50-50 ppb) while arsenic was removed from the water given to the second group (50-0 ppb). Controls received no arsenic throughout. Removal of arsenic from the water at 28 days of age did not return of pulmonary resistance to control levels. Both 50-50 animals and 50-0 animals showed increased resistance compares to controls. (controls, N=4; 50-50, N=7; 50-0, N=3). Values are mean ± sem. A=significantly different from Control (p<0.05).
Since functional alterations are not affected by removal of the arsenic, we investigated if arsenic exposure was resulting in airway anatomical structural changes. Changes in airway diameters and/or airway smooth muscle could account in part for the functional alterations. As a first measure of airway size, we evaluated the volume density of the airways (percentage of lung occupied by airways). There was no differences (data not shown), indicting no gross changes in airway development. Examination of airways from 28 day old animals that had been immunostained with smooth muscle actin (SMA) antibodies, however, did show an apparent increase in smooth muscle around airways of animals that had been exposed to arsenic (Figure 3).
Figure 3. Airway smooth muscle.
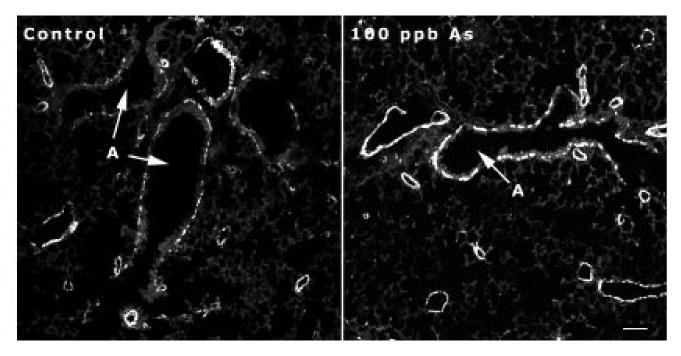
Photomicrograph of smooth muscle actin immunostaining in 28 day old mice. Mice that had received 100 ppb arsenic continuously during in utero and early postnatal development showed an apparent increase in the level of smooth muscle actin staining (white staining) around airways (A). Bar in lower right corner = 100 μm.
In order to determine whether quantifiable changes in SMA had occurred we determined the levels of mRNA and protein as a function of postnatal age (Figure 4). As has been previously shown (http://lungtranscriptome.bwh.harvard.edu/), mRNA expression of SMA peaked on postnatal day 7 and subsequently decreased at 12 and 28 days after birth. Exposure to arsenic during in utero and early postnatal development altered this expression pattern. High levels on day 7 remained elevated on postnatal days 12 and 28. Whole lung levels of protein expression followed a similar pattern, that is, elevation of day 7 with subsequent decreases in the control animals. SMA protein levels remained elevated in arsenic exposed pups.
Figure 4. Smooth muscle actin mRNA and protein levels from whole lung.
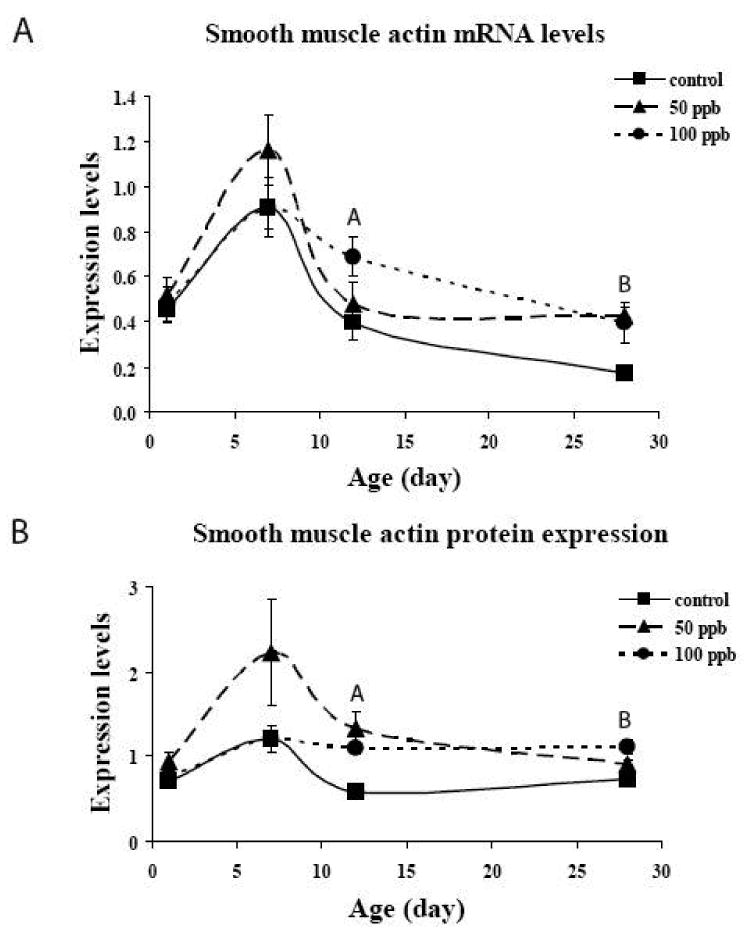
A – mRNA levels of SMA as a function of postnatal age are plotted from quantitative RT-PCR. Arsenic at 50 and 100 ppb led to increased levels of SMA gene expression in the whole lung. N=3 for each arsenic dose and postnatal age. Values are mean ± sem. A=100 ppb significantly different from control in 12 day old animals. B=50 and 100 ppb significantly different from controls in 28 day old animals. (p<0.05)
B – SMA protein levels as a function of postnatal age are plotted from Western blot analysis. During normal development, protein levels are increased on postnatal day 7 and then decrease on day 12 and 28. Arsenic increased the levels of SMA expression on day 12. These increases were maintained on day 28. N=3 for each arsenic dose and postnatal age. A=50 and 100 ppb significantly different from control in 12 day old animals. B=100 ppb significantly different from controls in 28 day old animals. (p<0.05).
