

Abstract
The generation of a series of analogs of Erythromycin A (EryA, 2) is described. In this study, we compared two peptide-based catalysts – one originally identified from a catalyst screen (5), and its enantiomer (ent-5) – for the selective functionalization of EryA. The semi-synthetic analogs were subjected to MIC evaluation with two bacterial strains and compared to unfunctionalized EryA.
The use of venerable natural product scaffolds as lead structures for new drugs and drug candidates has generally been a successful endeavor.1 In this spirit, molecules such as penicillin (1) and erythromycin (2), among others, have been used for decades as templates in the continuing battle against the numerous antibiotic resistant bacterial strains such as methicillin-resistant Staphylococcus aureus.2,3,4
The generation of analogs of natural products is challenging, in part due to the complexity of the interesting structures. If one were to desire analog compounds that are derived from natural products readily available from fermentation, then the direct, selective functionalization of the natural product could provide rapid access to site-specifically modified natural compounds without recourse to de novo synthesis or biosynthesis of each analog. In the case of polyol natural products, the specter of differentiating the same functional groups (R–OH, R′–OH, etc.) within the molecule provides a daunting challenge for catalysis. Polyketides present a particularly interesting class for this challenge, due to the complexity of structure and the typical presence of an abundance of hydroxyl groups available for synthetic derivatization. Differential functionalization of these polyol arrays has been met with limited success. Semi-synthetic techniques typically employ enzymatic catalysts.5 Natural product modification using small molecule catalysts is also possible.6,7
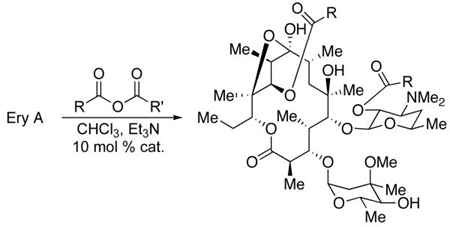
The inherent reactivity of erythromycin A (Ery A, 2) toward catalytic functionalization with achiral catalysts is known and had been previously employed to produce analogs that were then tested for activity against Staphylococcus aureus and Klebsiella pneumoniae.8 It was found that the 11-hydroxyl is the least reactive of the secondary hydroxyl groups; access to 11-acylated materials required protecting group strategies to block the 2′-position and 4″-position. The 2′-position is subject to autocatalytic acylation, owing to the vicinal dimethylamine. We focused our attention on overturning this reactivity pattern by employing peptide-based catalysts to produce 2′,11-diacylerythromycin analogs (3) directly, rather than via the 2′,4″-diacylerythromycin analogs 4, followed by an additional step involving potentially complicated ester hydrolyses.
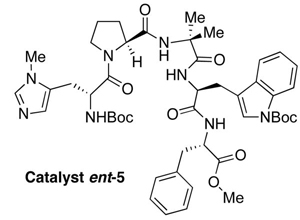
After careful screening of peptide-based catalysts, a lead (5, Figure 2) was identified by examination of product ratios achieved at a common level of reaction conversion.6 (1H NMR proved to be an effective technique for the initial determination of product ratios.) Acetic anhydride was utilized initially as the acyl donor with triethylamine employed as base. Catalyst loadings of 5-10 mol% were found to be satisfactory. Notably, catalyst 5 was effective at reversing the inherent selectivity observed with N-methylimidazole (NMI) as a catalyst. Product distributions with catalytic NMI typically produce 3 to 4 ratios ranging from 2:1 to >10:1, depending on the anhydride employed. Catalyst 5, on the other hand, exhibits reversals of this selectivity, with ratios of 3 to 4 ranging from 1:3.5 to 1:>10, depending on the nature of the anhydride.
Figure 2.

Catalyst-dependent functionalization of Ery A.
