

Abstract
Infection of macrophages with Leishmania parasites does not result in the production of IL-12. In addition, infection with Leishmania suppresses IL-12 production elicited by otherwise potent activators of IL-12. We provide evidence that engagement of phosphatidyl inositol-3 kinase (PI3K) signaling during Leishmania amazonensis infection leads to the prevention of IL-12 p70 production at the level of transcription of its p40 subunit in bone marrow derived macrophages (BMDMϕ). Inhibition of PI3K signaling with specific inhibitors of PI3K or the downstream kinase Akt, reverses the IL-12 blockade. Although the MAP kinase ERK (p44 and p42) was transiently activated by infection with L. amazonensis, inhibition of MEK, the kinase upstream of ERK, with PD98059, did not reverse the blockade of IL-12. Furthermore, inhibition of the other MAP kinases JNK and p38 as well as treatment of cells with pertussis toxin that blocks G protein mediated signaling, did not reverse the prevention of IL-12 production by Leishmania infection. Interestingly, activation of PI3K/Akt signaling had differential effects on ERK and p38 activation. Taken together we propose that infection of BMDMϕ with Leishmania promastigotes activates both positive and negative signaling pathways that control IL-12 production. PI3K signaling activated by the infection is the negative signaling pathway that prevents IL-12 production.
Keywords: Leishmania, Parasitic-Protozoan, Intracellular parasitism, Signal transduction, Cytokines, IL-12, Protein kinases, PI3K, Akt
1. Introduction
Approximately 12 to 15 million people are affected by leishmaniasis worldwide. This disease can manifest as cutaneous and mucocutaneous lesions or as visceral disease. Disease presentation depends upon multiple factors which includes the infecting Leishmania species and undefined host characteristics. Inflammatory cells of the macrophage and dendritic cell lineages are the primary host cells of Leishmania parasites. It is well established that the presence of cytokines such as IL-12, IFN-γ, IL-10 and IL-4 influences the clinical course of leishmaniasis (Reiner and Locksley, 1995, Jones et al, 1998, Belkaid et al, 2001, Kane and Mosser, 2001). In mouse models of leishmaniasis such as C57BL/6 mice infected with L. major where there is eventual control of the infection, the early production of IL-12 is important to help skew the immune response towards a TH1 type (Reiner and Locksley, 1995, Mattner et al, 1997). In experimental infections that do not exhibit a tendency to self cure, such as infection of C57BL/6 or BALB/c mice with L. mexicana, IL-12 has been shown to play a limited role in promoting parasite control (Torrentera et al, 2002, Buxbaum et al, 2002). Paradoxically though, infection of macrophages and some dendritic cells derived from most inbred mouse strains with the promastigote form of several Leishmania species does not result in the production of IL-12 (Reiner et al, 1994, Carrera et al, 1996, Bennet et al, 1999). This parasite effect on IL-12 production has been confirmed by in vitro and in vivo studies as well as by investigations in which IL-12 production was monitored at the single cell level (Belkaid et al, 1998). In addition to the prevention of IL-12 production during infection, these parasites also suppress infected macrophage IL-12 production in response to potent stimuli such as lipopolysaccharide (LPS) (Carrera el al, 1996, Cameron et al, 2004). Given that IL-12 plays an important role in the host’s control of Leishmania infections, it is imperative that the mechanisms that these parasites employ to modulate the production of this cytokine be completely elucidated.
IL-12 is composed of two covalently linked glycosylated chains, p40 and p35, which form the biologically active p70 heterodimer (Trinchieri and Scott, 1999). The p35 gene is ubiquitously expressed in most cells, whereas the p40 gene is primarily expressed by phagocytic cells, particularly in response to microbial agents and their products. Both positive and negative inducers of IL-12 have been described (Ma and Trinchieri, 2001). Whereas IFN-γ is a positive inducer of IL-12, phagocytic receptor co-ligation (e.g. Fc and complement receptors), engagement of G protein coupled receptors and IL-10 negatively regulate IL-12 production (Waggoner et al, 2005, Marth and Kelsall, 1997, Braun and Kelsall, 2001, D’Andrea et al, 1993).