We have previously shown in adults that chronic exposure to arsenic can lead to a significant reduction in expression of collagens and elastin in the lung (Lantz and Hays, 2006). Since these matrix molecules are important for appropriate lung development and since they can also control smooth muscle proliferation, we examined arsenic induced alterations in expression of Col1a1, Col1a2, Col3a1 and elastin. In utero and postnatal arsenic exposure resulted in dose and time dependent alterations in expression of Col1a2, Col3a1 and elastin (Figure 5). The expression patterns in animals that did not receive arsenic were similar to those that have previously been reported (http://lungtranscriptome.bwh.harvard.edu/). Rather than suppression of expression of these matrix genes, arsenic exposure resulted in increases in expression. Exposure to either 50 or 100 ppb arsenic resulted in increased Col1a2 expression on postnatal day 7 (Figure 5A). Col3a1 expression was increased on postnatal day 12 by 100 ppb (Figure 5B). The pattern of elastin expression was more complex but, in general, resulted in increased elastin expression at early postnatal times (Figure 5C). Levels of expression in arsenic exposed animals were decreased on day 28, similar to what we have previously seen in adults. Arsenic did not alter the expression pattern of Col1a1 during the early postnatal periods (data not shown).
Figure 5. Extracellular matrix gene expression.
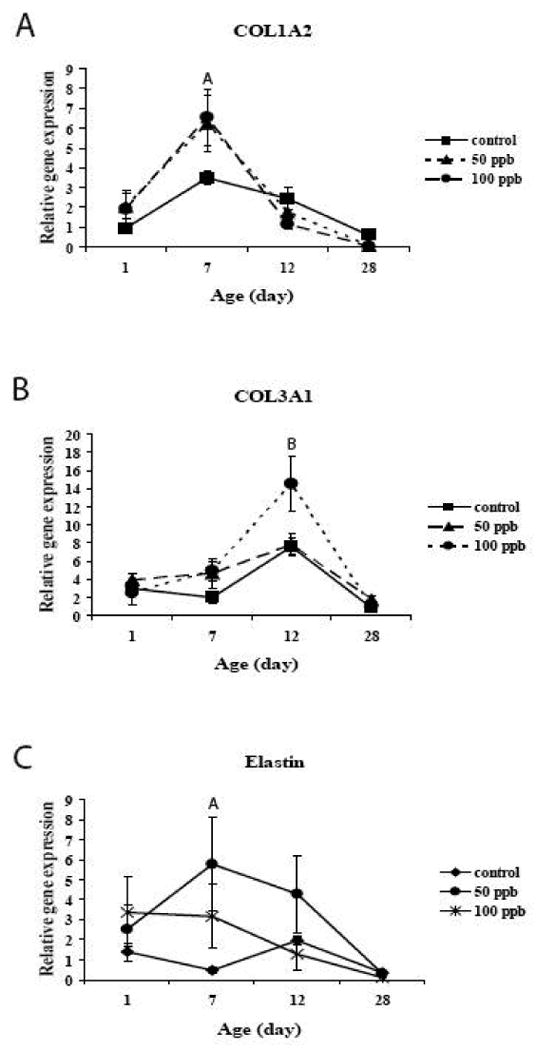
Quantitative RT-PCR for collagen 1a2 (A) and 3a1 (B) and elastin (C) show arsenic-induced changes in mRNA levels. For Col1a2, arsenic increased the levels of expression of postnatal day 7, while for col3a1, arsenic increased the levels of expression of postnatal day 12. Alterations in elastin expression were more complex but in general arsenic increased expression. On postnatal day 28, levels of expression in arsenic exposed animals were at or below control levels. Values are mean ± sem. N=3-6 animals/dose/age. Levels of expression were normalized to GAPDH expression. A=50 and 100 ppb significantly different from Control in 7 day old animals; B=100 ppb significantly different from Control in 12 day old animals. (p<0.05).
While levels of gene expression were altered by arsenic, whole lung levels of matrix protein were only marginally altered. Analysis of Western blot intensities, normalized to GAPDH expression levels, showed increased protein expression of Col1a2 on day 12 (control = 1.89 ± 0.09; 50 ppb arsenic = 2.18 ± 0.07; 100 ppb arsenic = 2.56 ± 0.48), decreased protein expression of Col3a1 on day 12 (control = 1.86 ± 0.09; 100 ppb arsenic = 1.26 ± 0.09) and decreased protein expression of elastin on day 7 (control = 2.20 ± 0.41; 50 ppb arsenic = 1.47 ± 0.58; 100 ppb arsenic = 0.61 ± 0.35). While these values show trends, only the arsenic-induced decrease in Col3a1 on day 12 reached significance. Whole lung protein levels for Col1a1, 1a2, 3a1 and elastin from arsenic exposed animals on day 28 were not different from controls (data not shown).
