

Abstract
We report a catalytic approach to the synthesis of a key intermediate on the synthetic route to a pharmaceutical drug candidate in single enantiomer form. In particular, we illustrate the discovery process employed to arrive at a powerful, peptide-based asymmetric acylation catalyst. The substrate this catalyst modifies represents a remarkable case of desymmetrization, wherein the enantiotopic groups are separated by nearly a full nanometer, and the distance between the reactive site and the pro-stereogenic element is nearly six Angstroms. Differentiation of enantiotopic sites within molecules that are removed from the prochiral centers by long distances presents special challenges to the field of asymmetric catalysis. As the distance between enantiotopic sites increases within a substrate, so too may the requirements for size and complexity of the catalyst. The approach presented herein contrasts enzymatic catalysts and small-molecule catalysts for this challenge. Ultimately, we report here a synthetic, miniaturized enzyme mimic that catalyzes a desymmetrization reaction over a substantial distance. In addition, studies relevant to mechanism are presented, including (a) the delineation of structure-selectivity relationships through the use of substrate analogs, (b) NMR experiments documenting catalyst-substrate interactions, and (c) the use of isotopically labeled substrates to illustrate unequivocally an asymmetric catalyst-substrate binding event.
Introduction
Situations of remote asymmetric induction present special challenges to the field of asymmetric catalysis. In particular, desymmetrization reactions wherein the reactive centers are substantially removed from pro-stereogenic centers are particularly challenging since the catalyst must simultaneously mediate bond formation or cleavage, while at the same time affording some type of meaningful interaction with pro-stereogenic functionality at a distance. Problems of this type in synthesis are often addressed through substrate control, with intramolecular relay of chiral information through the covalent framework of the molecule.1,2 When catalytic bond formation is targeted, across a truly substantial distance, enzymes are often used.3 Indeed, their macromolecular architecture offers the potential for simultaneous enantioselective recognition and functionalization over a considerable vector.4 Historically, selective C–H bond oxidations distal from functionality and stereogenic centers provide some of the most dramatic examples.5,6 Instances of the use of small molecules for remote catalytic asymmetric induction are rare, and much more typical are situations where the reacting site is removed from a pro-stereogenic element by just a few bonds.7
We recently confronted the desymmetrization of compound 1, a diphenyl methane bis(phenol), which was required to deliver, in optically pure form, an unsymmetrical derivative with distinct para-oxygenated substituents (e.g., 2; Figure 1a). Simple molecular mechanics calculations underscored the particular challenge of this desymmetrization; the desired site of functionalization is > 5.7 Å from the prochiral stereogenic center of the substrate (Figure 1b) and nearly a full nanometer spans the enantiotopic phenol oxygen atoms. We recently reported our discovery of a small-molecule peptide catalyst (4) that achieves this challenging desymmetrization with high levels of enantioselectivity.8 Herein, we describe in detail the process that led to the discovery of catalyst 4, in addition to experiments that shed some light on the mechanistic basis of this intriguing asymmetric reaction.
Figure 1.

(a) Compound 1 and its desymmetrized analog 2. (b) Substrate metrics for bis(phenol) 1, defining a 5.75 Å distance between desired site of functionalization and prochiral stereogenic center (MM calculations). 9.79 Å span the enantiotopic hydroxyl groups. (c) Catalyst 4 effects enantioselective desymmetrization with high enantiomeric excess.
Results and Discussion
Enzymatic desymmetrization of bis(phenol) 1
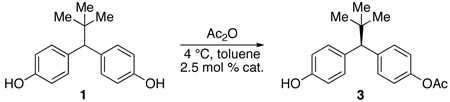
The initial approach to the desymmetrization of 1 examined enzymatic desymmetrization of bis(acetate) 5 (Scheme 1). After examination of a large lipase collection (>450 enzymes), the hydrolysis route was able to offer reasonable access to mono(acetate) 3 using Mucor miehei, a lipase composed of hundreds of amino acids.9 However, two main liabilities conspired to motivate an alternative strategy. First, high levels of throughput of bis(acetate) 5 to mono(acetate) 3 are difficult to achieve during individual batch runs, in part due to the substrate’s minimal solubility in the aqueous buffered media that allows maximum enzymatic efficiency. Furthermore, in order to obtain 3 with high levels of optical purity, substantial overhydrolysis to bis(phenol) 1 is necessary. The requirement for overhydrolysis of 5 to obtain high optical purity is a result of unremarkable enantiotopic group discrimination displayed by the enzyme, and a reflection of a secondary kinetic resolution that serves to “correct” the lack of stereochemical specificity exhibited by the enzyme.10 Indeed, mono(acetate) 3 is produced with only 50–55% ee at low conversion. Of course, overconversion of 5 to bis(phenol) 1 substantially reduces the overall yield of the process. Each of these factors contributed to a low 40% overall yield and 99% ee after recrystallization of mono(acetate) 3.
Scheme 1.