While various analogs of catalyst 5 were studied in the initial screen, we also seized the opportunity to perform an intriguing, targeted experiment. We wished to compare the performance of 5 to its enantiomer (ent-5), to see how its performance might differ. Typically, in studies of enantioselective catalysis, enantiomeric catalysts are employed to produce the opposite enantiomer of a product of interest. In addition, they may be employed as mechanistic probes of so-called “nonlinear effects.”9 In the present context, however, enantiomeric catalysts offer a special opportunity to probe robust, alternative (i.e., enantiomeric) catalyst architectures in the common spatial context of a stereochemically complex, single-enantiomer natural product substrate.10 Notably, when the enantiomer of peptide catalyst 5 (ent-5) was evaluated, it proved to be more selective than 5. Intriguingly, catalyst ent-5 also leads to preferential formation of 4 over 3. As shown in Table 1, catalyst ent-5 actually leads to an enhancement of selectivity for the production of compound 4 in all cases we examined, with reactions run to a common level of conversion. Most notably, with simple anhydrides, product ratios were enhanced from modest levels to more useful levels when catalyst 5 was exchanged for catalyst ent-5 (Acetate: 1:4.5 to 1:7, entry 1; Proprionate: 1:3.5 to 1:>10, entry 2). Presumably the molecular recognition issues leading to analogous sense of selectivity are different for the two enantiomeric catalysts, a situation that is mirrored in biological ligand-receptor interactions.11
Table 1.
Comparative selectivities obtained with catalysts 5 and ent-5.
 | ||||
|---|---|---|---|---|
| Cat. 5 | Cat. ent- 5 | |||
| Entry | Compound | R | Ratio 3 : 4 | Ratio 3 : 4 |
| 1 | 3 | Me | 1 : 4.5 | 1 : 7 |
| 2 | 6 | Et | 1 : 3.5 | 1 : >10 |
| 3 | 7 |
|
1 : 5 | 1 : 8 |
| 4 | 8 |
|
1 : >10 | 1 : >10 |
| 5 | 9 |
|
1 : 9 | 1 : >10 |
Operationally, catalyst ent-5 also leads to increased overall yield of the 2′,11-functionalized products (Table 2). Comparing the catalysts for the acetylation, it was found that the yield increased from 37% to 44% (entry 1). A more dramatic example of the effect of ent-5 was observed for the reaction of erythromycin A and propionic anhydride, with an increase in yield from 28% to 50% (entry 2). Yield increases were also observed for the β-alanyl (Boc) derivative (53 to 68%, entry 4) and the octanoyl derivative (58 to 71%, entry 5). Improved yields of natural product analogs in derivatization studies are no small matter, as isolation of pure materials is often difficult from complex mixtures of products, with each component present in minute and comparable quantities.
Table 2.
2′,11-Functionalized analogs of Ery A.
 | ||||
|---|---|---|---|---|
| Cat. 5 | Cat. ent- 5 | |||
| Entry | Compound | R | Yield (%) | Yield (%) |
| 1 | 3 | Me | 37 | 44 |
| 2 | 6 | Et | 28 | 50 |
| 3 | 7 |
|
56 | 52 |
| 4 | 8 |
|
53 | 68 |
| 5 | 9 |
|
58 | 71 |
Upon isolation of the 2′,11-functionalized substrates (i.e., compounds like 4), the corresponding 1H and 13C NMR spectra reveal a subtle molecular rearrangement involving the hemi-ketalization of the C9 ketone. As shown in Figure 3, functionalization of the C11-OH leads to the loss of a hydrogen bond between the 11-hydroxyl to the C9 ketone, and this event has been suggested as the basis for the rearrangement. The hemiketalization has furthermore been suggested to involve engagement of the C12-OH group,12 leading to the formation of structures like 4 (macrolide form B). The alternative possibility for hemi-ketalization, involving the C6-hydroxyl, has also been discussed. However, the 1H NMR data collected for the analogs in this study is most consistent with that reported by Everett et al for related compounds,10 suggesting preferential formation of the 9,12-hemiketal.
Figure 3.

Ketalization of 2′,11 functionalized Ery A.