Whole lung levels of expression are useful for determination of global expression changes. However, morphological targeting of analysis can better reflect region differences in expression. We therefore determined the levels of SMA protein expression around airways. Values were determined using image analysis as outlined in the methods. Figure 6A shows the results. Data are presented as total measurements of all airways. Data are also subdivided into data from airways with diameters less than or greater than 100 μm. When all airways are combined for analysis, there is an arsenic-induced dose dependent increase in the amount of SMA around airways of 28 day old mice. A breakdown of the data based on airway diameter indicates that the majority of this change is due to increased levels around airways that are less then 100 μm in diameter.
Figure 6. Quantitative image analysis of the levels of SMA and collagen staining.
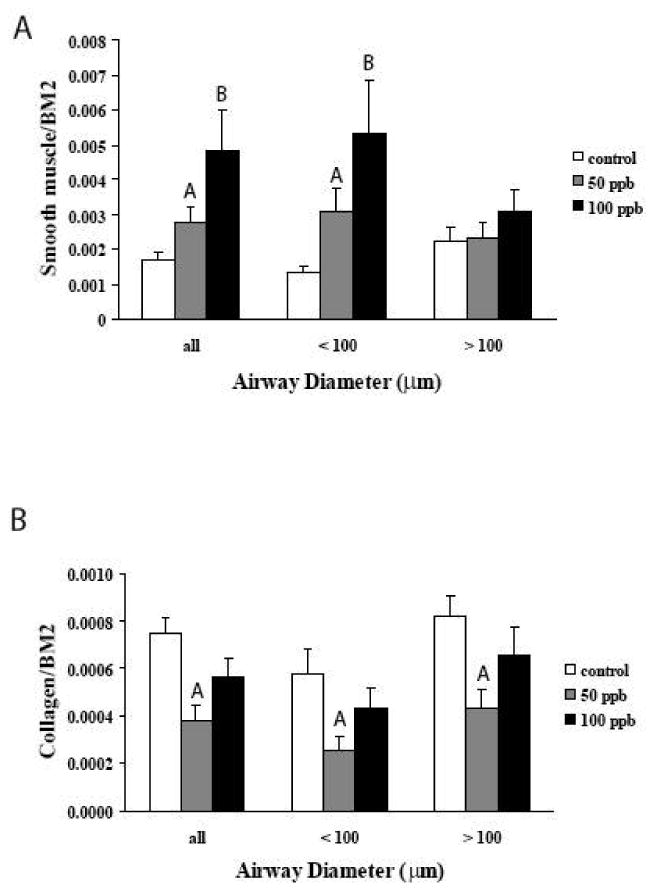
A – Quantitative image analysis of the levels of smooth muscle actin staining. The area of staining around airways was normalized to the area of the airway epithelial basement membrane. Combining data from all airways shows an arsenic-dependent increase in the levels of SMA. When the airways are segregated by diameter (< 100 μm and > 100 μm), the majority of the changes in SMA are seen in the smaller airways. (Control, 12 airways from 4 mice; 50 ppb, 17 airways from 4 mice; 100 ppb, 13 airways from 4 mice). Values are mean ± sem. A=significantly different from Controls; B=significantly different from Controls and 50 ppb. (p<0.05).
B - Quantitative image analysis of the levels of collagen around airways. The area of staining around airways was normalized to the area of the airway epithelial basement membrane. Combining data from all airways shows an arsenic-dependent increase in the levels of collagen. The effects of arsenic are independent of the airway diameter. (< 100 μm and > 100 μm), (Control, 12 airways from 4 mice; 50 ppb, 17 airways from 4 mice; 100 ppb, 13 airways from 4 mice). Values are mean ± sem. A=significantly different from Controls. (p<0.05).
Since we have seen arsenic-induced alterations in SMA expression around airways, we also examined the levels of collagen around airways as well. Sections were stained with sirius red and levels of collagen were determined by image analysis as outlined in the methods. This stain does not differentiate collagen subtypes so data reflect total collagen around airways. Data were also subdivided based on airway diameters. As can be seen in Figure 6B, arsenic exposures resulted in reduced levels of collagen expression around airways. The suppression of collagen expression was more evident at 50 ppb exposure levels. In addition, the relative decrease in collagen expression did not appear to be related to airway diameters, 50 ppb arsenic resulted in 44,5% decrease in airways with diameters < 100 μm and a 53.1% decrease in airways with diameters > 100 μm.
Discussion
Using environmentally relevant levels of exposure to arsenic in drinking water, we have shown that continuous in utero and early postnatal exposure results in both functional and structural alterations in the lung. Arsenic exposure resulted in increased bronchoconstriction following methacholine challenge. This alteration in responsiveness could not be reversed by removal from arsenic exposure. In addition, the alterations in function were the result of exposure during in utero and early postnatal lung development. Exposure to adults only did not result in the changes. Associated with these functional changes were alterations in lung structure. Airway smooth muscle content was increased with increasing arsenic exposure. These changes were most prominent in airways less than 100 μm in diameter. In addition, arsenic exposure also altered the expression patterns of several matrix genes that can regulate smooth muscle proliferation.