Enzymatic Desymmetrization of Bis(acetate)
It is likely that further and intensive study of enzymatic processes could well lead to an effective biocatalytic solution to the desymmetrization of 5.11 Yet, the inherently low stereospecificity exhibited by the enzymes, in combination with the low volumetric throughput encountered in the enzymatic studies,12 stimulated the pursuit of an alternative strategy. In particular, we elected to pursue a small-molecule asymmetric acylation approach,13 with peptide-based catalysts as the cornerstone of the strategy.14
Directed library design
The presence of the phenol functionality in the substrate, as well as the large span between the hydroxyls made it necessary to generate some lead catalysts by preparing a library of peptides. This peptide library was biased to mimic the functional group array presented by the substrate. Figure 2 illustrates our initial thinking, wherein incorporation of a “linear” aromatic-aliphatic-aromatic motif was targeted to mimic the substrate architecture of bis(phenol) 1. Hexapeptides were initially chosen with the hypothesis that the bis(phenol) could be a bifunctional substrate – one that would present one hydroxyl group for derivatization, while another might form a noncovalent contact with the catalyst in the transition state.15 We speculated, on the basis of hand-held models, that a six-residue peptide could easily interact with both termini of the substrate. If such a bifunctional catalyst-substrate complex could form, then we speculated that the twists and turns of peptide secondary structure in these transition states could lead to substantial energetic differentiation of diastereomeric transition states that would result from interaction of each phenolic group as either a nucleophile, or anchor for hydrogen bond contact to catalyst.
Figure 2.

Initial peptide design influenced by target structure
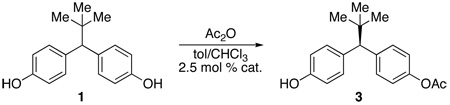
The first 42 hexapeptides were produced using solid-phase peptide synthesis and contained both directed aromatic-aliphatic-aromatic as well as random sequences. The initial conditions employed were 2.5 mol % peptide catalyst in toluene at 4 °C with two equivalents of acetic anhydride. Nonpolar solvents (e.g., PhCH3) were found to be optimal, although in the initial studies small aliquots of EtOH were included in the reaction mixture. This modification was identified as beneficial in terms of maintaining homogeneous solutions throughout the course of the reaction. In later studies, EtOH was omitted in favor of a small quantity of THF, which had the same effect (vide infra). The addition of tertiary amine bases to scavenge the acetic acid was found to catalyze the reaction and produce racemic product, and was not pursued any further.16 The most selective peptide catalysts are shown in Table 1 (entries 1–7).17 A range in enantiomeric excesses was observed with this catalyst set (21% ee to 40% ee), and yields of 3 were modest (12% to 46%), due primarily to overacylation of the product. We note that our early analysis of the results assumed that there was only minimal modulation of product enantioselectivity as a function of secondary kinetic resolution. While we did not verify this assertion in all instances, we did show in selected cases that the results were a good indicator of raw enantiotopic group discrimination in the catalysis. Subsequent experiments with the best catalyst confirmed that kinetic resolution of (±)-3 occurs to only a minimal extent with many of these catalysts (vide infra).
Table 1.
Peptide Results for First Three Libraries
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | i | i+1 | i+2 | i+3 | i+4 | i+5 | 1 (%) | 3 (%) | 5 (%) | ee (%) |
| Library 1 | ||||||||||