Utilizing peptide catalysts 5 and ent-5 for derivatization of EryA with other anhydrides allowed straightforward access to a number of analogs directly, and differentially functionalized at the 2′- and 11-positions (Table 2). Synthesis of these derivatives in sufficient quantities (at least 8.0 mg, each) permitted biological testing of the 1st generation analogs. In addition, each provides a valuable precursor for further functionalization reactions. The isolated 2′,11-diacylated products could be either hydrolyzed to the 11-functionalized products, or the 4″-position could be phosphorylated to provide tris-functionalized erythromycins. Resistance to EryA has been associated with the phosphorylation of the natural product by a number of bacteria.13 Phosphorylation of the 4″-position was easily achieved with the chemical methods reported in this study. A representative list of the trifunctionalized analogs we produced is shown in Table 3.
Table 3.
Synthesis of trisfunctionalized Ery A.
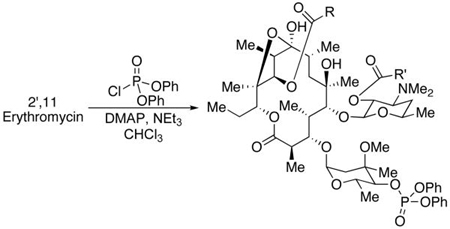 | ||||
|---|---|---|---|---|
| Entry | Compound | R | R′ | Yield (%) |
| 1 | 10 | Et | Et | 82 |
| 2 | 11 |
|
|
52 |
| 3 | 12 |
|
Me | 69 |
| 4 | 13 |
|
|
58 |
These analogs, along with those noted above, were evaluated for their activity against Staphylococcus aureus and Enterococcus faecalis. Utilizing the MIC method (minimum inhibitory concentration),14 the performance of analogs is summarized in Table 4.
Table 4.
MIC testing of erythromycin analogs.
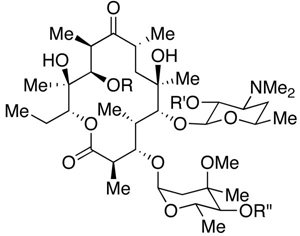 | ||||||
|---|---|---|---|---|---|---|
| MIC (μg/mL) | ||||||
| Entry | Compound | R | R′ | R′′ | S. aureus 29213 | E. faecalis 29212 |
| 1 | 6 |
|
|
H | 32 | 32 |
| 2 | 7 |
|
|
H | 32 | >32 |
| 3 | 8 |
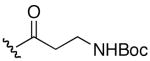
|
|
H | >32 | >32 |
| 4 | 9 |
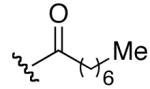
|
|
H | >32 | >32 |
| 5 | 10 |
|
|
P(O)(OPh)2 | >32 | >32 |
| 6 | 11 |
|
|
P(O)(OPh)2 | >32 | >32 |
| 7 | 12 |
|
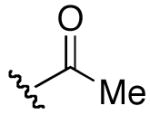
|
P(O)(OPh)2 | >32 | >32 |
| 8 | 13 |
|
|
P(O)(OPh)2 | >32 | >32 |
| 9 | 14 |
|
|
H | 32 | >32 |
| 10 | 15 |
|
H |
|
>32 | >32 |
| 11 | 16 |
|
H | H | 16 | 32 |
| 12 | 17 |
|
H | H | 16 | 32 |
| 13 | 18 |
|
H | H | 32 | >32 |
| 14 | 19 |
|
H | H | 32 | 32 |
| 15 | 2 | H | H | H | 1 | 2 |
In comparison to EryA (entry 15), the analogs generally did not exhibit the impressive levels of activity against the bacterial strains. Two analogs that retained some of the activity of EryA against S. aureus were the propionate analog 16 (entry 11, 16 μg/mL) and the 4-pentenoate derivative 17 (entry 12, 16 μg/mL). The presence of esters at the 2′-position has been previously reported to require hydrolysis for activity.15 Consistent with these findings, the analogs with free 2′-hydroxyl did exhibit some increase in effectiveness (e.g., entry 1 versus entry 11; entry 2 versus entry 12).