Exposure to toxicants during sensitive times of development can produce adverse health outcomes. For arsenic, this has been demonstrated by Smith et al (2006), who have studied a human population from Chile that received in utero and early postnatal exposures to high environmental levels (800 – 900 ppb) of arsenic. These developmental exposures greatly increased the incidence of lung disease associated with mortalities later in life. Standard mortality ratios (SMR) from lung cancers, bronchiectasis and other chronic lung diseases were increased five to ten fold with early postnatal exposure alone. Combined in utero and postnatal exposures increased the SMR even higher. Our data reported here using 50 to 100 ppb arsenic exposures are therefore relevant for providing potential mechanisms that can lead to human diseases.
A significant proportion of adult lung disease originates in utero or early infancy (Merkus, 2003). A number of agents have been shown to affect normal lung growth and development when administered in utero or in the first month after birth. Maternal smoking has been associated with decreased elastic tissue and increased alveolar size (Collins et al, 1985). For postnatal exposures, the more extensively studied compounds are dexamethasone, hyperoxia and Sendai viral infections. Dexamethasone, administered in a critical time period between 4 and 14 days after birth, resulted in larger alveoli on day 14. This enlargement was still present on day 60 after birth (Blanco et al, 1989). Dexamethasone had no effect when given after day 14 (Blanco and Frank, 1993). Similarly, the postnatal age at the onset of hyperoxic exposure was found to affect the expression of tropoelastin (Bruce et al, 1996). Exposure from 3 to 13 days of age interfered with the normal increase in tropoelastin expression seen during this time. Hyperoxic exposure in weeks 3 and 4 was still capable of increasing alveolar size and decreasing alveolar number (Blanco and Frank, 1993). In these experiments, decreases in septation were associated with decreases in elastin fiber length and enlarged alveoli.
A similar sensitive period was noted in rats that had been infected with Sendai virus (Castleman et al, 1988). Exposure of 5-day-old rats produced more severe changes in structure and function than inoculations on day 25. In these studies, lungs from virus exposed rats appeared to have mild alveolar emphysema. Lung function revealed elevated resistance and decreased compliance, and increased airway hyperresponsiveness (Sorkness et al, 1991). These alterations were not compensated for when rats were tested at 39 days after inoculation.
In addition to alterations caused by postnatal exposure to toxicants and infectious agents, prenatal exposure to cigarette smoke results in increased airway hyperreactivity (AHR) (Singh et al, 2003). The alterations in lung function occurred only in animals that were exposed in utero. Postnatal exposures or exposures in adults did not lead to increases in AHR.
In the current report, we have provided continual in utero and postnatal exposures similar to what would be experienced in the real world. As such we can not differentiate between the effects of in utero or postnatal alone. Defining the sensitive exposure window will be important in future work. Our results however, are similar to other agents that act either in utero or postnatally. Alterations in function are caused by exposures during these developmental times and are irreversible even with withdrawal of the arsenic.
Effects of agents that lead to structural and functional alterations on gene expression have only been examined in a limited number of studies in neonatal lung. Hyperoxia in neonates has been shown to induce antioxidant enzymes (Clerch and Massaro, 1992) and surfactant protein A (D'Angio et al, 1997) while down-regulating tropoelastin expression (Bruce, 1991). Treatment of neonatal lung with dexamethasone has also been shown to down-regulate cellular retinol binding protein 1 (Whitney et al, 1999).
We have previously shown down regulation of extracellular matrix gene expression in the lung following chronic exposure in adult mice (Lantz and Hays, 2006). Based on the importance of these proteins in lung development we tested whether arsenic could down regulate collagen and elastin expression during postnatal lung development. However, rather than inhibiting the expression, arsenic enhanced the levels of whole lung gene expression for Col1a2 and Col3a1 in a time dependent manner. This suggests that arsenic is interacting with the normal developmental process to alter expression of these matrix genes. Whole lung protein levels however were not significantly altered. One explanation is that mRNA levels of expression may be increasing to compensate for losses of proteins, so that normal levels of matrix protein required for developmental processes are maintained. We have previously demonstrated that arsenic can induce increased expression of matrix metalloproteinase-9 (MMP-9) (Olsen et al, 2008). Similar arsenic-induced changes in MMP-9 during development would degrade matrix. Increases in mRNA expression could be a compensatory response.
While whole lung levels of matrix proteins were unchanged, regional decreases in total collagen in adventia around airways was seen in 28 day old mice exposed to arsenic during development. This localized regional decrease in collagen could contribute to the increased smooth muscle around airways (Dekkers et al, 2007; Parameswaran et al, 2006), similar to collagen knockout mice. Elastin and collagen knockout mice both show hyperproliferation of smooth muscle around blood vessels (Karnik et al, 2003, Liu et al, 1997). Therefore decreased expression of collagen around airways may also contribute to the increased levels of smooth muscle.
Increases in smooth muscle are associated with increases in airway hyperresponsiveness (Martin et al, 2000; Cockcroft and Davis, 2006). Alterations in smooth muscle have best been characterized in a primate model (Plopper et al, 2007). Exposure to ozone during postnatal lung development led to alterations in airway smooth muscle orientation (Fanucchi et al, 2006). However, there was no change in the thickness or abundance of smooth muscle around the airways following ozone only exposures. Alterations of function were only noted during a subsequent allergen challenge, with no changes in baseline lung function (Tran et al, 2004). Exposure to house dust mite allergen in the same developmental primate model did result in increased abundance and mass of smooth muscle around airways. Rodents sensitized with ovalbumin also show increased airway hyper-responsiveness to challenge, which is associated with increased airway smooth muscle (Moir et al, 2003). Arsenic-induced increases in smooth muscle mass, particularly around small airways is therefore more like alterations seen with allergen senitization.