| 1 | Pmh | Asn(Trt) | Leu | Val | DPhe | Phe | 0.2 | 27.2 | 72.6 | 40 |
| 2 | Pmh | Phe | Gly | Leu | Phe | Phe | 0.3 | 19.5 | 80.2 | 29 |
| 3 | Pmh | Dbg | DPhe | DPro | Val | Leu | 4.7 | 46.3 | 49.0 | 28 |
| 4 | Pmh | Tyr(t-Bu) | DPhe | DHis(Trt) | Tyr(t-Bu) | Phe | 0.2 | 16.2 | 83.6 | 25 |
| 5 | Pmh | Leu | Aib | Phe | DVal | Phe | 0.2 | 12.1 | 87.7 | 23 |
| 6 | Pmh | Phe | Gly | Pro | Phe | Phe | 0.4 | 18.4 | 81.2 | 21 |
| 7 | Pmh | Thr(t-Bu) | DPro | Ser(t-Bu) | Phe | Phe | 0.5 | 30.4 | 69.1 | 21 |
| Library 2 | ||||||||||
| 8 | Pmh | Asn(Trt) | Leu | Thr(t-Bu) | DPhe | Phe | 0.1 | 25.4 | 74.5 | 49 |
| 9 | Pmh | Asn(Trt) | Leu | Ser(t-Bu) | DPhe | Phe | 0.2 | 27.3 | 72.5 | 47 |
| 10 | Pmh | Asn(Trt) | Leu | Ala | DPhe | Phe | 0.2 | 14.7 | 85.1 | 43 |
| 11 | Pmh | Asn(Trt) | Aib | Val | DPhe | Phe | 0.2 | 20.2 | 79.6 | 41 |
| 12 | Pmh | Asn(Trt) | Dbg | Val | DPhe | Phe | 0.2 | 17.8 | 82.0 | 39 |
| 13 | Pmh | Asn(Trt) | Leu | Val | DPhe | DPhe | 0.2 | 15.6 | 84.2 | 38 |
| 14 | Pmh | Asn(Trt) | Ala | Val | DPhe | Phe | 0.2 | 70.6 | 29.2 | 32 |
| Library 3 | ||||||||||
| 15 | Pmh | DAsp(Ot-Bu) | Leu | Thr(t-Bu) | DPhe | Phe | 1.3 | 25.0 | 73.7 | 42 |
| 16 | Pmh | DAsp(Ot-Bu) | Leu | Ser(t-Bu) | DPhe | Phe | 0.2 | 16.6 | 83.2 | 42 |
| 17 | Pmh | Asp(OBzl) | Leu | Val | DPhe | Phe | 0.2 | 9.4 | 90.4 | 40 |
| 18 | Pmh | Asn(Trt) | Leu | Tle | DPhe | Phe | 0.1 | 25.4 | 74.5 | 36 |
| 19 | Pmh | Asn(Trt) | Leu | Val | DVal | Phe | 0.4 | 40.0 | 59.6 | 34 |
| 20 | Pmh | DAsp(Ot-Bu) | Leu | Val | DPhe | Phe | 0.3 | 16.6 | 83.1 | 32 |
The most promising catalyst of the first peptide library possessed the aromatic-aliphatic-aromatic motif with the sequence of Boc-Pmh-Asn(Trt)-Leu-Val-d-Phe-Phe-OMe (catalyst 6; 40% ee, entry 1, Table 1). Although the amount of bis(acetate) 5 that was produced is high (73%), the 40% enantiomeric excess represented a reasonable starting place for further catalyst optimization. A second library was synthesized (entries 8–14, 24 members) to optimize the i+2 and i+3 residues within the catalyst. Residues were selected at random and included some stereochemical perturbations to assess any variation in selectivity as a function of diastereomeric catalyst structures. Indeed, some improvements over the first library were observed, with enantioselectivities of up to 49% documented (25% yield, entry 8, Table 1). Most notably, alteration of the valine residue (i+3) within catalyst 6 to protected serine or threonine led to enhanced selectivity (47% ee, entry 9; 49% ee, entry 8, Table 1). The third library (entries 16–20, 24 members) examined variation of the i+1 site (occupied by tritylated asparagine in catalyst 6). Most perturbations at this position were deleterious to the selectivity (e.g., 32–42% ee, entries 15, 16, 17 and 20).

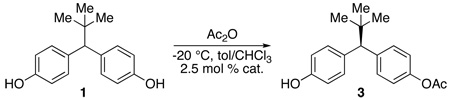
The fourth library altered and optimized the aliphatic and aromatic portion (i+3, 4, 5, 6) of the peptide while maintaining the i+1 position as either a tritylated histidine or asparagine (Table 2). We also explored variations in the reaction conditions. For example, the optimal solvent system was found to be a blend of toluene and chloroform (1/1 v/v). At this stage, the inclusion of 1% THF (by volume) was also identified as beneficial for complete solubilization of materials, and homogeneous conditions throughout the course of the reaction. Modulation of temperature also resulted in a dramatic effect. Performing reactions at −20 °C led to an improved yield of the monoacylated material 3, with substantial diminution of bis(acetate) 5. At the same time, enantioselectivity was markedly improved. The results of the studies of the fourth library are shown in Table 2 and culminated in lead catalyst 7 (72% yield, 79% ee, entry 7, Table 2).
Table 2.