In summary, we have used two peptide-based catalysts to reorder the inherent reactivity offered by the natural product scaffold provided by EryA. These catalysts have allowed the straightforward access to useful quantities of a number of natural product analogs, which were then evaluated against two strains of bacteria. The ability to use chiral catalysts for selective modification of complex molecules could prove to be a useful tool in the direct modification of natural products quite broadly. Such experiments also raise fascinating issues in the area of asymmetric catalysis, which are aggressively under study in our laboratory at the present time.
Supplementary Material
Figure 1.

Penicillin G and Erythromycin A (Ery A).
Acknowledgments
We are grateful to the NIH/NIGMS (GM-068649) for generous support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Wilson RM, Danishefsky SJ. J Org Chem. 2006;71:8329. doi: 10.1021/jo0610053. [DOI] [PubMed] [Google Scholar]
- 2.Walsh CT, Wright GD. Chem Rev. 2005;105:391. doi: 10.1021/cr030100y. [DOI] [PubMed] [Google Scholar]
- 3.Fisher JF, Meroueh SO, Mobashery S. Chem Rev. 2005;105:395. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 4.McDaniel R, Welch M, Hutchinson CR. Chem Rev. 2005;105:543. doi: 10.1021/cr0301189. [DOI] [PubMed] [Google Scholar]
- 5.(a) Abel U, Koch C, Speitling M, Hansske FG. Curr Opin Chem Biol. 2002;6:453. doi: 10.1016/s1367-5931(02)00338-1. [DOI] [PubMed] [Google Scholar]; (b) Gu J, Ruppen ME, Cai P. Org Lett. 2005;7:3945. doi: 10.1021/ol0514395. [DOI] [PubMed] [Google Scholar]
- 6.Lewis CA, Miller SJ. Angew Chem Int Ed. 2006;45:5616. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- 7.(a) Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; (b) Peddibhotla S, Dang Y, Liu JO, Romo D. J Am Chem Soc. 2007;129:12222. doi: 10.1021/ja0733686. [DOI] [PubMed] [Google Scholar]
- 8.(a) Jones PH, Perun TJ, Rowley EK, Baker EJ. J Med Chem. 1972;15:631. doi: 10.1021/jm00276a017. [DOI] [PubMed] [Google Scholar]; (b) Martin YC, Jones PH, Perun TJ, Grundy WE, Bell S, Bower RR, Shipkowitz NL. J Med Chem. 1972;15:635. doi: 10.1021/jm00276a018. [DOI] [PubMed] [Google Scholar]
- 9.Blackmond DG. Acc Chem Res. 2000;33:402. doi: 10.1021/ar990083s. [DOI] [PubMed] [Google Scholar]
- 10.The issue is related to that of “double diastereoselectivity.” See: Masamune S, Choy W, Petersen JS, Sita LR. Angew Chem Int Ed. 1985;97:1.
- 11.Feng S, Schreiber SL. J Am Chem Soc. 1997;119:10873. [Google Scholar]
- 12.Everett JR, Hunt E, Tyler JW. J Chem Soc, Perkin Trans 2. 1991:1481–1487. [Google Scholar]
- 13.Kaltz L, Ashley GW. Chem Rev. 2005;105:499. doi: 10.1021/cr030107f. and references therein. [DOI] [PubMed] [Google Scholar]
- 14.(a) Bou G. Methods Mol Biol. 2007;391:29. doi: 10.1007/978-1-59745-468-1_3. [DOI] [PubMed] [Google Scholar]; (b) Lorian V. Antibiotics in Laboratory Medicine. Fifth. Lippincott Williams and Wilkins; 2005. [Google Scholar]
- 15.Tardrew PL, Mao JCH, Kenney D. Appl Microbiol. 1969;18:159. doi: 10.1128/am.18.2.159-165.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.