Alterations in collagen expression can not be the only factor leading to increased smooth muscle mass. Smooth muscle alterations were greatest in the smaller airways, while decreases in collagen did not show an airway size preference. While these results are not as we expected from our adult exposures, they do demonstrate that arsenic is interacting with the expression of important developmental genes, which contributes to alterations in lung structure and function.
There is concern about arsenic levels in laboratory diets affecting and masking results obtained with animals exposed to low dose arsenic in drinking water (Kozul et al, 2008). Differences in gene expression were evident when diets containing 400 ppb arsenic were compared with diets that had low levels of arsenic (20 ppb). Our diets contained 40 ppb total arsenic, which is higher than the AIN-76A diet but an order of magnitude lower than the LRD-5001 diet studied by Kozul. The diet we selected was based on the ability to alter nutritional items for future studies. Based on the NHEXAS data (Pellizzari and Clayton, 2006), the average human adult arsenic exposure levels are around 20 ppb. Children's exposure levels were about twice as high. The levels found in our diets were close to the human exposure levels. It is difficult to calculate the daily intake of arsenic during development, due to multiple sources of exposure (i.e. in utero, nursing after birth, weaned pups). However, assuming total bioavailablity from both water and food, an assumption that is probably an over estimation of the actual exposure levels, we would expect the arsenic food levels to raise the total arsenic exposure by around 20 ppb. This would elevate the total arsenic intake by about 25% and 15% for 50 and 100 ppb water, respectively. However, since both the food and water intake levels are similar to those seen in real world situations, we feel that our data still represent arsenic-induced changes that are seen at environmentally relevant levels.
It is interesting to speculate on which developmental pathways may be targeted by the arsenic exposures. The results from our current experiments show altered matrix expression and increased levels of smooth muscle around airways and increases in smooth muscle actin in whole lung. Our previous research has shown that arsenic administered at levels similar to those used in these experiments, increased MMP-9 expression and altered wound repair processes (Olsen et al, 2008). One growth factor that plays a role in all of these processes is TGF-β (Xie et al, 2007; Ohbayashi and Shimokata, 2005). Arsenic-induced increases in TGF-β1 have been reported for both acute and chronic exposures to arsenite. Injection of arsenite resulted in increased levels of TGF- β1 in kidney (Kimura et al, 2006). Long term ingestion of 200 ppm arsenite for 10 months increased TGF- β1 gene expression in the liver (Wu et al, 2008). In this case, gene expression for procollagen 1 and 3 and for smooth muscle actin were also increased. Therefore, arsenic-induced changes in TGF- β1 signaling may contribute to the alterations during development. We have previously identified the β-catenin pathway as an important target of arsenic in the lung during in utero development (Petrick et al, 2008). Wnt and β-catenin signaling, which can be modulated by TGF-β (Caraci et al, 2008), can also lead to increases in airway smooth muscle (Nunes et al, 2008). The effects of arsenic on TGF-β expression during development are currently under investigation in our laboratory.
We have shown continuous in utero and early postnatal exposure to arsenic at environmentally relevant levels results in irreversible functional and structural alterations in the lung. These results demonstrate the importance of exposures during sensitive developmental time points and suggest that interventions must also occur during these developmental time points in order to be effective.
Acknowledgments
This work was supported in part by the Superfund Basic Research Program NIEHS Grant Number P42-ES-04940 and the Southwest Environmental Health Sciences Center P30-ES-06694.