Fourth Peptide Library with Varying Temperature
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 4 °C Reactions | ||||||||||
| Entry | i | i+1 | i+2 | i+3 | i+4 | i+5 | 1 (%) | 3 (%) | 5 (%) | ee (%) |
| 1 | Pmh | Asn(Trt) | Aib | Ser(t-Bu) | DTyr(t-Bu) | Phe | 0.2 | 24.8 | 75.0 | 56 |
| 2 | Pmh | Asn(Trt) | Aib | Ser(t-Bu) | DPhe | Phe | 0.2 | 24.1 | 75.7 | 52 |
| 3 | Pmh | His(Trt) | Aib | Ser(t-Bu) | DPhe | Phe | 0.2 | 21.9 | 77.9 | 51 |
| 4 | Pmh | Asn(Trt) | Leu | Ser(t-Bu) | DTyr(t-Bu) | Phe | 0.1 | 26.4 | 73.5 | 51 |
| 5 | Pmh | Asn(Trt) | Leu | Thr(t-Bu) | DTyr(t-Bu) | Phe | 0.2 | 23.3 | 76.5 | 51 |
| 6* | Pmh | Asn(Trt) | Leu | Ser(t-Bu) | DPhe | Phe | 0.1 | 11.7 | 88.2 | 51 |
| *Entry 9, Table 1 | ||||||||||
| −20 °C Reactions | ||||||||||
| 7 | Pmh | Asn(Trt) | Aib | Ser(t-Bu) | DTyr(t-Bu) | Phe | 3.2 | 71.9 | 24.9 | 79 |
| 8 | Pmh | His(Trt) | Aib | Ser(t-Bu) | DPhe | Phe | 2.4 | 73.5 | 24.1 | 78 |
| 9 | Pmh | Asn(Trt) | Aib | Thr(t-Bu) | DPhe | Phe | 0.2 | 53.5 | 46.3 | 78 |
| 10 | Pmh | Asn(Trt) | Aib | Ser(t-Bu) | DPhe | Phe | 3.5 | 74.8 | 21.7 | 77 |
| 11* | Pmh | Asn(Trt) | Leu | Ser(t-Bu) | DPhe | Phe | 0.6 | 62.8 | 36.6 | 70 |
| 12 | Pmh | Asn(Trt) | Leu | Ser(t-Bu) | DTyr(t-Bu) | Phe | 12.6 | 73.2 | 14.2 | 68 |
| *Entry 9, Table 1 | ||||||||||

At this stage, further variation in peptide sequence did not lead to dramatic changes in catalyst performance. As shown in Table 3, additional variations at the i+1, 5 or i+1, 4 residues did not lead to improvements (75–78% yield, 81% ee, entry 1 and 2, Table 3). Strikingly, changes at the i+4, 5 positions (73–78% yield, 72–80% ee, entries 3–6, Table 3) also had little effect. As a result, we wondered if the residues were necessary at all, and they were thus targeted for deletion. Truncation of the most selective hexapeptide catalyst (7; Table 2, entry 1 and 7) was examined to determine the number of residues required for enantioselectivity. Indeed, we found that the first four residues were most critical (i.e., catalyst 8: Boc-Pmh-Asn(Trt)-Aib-Ser(t-Bu), entry 9, Table 3) and the last two residues are nearly superfluous (66–68% yield, 68–72% ee, entry 7–8, Table 3).
Table 3.
Final Library and Truncation Studies
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | i | i+1 | i+2 | i+3 | i+4 | i+5 | 1 (%) | 3 (%) | 5 (%) | ee (%) |
| 1 | Pmh | His(Trt) | Aib | Ser(t-Bu) | DTyr(t-Bu) | Phg | 5.1 | 77.8 | 17.1 | 81 |
| 2 | Pmh | His(Trt) | Aib | Ser(t-Bu) | DLeu | Phe | 15.5 | 75.2 | 9.3 | 81 |
| 3 | Pmh | Asn(Trt) | Aib | Ser(t-Bu) | DLeu | Phe | 3.4 | 77.7 | 18.9 | 80 |
| 4 | Pmh | Asn(Trt) | Aib | Ser(t-Bu) | DTyr(t-Bu) | Phg | 8.2 | 76.9 | 14.9 | 80 |
| 5 | Pmh | Asn(Trt) | Aib | Ser(t-Bu) | DTyr(t-Bu) | Ala | 18.2 | 74.4 | 7.4 | 78 |
| 6 | Pmh | Asn(Trt) | Aib | Ser(t-Bu) | DTyr(t-Bu) | Ile | 6.1 | 73.4 | 20.5 | 72 |
| 7* | Pmh | Asn(Trt) | Aib | Ser(t-Bu) | DTyr(t-Bu) | Phe | 13.9 | 66.0 | 20.1 | 68 |
| 8 | Pmh | Asn(Trt) | Aib | Ser(t-Bu) | DTyr(t-Bu) | 8.5 | 68.0 | 23.5 | 72 | |
| 9 | Pmh | Asn(Trt) | Aib | Ser(t-Bu) | 3.3 | 68.8 | 27.9 | 71 | ||
| 10 | Pmh | Asn(Trt) | Aib | 10.4 | 64.2 | 25.4 | 54 | |||
| 11 | Pmh | Asn(Trt) | 20.4 | 52.1 | 27.5 | 11 | ||||
| *Entry 7, Table 2 | ||||||||||

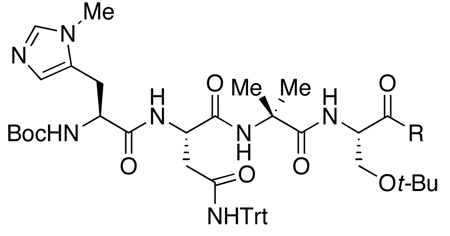
We then undertook studies of optimization of the C-terminal substituent of tetrameric peptide catalyst 8. In particular, we explored changing the C-terminus group from a methyl ester (8) to a number of amides (9–14, 4, Table 4). The simple exchange of the ester for benzyl amide 9 increased the selectivity marginally (61% yield, 68% ee, entry 2, Table 4). The use of chiral methylated benzyl amide 10 and 11 maintained similar results (60–68% yield, 69–70% ee, entry 3–4, Table 4). Extension of the aromatic system to chiral naphthyl amide 13 increased the ee to 79%, and it was found that tosyldiamine 4 (Table 4, entry 8) was even more beneficial, delivering catalyst 4 that afforded the product with 84% ee, and 89% isolated yield.
Table 4.
C-Terminus Optimization
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R | 3 (%) | ee (%) | Entry | R | 3 (%) | ee (%) | ||
| 1 | 8 | OMe | 62 | 65 | 5 | 12 | 76 | 67 | |
| 2 | 9 | 61 | 68 | 6 | 13 | 71 | 79 | ||
| 3 | 10 | 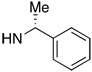 |
60 | 70 | 7 | 14 | 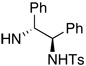 |
82 | 64 |
| 4 | 11 | 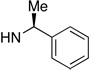 |
68 | 69 | 8 | 4 | 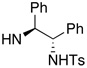 |
89 | 84 |
All reactions were run at −30 °C in CHCl3 with 2.5 mol % catalyst loading.
At this stage, we endeavored to further optimize the reaction conditions for the best catalyst. Indeed, we found that performing the reaction at −30 °C in chloroform, with a mere 2.5 mol % of catalyst 4 led to isolation of 3 in 80% yield, with optical purity of 95% ee (Scheme 2). Overall, 138 peptides were synthesized in various sub-libraries to deliver a lead peptide-based hexameric catalyst (7; Table 2, entry 1 and 7). The peptide was then truncated to a tetrapeptide catalyst (8; Table 3, entry 9). Optimization of the C-terminal substituent afforded the final catalyst 4, which provides excellent results for the desymmetrization.
Scheme 2.

Final Optimized Peptide
Substrate analog studies – variation of the prochiral center substituent
The role of the prochiral center of bis(phenol) 1 in this unique desymmetrization is intriguing to consider. On the one hand, it could impart steric demands on bond rotations that dictate the relative disposition of the aryl rings (e.g., note arrows in structure 15). On the other hand, there is the possibility that the substituent actually is involved in catalyst-substrate contacts. Varying the nature of this substituent would allow examination of the subtle effects of substrate-catalyst interactions. A series of bis(phenol) analogs were generated and the peptide catalyst was applied under the standard conditions (Table 5). The systematic reduction of the steric bulk of the t-Bu group (1) to isopropyl decreased the selectivity (substrate 15a, 73% ee, entry 1). Further decreasing the size of the R-group to ethyl (15b), methyl (15c) led to lower enantioselectivities (63% and 52% ee, entries 2 and 3). Quite notably, phenyl-bearing substrate 15d –which is nearly C3-symmetric – still leads to an observed enantiomeric excess of 50% (entry 4). Thus, the absolute steric size of the substituent (as defined by A-values for example; t-Bu >4.5, i-Pr 2.2, Et 1.8, Ph 2.8 kcal/mol)18 do not correlate well with the magnitude of the enantioselectivities. These observations may suggest that an explicit contact between the prochiral center substituent and the catalyst may not be stereochemistry determining. Rather, these observations suggest that the role of the substituent may be more critical in creating a propeller-like twist between the aryl rings of 1, and that the barrier to interconversion between propeller twists may be significant for enantioselective catalysis.
Table 5.
Variation of Prochiral Center
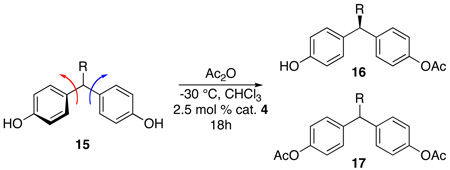 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | Recovered 15 (%) | 16 (%) | ee 16 (%) | 17 (%) | |
| 1 | i-Pr | a | 22 | 62 | 73 | 7 |
| 2 | Et | b | 22 | 60 | 63 | 10 |
| 3 | Me | c | 50 | 42 | 52 | 7 |
| 4 | Ph | d | 32 | 40 | 50 | 11 |
Kinetic resolution studies
Effective desymmetrization of 1 could derive from a) enantiotopic group selection and/or b) secondary kinetic resolution of monofunctionalized product.19 Each catalyst could rely on varying contributions from these two situations, which influence product ratios as well as enantioselectivity. In an optimally efficient desymmetrization, the selective catalyst will afford high enantiotopic group selection and rely less on the secondary kinetic resolution.20 In this limit, the recovery of monofunctionalized product approaches 100%. However, for less selective catalysts, such as the enzymes initially studied above, the enantiotopic group selection may be poor, but the secondary kinetic resolution may provide good enantioselectivity. In this scenario, yield suffers.
The acylation utilizing successful catalyst 4 provided 80% yield and 95% ee in the desymmetrization of 1, which suggest a low level of reliance on secondary kinetic resolution. The peptide was tested against racemic mono(acetate) material (3) and, indeed, it was found to produce only 11% ee material with 52% recovery of the mono(acetate) (krel = 1.4,21 entry 1, Table 6).