Footnotes
Conflict of Interest: The authors declare that there is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blanco LN, Frank L. The formation of alveoli in rat lung during the third and fourth postnatal weeks: effect of hyperoxia, dexamethasone, and deferoxamine. Ped Res. 1993;34:334–340. doi: 10.1203/00006450-199309000-00019. [DOI] [PubMed] [Google Scholar]
- Blanco LN, Massaro GD, Massaro D. Alveolar dimensions and number, developmental and hormonal regulation. Am J Physiol. 1989;257:L240–L247. doi: 10.1152/ajplung.1989.257.4.L240. [DOI] [PubMed] [Google Scholar]
- Bruce MC. Developmental changes in tropoelastin mRNA levels in rat lung: evaluation by in situ hybridization. Amer J Respir Cell Mol Biol. 1991;5:344–350. doi: 10.1165/ajrcmb/5.4.344. [DOI] [PubMed] [Google Scholar]
- Bruce MC, Honaker C, Karathanasis P. Postnatal age at onset of hyperoxic exposure influences developmentally regulated tropoelastin gene expression in the neonatal rat lung. Am J Respir Cell Mol Biology. 1996;14:177–185. doi: 10.1165/ajrcmb.14.2.8630268. [DOI] [PubMed] [Google Scholar]
- Camateros P, Tamaoka M, Hassan M, Marino R, Moisan J, Marion D, Guiot MC, Martin JG. Chronic asthma-induced airway remodeling is prevented by toll-like receptor-7/8 ligand S28463. Am J Respir Crit Care Med. 2007;175:1241–1249. doi: 10.1164/rccm.200701-054OC. [DOI] [PubMed] [Google Scholar]
- Caraci F, Gili E, Calafiore M, Failla M, La Rosa C, Crimi N, Sortino MA, Nicoletti F, Copani A, Vancheri C. TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol Res. 2008;57:274–282. doi: 10.1016/j.phrs.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Castleman WL, Sorkness RL, Lemanske RF, Grasee G, Suyemoto MM. Neonatal viral bronchiolitis and pneumonia induces bronchiolar hypoplasia and alveolar dysplasia in rats. Lab Invest. 1988;59:387–396. [PubMed] [Google Scholar]
- Chen CJ, Kuo TL, Wu MM. Arsenic and cancers (letter) Lancet. 1988;1988:414–415. doi: 10.1016/s0140-6736(88)91207-x. [DOI] [PubMed] [Google Scholar]
- Chiou HY, Hsueh YM, Liaw KF, Horng SF, Chiang MH, Pu YS, Lin JS, Huang CH, Chen CJ. Incidence of internal cancers and ingested inorganic arsenic: a seven-year follow-up study in Taiwan. Cancer Res. 1995;55:1296–1300. [PubMed] [Google Scholar]
- Clerch LB, Massaro D. Rat lung antioxidant enzymes: differences in perinatal gene expression and regulation. Am J Physiol. 1992;263:L466–L470. doi: 10.1152/ajplung.1992.263.4.L466. [DOI] [PubMed] [Google Scholar]
- Cockcroft DW, Davis BE. Mechanisms of airway hyperresponsiveness. J Allergy Clin Immunol. 2006;118:551–559. doi: 10.1016/j.jaci.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Collins MH, Moessinger AC, Kleinerman J, Bassi J, Rosso P, Collins AM, James S, Blanc WA. Fetal lung hypoplasia associated with maternal smoking: a morphometric analysis. Ped Res. 1985;19:408–412. doi: 10.1203/00006450-198519040-00018. [DOI] [PubMed] [Google Scholar]
- Concha G, Vogler G, Lezcano D, Nermell B, Vahter M. Exposure to inorganic arsenic metabolites during early human development. Toxicol Sci. 1998;44:185–190. doi: 10.1006/toxs.1998.2486. [DOI] [PubMed] [Google Scholar]
- D'Angio CT, Finkelstein JN, Lomonaco MB, Paxhia A, Wright SA, Baggs RB, Notter RH, Ryan RM. Changes in surfactant protein gene expression in a neonatal rabbit model of hyperoxia-induced fibrosis. Am J Physiol. 1997;272:L720–L730. doi: 10.1152/ajplung.1997.272.4.L720. [DOI] [PubMed] [Google Scholar]
- Dekkers BGJ, Schaafsma D, Nelemans SA, Zaagsma J, Meurs H. Extracellular matrix proteins differentially regulate airway smooth muscle phenotype and function. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1405–L1413. doi: 10.1152/ajplung.00331.2006. [DOI] [PubMed] [Google Scholar]
- Fanucchi MV, Plopper CG, Evans MJ, Hyde DM, Van Winkle LS, Gershwin LJ, Schelegle ES. Cyclic exposure to ozone alters distal airway development in infant rhesus monkeys. Am J Physiol Lung Cell Mol Physiol. 2006;291:L644–L650. doi: 10.1152/ajplung.00027.2006. [DOI] [PubMed] [Google Scholar]
- Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen GL, Irvin CG, Gelfand EW. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med. 1997;156:766–775. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- Hopenhayn-Rich C, Biggs ML, Smith AH. Lung and kidney cancer mortality associated with arsenic in drinking water in Cordoba, Argentina. Int J Epidemiol. 1998;27:561–569. doi: 10.1093/ije/27.4.561. [DOI] [PubMed] [Google Scholar]
- Hopenhayn-Rich C, Browning SR, Hertz-Picciotto I, Ferreccio C, Gibb H. Chronic arsenic exposure and risk of infant mortality in two areas of Chile. Environ Health Perspect. 2000;108:667–673. doi: 10.1289/ehp.00108667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopenhayn C, Ferreccio C, Browning SR, Huang B, Peralta C, Gibb H, Hertz-Picciotto I. Arsenic exposure from drinking water and birth weight. Epidemiol. 2003;14:593–602. doi: 10.1097/01.ede.0000072104.65240.69. [DOI] [PubMed] [Google Scholar]
- Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY. Development and disease: a critical role for elastin signaling in vascular morphogenesis and disease. Develop. 2003;130:411–423. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- Kimura A, Ishida Y, Hayashi T, Wada T, Yokoyama H, Sugaya T, Mukaida N, Kondo T. Interferon-gamma plays protective roles in sodium arsenite-induced renal injury by up-regulating intrarenal multidrug resistance-associated protein 1 expression. Am J Pathol. 2006;169:1118–1128. doi: 10.2353/ajpath.2006.060024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozul CD, Nomikos AP, Hampton TH, Warnke LA, Gosse JA, Davey JC, Thorpe JE, Jackson BP, Ihnat MA, Hamilton JW. Laboratory diet profoundly alters gene expression and confounds genomic analysis in mouse liver and lung. Chem Biol Interact. 2008;173:129–140. doi: 10.1016/j.cbi.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Lantz RC, Hays AM. Role of oxidative stress in arsenic-induced toxicity. Drug Metab Rev. 2006;38:791–804. doi: 10.1080/03602530600980108. [DOI] [PubMed] [Google Scholar]
- Last JA, Ward R, Temple L, Pinkerton KE, Kenyon NJ. Ovalbumin-induced airway inflammation and fibrosis in mice also exposed to ultrafine particles. Inhal Toxicol. 2004;16:93–102. doi: 10.1080/08958370490265077. [DOI] [PubMed] [Google Scholar]
- Liu J, Xie Y, Ducharme MK, Shen J, Tennant R, Diwan BA, Waalkes MP. Global gene expression associated with hepatocarcinogenesis in adult male mice induced by in utero arsenic exposure. Environ Health Perspect. 2006;114:404–411. doi: 10.1289/ehp.8534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu H, Byrne M, Krane S, Jaenisch R. Type III collagen is critical for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci. 1997;94:1852–1856. doi: 10.1073/pnas.94.5.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani TJ, Reed JJ, Shapiro SD. Expression profiling of the developing mouse lung: insights into the establishment of the extracellular matrix. Am J Respir Cell Mol Biol. 2002;26:541–548. doi: 10.1165/ajrcmb.26.5.2001-00080c. [DOI] [PubMed] [Google Scholar]
- Martin JG, Duguet A, Eidelman DH. The contribution of airway smooth muscle to airway narrowing and airway hyperresponsiveness in disease. Eur Respir J. 2000;16:349–354. doi: 10.1034/j.1399-3003.2000.16b25.x. [DOI] [PubMed] [Google Scholar]
- Mazumder DN. Effect of drinking arsenic contaminated water in children. Indian Pediatr. 2007;44:925–927. [PubMed] [Google Scholar]
- Mazumder DN, Haque R, Ghosh N, De BK, Santra A, Chakraborti D, Smith AH. Arsenic in drinking water and the prevalence of respiratory effects in West Bengal, India. Int J Epidemiol. 2000;29:1047–1052. doi: 10.1093/ije/29.6.1047. [DOI] [PubMed] [Google Scholar]
- Merkus PJ. Effects of childhood respiratory disease on the anatomical and functional development of the respiratory system. Paed Respir Reviews. 2003;4:28–39. doi: 10.1016/s1526-0542(02)00311-1. [DOI] [PubMed] [Google Scholar]
- Moir LM, Leung SY, Eynott PR, McVicker CG, Ward JP, Chung KF, Hirst SJ. Repeated allergen inhalation induces phenotypic modulation of smooth muscle in bronchioles of sensitized rats. Am J Physiol Lung Cell Mol Physiol. 2003;284:L148–L159. doi: 10.1152/ajplung.00105.2002. [DOI] [PubMed] [Google Scholar]
- Nunes RO, Schmidt M, Dueck G, Baarsma H, Halayko AJ, Kerstjens HA, Meurs H, Gosens R. GSK-3/beta-catenin signaling axis in airway smooth muscle: role in mitogenic signaling. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1110–L1118. doi: 10.1152/ajplung.00500.2007. [DOI] [PubMed] [Google Scholar]
- Ohbayashi H, Shimokata K. Matrix metalloproteinase-9 and airway remodeling in asthma. Current Drug Targets - Inflammation Allergy. 2005;4:177–181. doi: 10.2174/1568010053586246. [DOI] [PubMed] [Google Scholar]
- Olsen C, Liguori A, Zong Y, Lantz RC, Burgess J, Boitano S. Arsenic upregulates MMP-9 and inhibits wound repair in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;295:L293–L302. doi: 10.1152/ajplung.00134.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran K, Willems-Widyastuti A, Alagappan VKT, Radford K, Kranenburg AR, Sharma HS. Role of extracellular matrix and its regulators in human airway smooth muscle biology. Cell Biochem Biophys. 2006;44:139–146. doi: 10.1385/CBB:44:1:139. [DOI] [PubMed] [Google Scholar]
- Pellizzari ED, Clayton CA. Assessing the measurement precision of various arsenic forms and arsenic exposure in the National Human Exposure Assessment Survey (NHEXAS), Environ. Health Perspect. 2006;114:220–227. doi: 10.1289/ehp.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrick JS, Blachere FM, Selmin O, Lantz RC. Inorganic arsenic as a developmental toxicant: in utero exposure and alterations in developing lung. Mol Nut Food Res. 2008 doi: 10.1002/mnfr.200800019. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plopper CG, Smiley-Jewell SM, Miller LA, Fanucchi MV, Evans MJ, Buckpitt AR, Avdalovic M, Gershwin LJ, Joad JP, Kajekar R, Larson S, Pinkerton KE, Van Winkle LS, Schelegle ES, Pieczarka EM, Wu R, Hyde DM. Asthma/allergic airways disease: does postnatal exposure to environmental toxicants promote airway pathobiology? Toxicol Pathol. 2007;35:97–110. doi: 10.1080/01926230601132030. [DOI] [PubMed] [Google Scholar]