Table 6.
Kinetic Resolution
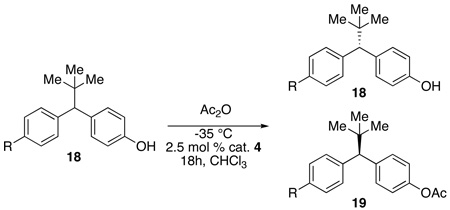 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | Recovered 18 (%) | ee 18 (%) | krel 18 | 19 (%) | ee 19 (%) |
| 1a | OAc, 3 | 52c | 11c | 1.4 | 48d | -- |
| 2a | H, a | 52 | 16 | 1.7 | 46 | 22 |
| 3b | OMe, b | 55 | 5 | 1.2 | 44 | 13 |
| 4b | OTBS, c | 62 | 5 | 1.2 | 38 | 5 |
2 equiv Ac2O.
4 equiv Ac2O.
Mono(acetate) 3.
Bis(acetate) 5.
Nevertheless, further studies of kinetic resolution of substrate analogs provided additional mechanistic insight. For example, monodeoxy substrate (18a, entry 2, Table 6) also proves a poor substrate for kinetic resolution (22% ee at 46% conversion, krel = 1.7). One possible implication is that the “non-reacting” free phenol of 1 may be involved in a catalyst-substrate contact, perhaps by H-bonding, during the stereodefining transition state. Similarly, neither methyl ether nor silyl protected substrates exhibit significant kinetic resolution (18b and 18c; krel = 1.2 and 1.2, entry 3 and 4, Table 6). Taken together, these data may imply a bifunctional transition state with a role for each substrate phenol – a situation in which one undergoes bond formation, while the other serves as an anchor for a catalyst-substrate interaction nearly a full nanometer removed from the reacting center.
Spectroscopic studies related to mechanism
If indeed there is a bifunctional mechanism at play during the desymmetrization of 1, it could be possible to observe a catalyst-substrate complex that reflects key interactions. Such a complex could also have implications for the mechanism of bond-formation in the reaction. The basis of N-methylimidazole-catalyzed acylation of alcohols with anhydrides is something of a matter of debate, with studies supporting both nucleophilic and general base catalysis mechanisms described in the literature.22 In fact, certain alcohols such as aliphatic alcohols may proceed by a nucleophilic catalysis mechanism (Scenario 1, Figure 3) due to the high pKa of the hydroxyl. Phenolic analogs such as 1 may proceed by general base catalysis (Scenario 2, Figure 3) since the pKa of the hydroxyl is substantially lower. Peptide catalyst 4 possesses a modified histidine amino acid, which may act as a nucleophilic catalyst or a general base type catalyst (Figure 3). Thus, any observation of a catalyst-substrate complex does not definitively establish the role of an individual phenolic site as either a locus of general base-catalyzed bond formation, or alternatively as an anchor for H-bonding contemporaneous with bond formation at a remote site. Nonetheless, the observation of a catalyst-substrate complex could provide evidence for differentiation of the two phenols on the time scale of an NMR experiment, supporting the notion that each phenol fulfills a different role in the transition state.
Figure 3.

Possible mechanistic roles of phenols during 4-catalyzed desymmetrization of 1.
If substrate 1 and catalyst 4 form a pre-acylation complex on the NMR time scale, observation of a catalyst-substrate complex should be possible.23 Indeed, co-mixing substrate 1 and catalyst 4 in a 1:1 ratio in CDCl3 resulted in an 1H NMR spectrum (500 MHz) consistent with non-degenerate aromatic rings in the bis(phenol) region (Ha ~ 6.67 and Hb ~ 7.16 ppm; Figure 4). This lack of degeneracy suggests the formation of a catalyst-substrate complex that is in slow exchange on the NMR time scale. Such differentiation of the two rings could then be manifested in the catalytic reactions through either ground state destabilization, or alternatively the loss of degeneracy could also be manifested in a transition state involving the participation of acetic anhydride as well.
Figure 4.

1H NMR of peptide-bis(phenol) complex
In order to further understand these catalyst-substrate complexes, we also performed related experiments with 13C-labeled substrates. Simulating the reaction conditions, a 13C-labeled substrate 1 was produced and mixed with the different catalysts at −30 °C in CDCl3 and their 13C NMR spectra recorded (500 MHz). Strikingly, when the most effective catalyst 4 is employed in this experiment, an unambiguous loss of aryl ring degeneracy is observed (Figure 5e). However, when less selective catalysts were co-mixed with 1, the effect is much less striking, and even undetectable with catalysts that are quite unselective in the catalytic reaction. For example, when N-methylimidazole (NMI) is mixed with 1, a unique and sharp singlet is observed (Figure 5a). Of course, when NMI catalyzes acylation of 1, a racemic (0% ee) product is formed. Less selective peptide-based catalysts provided an intermediate result, with catalysts that afford moderate ee (Figure 5b–d, 54%–72%) producing 13C NMR spectra that exhibit line-broadening, reflecting weak interactions between catalyst and substrate. Strikingly, only the best catalyst (4, 95% ee for desymmetrization of 1) reveals a doubling of signals upon exposure to the carbon-labeled analog of 1. These observations suggest a correlation between significant catalyst-substrate binding, and ultimate catalyst performance. Indeed, the specific interaction of 1 with 4 suggests that this unique catalyst is able to associate with unique substrate 1, breaking the symmetry of the phenolic rings, and that this capacity is also manifested kinetically in this remarkable desymmetrization.
Figure 5.

13C Spectra for catalysts and labeled bis(phenol). (a) NMI, 0% ee; (b) Table 3, entry 10, 54% ee; (c) Table 3, entry 8, 72% ee; (d) Table 3, entry 9, 71% ee; (e) Peptide catalyst 4, 95% ee.
Conclusions
The catalyst discovery protocol employed for this study culminated, after the synthesis of fewer than 150 simple peptides, in tetrameric catalyst 4 that is highly effective for a desymmetrization reaction involving a substrate with substantial enantiotopic group separation. It is also notable that substrate 1 bears no resemblance to any class of alcohol substrate that had been previously studied with simple peptide-based catalysts. Since peptide 4 is simpler than a macromolecular enzyme, its performance establishes that small molecules may supplement enzymes as potential catalysts for reactions involving the extremes of remote asymmetric induction. Furthermore, the collection of evidence supporting specific catalyst-substrate interactions suggests that the determination of a three-dimensional solution structure of a complex might be possible, and that such a structure could be correlated to the outcome of the catalytic reaction.
Finally, since target-oriented natural products syntheses and process-oriented pharmaceutical syntheses often present tantalizing and unique structures that have not been previously encountered in synthetic methodology development, the rapid, protocol-based approach to the discovery of effective specific catalysts may be of particular value in these arenas.
Supplementary Material
Experimental procedures and data. This material is available via the Internet at http://pubs.acs.org.
Acknowledgements
This work was supported by the NIH (GM-068649) and Merck Research Laboratories.
References
- 1.(a) Linnane P, Magnus N, Magnus P. Nature. 1997;385:799. [Google Scholar]; (b) Shimizu M, Mikami K. J. Org. Chem. 1992;57:6105. [Google Scholar]; (c) Mikami K, Narisawa S, Shimizu M, Terada M. J. Am. Chem. Soc. 1992;114:6566. [Google Scholar]
- 2.Clayden J, Lund A, Vallverdu L, Helliwell M. Nature. 2004;431:966. doi: 10.1038/nature02933. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Urdiales E, Alfonso I, Gotor V. Chem. Rev. 2005;105:313. doi: 10.1021/cr040640a. [DOI] [PubMed] [Google Scholar]
- 4.Hughes DL, Bergan JJ, Amato JS, Bhupathy M, Leazer JL, McNamara JM, Sidler DR, Reider PJ, Grabowski EJJ. J. Org. Chem. 1990;55:6252. [Google Scholar]
- 5.Capdevila JH, Wei S, Helvig C, Falck JR, Belosludtsev Y, Truan G, Graham-Lorence SE, Peterson JA. J. Biol. Chem. 1996;37:22663. doi: 10.1074/jbc.271.37.22663. [DOI] [PubMed] [Google Scholar]
- 6.Yang J, Breslow R. Angew. Chem. Int. Ed. 2000;39:2692. doi: 10.1002/1521-3773(20000804)39:15<2692::aid-anie2692>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 7.(a) Mikami K, Narisawa S, Shimizu M, Terada M. J. Am. Chem. Soc. 1992;114:6566. [Google Scholar]; (b) Trost BM, Mino T. J. Am. Chem. Soc. 2003;125:2410. doi: 10.1021/ja029708z. [DOI] [PubMed] [Google Scholar]; (c) Lewis CA, Sculimbrene BR, Xu Y, Miller SJ. Org. Lett. 2005;7:3021. doi: 10.1021/ol051061a. [DOI] [PubMed] [Google Scholar]; (d) Zhao Y, Rodrigo J, Hoveyda AH, Snapper ML. Nature. 2006;443:67. doi: 10.1038/nature05102. [DOI] [PubMed] [Google Scholar]
- 8.Lewis CA, Chiu A, Kubryk M, Balsells J, Pollard D, Esser CK, Murry J, Reamer RA, Hansen KB, Miller SJ. J. Am. Chem. Soc. 2006;128:16454. doi: 10.1021/ja067840j. Also see “Research Highlights” Nature 2006, 444, 974. [DOI] [PubMed] [Google Scholar]