- Robledo RF, Young RS, Lantz RC, Witten ML. Short-term pulmonary response to inhaled JP-8 jet fuel aerosol in mice. Toxicol Pathol. 2000;28:656–63. doi: 10.1177/019262330002800504. [DOI] [PubMed] [Google Scholar]
- Rosenberg HG. Systemic arterial disease and chronic arsenicism in infants. Arch Pathol. 1974;97:360–365. [PubMed] [Google Scholar]
- Shen J, Liu J, Xie Y, Diwan BA, Waalkes MP. Fetal onset of aberrant gene expression relevant to pulmonary carcinogenesis in lung adenocarcinoma development induced by in utero arsenic exposure. Toxicol Sci. 2007;95:313–320. doi: 10.1093/toxsci/kfl151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SP, Barrett EG, Kalra R, Razani-Boroujerdi S, Langley RJ, Kurup V, Tesfaigzi Y, Sopori ML. Prenatal cigarette smoke decreases lung cAMP and increases airway hyperresponsiveness. Am J Respir Crit Care Med. 2003;168:342–347. doi: 10.1164/rccm.200211-1262OC. [DOI] [PubMed] [Google Scholar]
- Smith AH, Goycolea M, Haque R, Biggs ML. Marked changes in bladder and lung cancer mortality in a region of northern Chile due to arsenic in drinking water. Am J Epidemiol. 1998;147:660–669. doi: 10.1093/oxfordjournals.aje.a009507. [DOI] [PubMed] [Google Scholar]
- Smith AH, Marshall G, Yuan Y, Ferreccio C, Liaw J, von Ehrenstein O, Steinmaus C, Bates MN, Selvin S. Increased mortality from lung cancer and bronchiectasis in young adults after exposure to arsenic in utero and in early childhood. Environ Health Perspect. 2006;114:1293–1296. doi: 10.1289/ehp.8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkness R, Lemanske RF, Jr, Castleman WL. Persistent airway hyperresponsiveness after neonatal bronchiolitis in rats. J Appl Physiol. 1991;70:375–383. doi: 10.1152/jappl.1991.70.1.375. [DOI] [PubMed] [Google Scholar]
- Tran MU, Weir AJ, Fanucchi MV, Rodriguez AE, Pantle LM, Smiley-Jewell SM, Van Winkle LS, Evans MJ, Miller LA, Schelegle ES, Gershwin LJ, Hyde DM, Plopper CG. Smooth muscle hypertrophy in distal airways of sensitized infant rhesus monkeys exposed to house dust mite allergen. Clin Exp Allergy. 2004;34:1627–1633. doi: 10.1111/j.1365-2222.2004.02057.x. [DOI] [PubMed] [Google Scholar]
- Vahter M. Health effects of early life exposure to arsenic. Basic Clin Pharmacol Toxicol. 2008;102:204–211. doi: 10.1111/j.1742-7843.2007.00168.x. [DOI] [PubMed] [Google Scholar]
- von Ehrenstein OS, Mazumder DN, Yuan Y, Samanta S, Balmes J, Sil A, Ghosh N, Hira-Smith M, Haque R, Purushothamam R, Lahiri S, Das S, Smith AH. Decrements in lung function related to arsenic in drinking water in West Bengal, India. Am J Epidemiol. 2005;162:533–541. doi: 10.1093/aje/kwi236. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Ward JM, Liu J, Diwan BA. Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice, Toxicol. Appl Pharmacol. 2003;186:7–17. doi: 10.1016/s0041-008x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- Wei S, Bellusci S, Warburton D. Lung development and adult lung diseases. Chest. 2007;132:651–656. doi: 10.1378/chest.06-2663. [DOI] [PubMed] [Google Scholar]
- Weibel ER. Stereological Methods Vol 1: Practical Methods for Biological Morphometry. Academic Press; London: 1979. [Google Scholar]
- Whitney D, Massaro GD, Massaro D, Clerch LB. Gene expression of cellular retinoid-binding proteins: modulation by retinoic acid and dexamethasone in postnatal rat lung. Ped Res. 1999;45:2–7. doi: 10.1203/00006450-199901000-00002. [DOI] [PubMed] [Google Scholar]
- Winer BJ, Brown DR, Michels KM. Statistical Principles in Experimental Design. McGraw-Hill; New York: 1991. [Google Scholar]
- Wlodarczyk B, Bennett GD, Calvin JA, Craig JC, Finnell RH. Arsenic-induced alterations in embryonic transcription factor gene expression: implications for abnormal neural development. Dev Genet. 1996;18:306–315. doi: 10.1002/(SICI)1520-6408(1996)18:4<306::AID-DVG4>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Wu J, Liu J, Waalkes MP, Cheng ML, Li L, Li CX, Yang Q. High dietary fat exacerbates arsenic-induced liver fibrosis in mice. Exp Biol Med. 2008;233:377–384. doi: 10.3181/0710-RM-269. [DOI] [PubMed] [Google Scholar]
- Wu MM, Kuo TL, Hwang YH, Chen CJ. Dose–response relation between arsenic concentration in well water and mortality from cancers and vascular disease. Am J Epidemiol. 1989;130:1123–1132. doi: 10.1093/oxfordjournals.aje.a115439. [DOI] [PubMed] [Google Scholar]
- Xie S, Sukkar MB, Issa R, Khorasani NM, Chung KF. Mechanisms of induction of airway smooth muscle hyperplasia by transforming growth factor-β. Am J Physiol Lung Cell Mol Physiol. 2007;293:L245–L253. doi: 10.1152/ajplung.00068.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]