- 9.(a) Tonouchi N, Shoun H, Uozumi T, Beppu T. Nucleic Acids Res. 1986;14:7557. doi: 10.1093/nar/14.19.7557. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fatima S, Ahmad B, Khan RH. Life. 2007;59:179–186. doi: 10.1080/15216540701335716. [DOI] [PubMed] [Google Scholar]
- 10.Schreiber SL, Schreiber TS, Smith DB. J. Am. Chem. Soc. 1987;109:1525. [Google Scholar]
- 11.Klibanov AM. Acc. Chem. Res. 1990;23:114. [Google Scholar]
- 12.Jacobsen EN, Finney NS. Chem. Biol. 1994;1:85. doi: 10.1016/1074-5521(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 13.(a) Vedejs E, Chen X. J. Am. Chem. Soc. 1996;118:1809–1810. [Google Scholar]; (b) Ruble JC, Latham HA, Hu GC. J. Am. Chem. Soc. 1997;119:1492–1493. [Google Scholar]; (c) Spivey AC, Fekner T, Spey SE, Adams H. J. Org. Chem. 1999;64:9430–9443. [Google Scholar]; (d) Spivey AC, Maddaford A, Redgrave AJ. Org. Prep. Proced. Int. 2000;32:333. [Google Scholar]; (e) France S, Guerin DJ, Miller SJ, Lectka T. Chem. Rev. 2003;103:2985. doi: 10.1021/cr020061a. [DOI] [PubMed] [Google Scholar]; (f) Ishihara K, Kosugi Y, Akakura M. J. Am. Chem. Soc. 2004;126:12212. doi: 10.1021/ja045850j. [DOI] [PubMed] [Google Scholar]; (g) Connon SJ. Lett. Org. Chem. 2006;3:333. [Google Scholar]; (h) MacKay JA, Vedejs E. J. Org. Chem. 2006;71:498. doi: 10.1021/jo0519155. [DOI] [PubMed] [Google Scholar]; (i) Wurz RP. Chem. Rev. 2007;107:5570. doi: 10.1021/cr068370e. [DOI] [PubMed] [Google Scholar]
- 14.(a) Colby Davie E, Mennen SM, Xu Y, Miller SJ. Chem. Rev. 2007;107:5759. doi: 10.1021/cr068377w. [DOI] [PubMed] [Google Scholar]; (b) Miller SJ. Acc. Chem. Res. 2004;37:601. doi: 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]
- 15.Vasbinder MM, Jarvo ER, Miller SJ. Angew. Chem. Int. Ed. 2001;40:2824. doi: 10.1002/1521-3773(20010803)40:15<2824::aid-anie2824>3.3.co;2-a. [DOI] [PubMed] [Google Scholar]
- 16.Zhu J, Chastanet J, Beugelmans R. Synth. Commun. 1996;26:2479. [Google Scholar]
- 17.HPLC product ratios
- 18.Eliel EL, Wilen SH, Mander LN. Stereochemistry of Organic Compounds. New York: Wiley & Sons; 1994. p. 696. [Google Scholar]
- 19.(a) Keith JM, Larrow JF, Jacobsen EN. Adv. Synth. Catal. 2001;343:5. [Google Scholar]; (b) Robinson DEJE, Bull SD. Tetrahedron: Asymmetry. 2003;14:1407. [Google Scholar]; (c) Vedejs E, Jure M. Angew. Chem. Int. Ed. 2005;44:3974. doi: 10.1002/anie.200460842. [DOI] [PubMed] [Google Scholar]
- 20.Willis MC. J. Chem. Soc., Perkin Trans. 1999;1:1765. [Google Scholar]
- 21.Kagan HB, Fiaud JC. Top. Stereochem. 1988;18:249. [Google Scholar]
- 22.Guibe-Jampel E, Bram G, Vilkas M. Bull. Soc. Chim. Fr. 1973:1021. [Google Scholar]; (b) Höfle G, Steglich W, Vorbrüggen H. Angew. Chem. Int. Ed. Engl. 1978;17:569. [Google Scholar]; (c) Pandit NK, Connors KA. J. Pharm. Sci. 1982;71:485. doi: 10.1002/jps.2600710503. [DOI] [PubMed] [Google Scholar]; (d) Hiratake J, Inagaki M, Yamamoto Y, Oda J. J. Chem. Soc. Chem. Commun. 1985:1717. [Google Scholar]; (e) Hiratake J, Inagaki M, Yamamoto Y, Oda J. J. Chem. Soc., Perkin Trans. 1987;1:1053. [Google Scholar]; (f) Savelova VA, Belousova IA, Simanenko YS. Zh. Org. Khim. 1994;30:246. [Google Scholar]; (g) Denmark SE, Beutner GL. Angew. Chem. Int. Ed. 2008;47:2. [Google Scholar]
- 23.Although we have not established the kinetic order of these reactions explicitly, in related studies a 1st-order dependence on substrate and catalyst was found. It is, of course, possible that an alternative kinetic order ensues in the present cases. see: Jarvo ER, Copeland GT, Papaioannou N, Bonitatebus PJ, Jr, Miller SJ. J. Am. Chem. Soc. 1999;121:11638–11643.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and data. This material is available via the Internet at http://pubs.acs.org.