Recent studies on the intracellular events that regulate IL-12 production by macrophages have identified the activation of phosphatidyl inositol-3 kinase (PI3K) as a signal transducer that negatively regulates IL-12 production (Fukao et al, 2002, Martin et al, 2003, Waggoner et al, 2005). Ruhland et al, (2007) recently found that infection of macrophages with L. major, L. pifanoi and L. amazonensis, results in the engagement and sustained activation of the PI3K/Akt signaling pathway. The involvement of PI3K/Akt signaling in the prevention of IL-12 production by infected parasites or suppression of IL-12 production by Leishmania infected macrophages in response to otherwise potent stimuli has not been addressed. In this study, we provide evidence that although L. amazonensis parasites engage PI3K/Akt and MAPK signaling pathways in bone marrow derived macrophages, it is only the activation of PI3K/Akt which results in the prevention of IL-12 production and that inhibition of the PI3K/Akt pathway relieves this suppression.
2. Materials and Methods
2.1 Parasites, Macrophages (BMDMϕ) and Infections
Leishmania amazonensis (MHOM/BR/77/LTB0016) promastigotes were grown in Schneider’s Drosophila medium (GIBCO BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville GA) and 10μg/ml gentamicin at 23°C. Infectivity of parasites was maintained by periodic passage through BALB/c mice as reported previously (Soong et al, 1996). Parasites were used in the late stationary phase.
Pathogen free BALB/C or C57Bl/6 mice were obtained from the University of Florida’s Department of Pathology. Bone marrow derived macrophages (BMDMϕ) were obtained from mouse femurs and plated directly into 60 or 100 mm tissue culture plastic dishes (Falcon, Becton Dickinson Labware, Franklin Lakes, NJ) at a cell density of 7.5 × 105 and 5 × 106 cells per dish, respectively. Bone marrow cells were cultured in DMEM containing 10% fetal bovine serum (FBS) supplemented with 20% L929 cell-conditioned medium with a medium change at day 5. After 7 days, BMDMϕ medium was replaced with complete DMEM (without L929 cell conditioned medium) for use in the experiments described. These studies were approved by the Institutional Animal Care and Use Committee at the University of Florida. The murine macrophage RAW 264.7 cell line was maintained in RPMI 1640 supplemented with 10% FBS.
To assess cytokine production by BMDMϕ in response to infection, BMDMϕ were incubated with Leishmania promastigotes at 1:10 ratio (macrophages: parasites) and incubated at 34 °C for 24 hours at which time the culture medium was recovered. Internalization of parasites was microscopically confirmed. In experiments to assess the effect of inhibiting signaling pathways, the inhibitors were added at the time of infection and left in the cultures for the duration of the experiment. All inhibitors were initially dissolved in DMSO and diluted into culture medium. Inhibition studies were carried out with these specific inhibitors: Akt inhibitor IV (10μM), wortmannin (100nM), pertussis toxin (1μg/ml), JNK inhibitor II (20μM), PD 98059 (MEK inhibitor) (50μM), and SB203580 (p38 MAPK inhibitor) (20μM) (EMD Biosciences, San Diego, CA). Control infections and treatment of resting macrophages were set up with vehicle (DMSO). The effect of inhibitors on parasite viability was assessed by evaluating the course of infections on coverslips.
Lipopolysaccharide from E. coli was used at a final concentration of 500ng/ml (Sigma Aldrich, St. Louis, MO). IFN-γ was purchased from BD Biosciences (San Jose, CA) and used at a concentration of 100U/ml. Cells that were stimulated with LPS/IFN-γ were first primed with IFN-γ for twelve hours prior to LPS/IFN-γ treatment.
2.2 Phospho-protein Analysis
Activation of specific signaling intermediates was determined by probing of Western blots for phosphorylated proteins. BMDMϕ plated in 100mm dishes were infected with L. amazonensis promastigotes at a parasite to macrophage ratio of 20:1 to ensure that there was a high percentage of infected macrophages at early times after infection. At the times indicated in the results section, parasites were removed; cell monolayers washed twice with PBS and the BMDMϕ were lysed directly in the plate with lysis buffer (25mM HEPES, 150 mM NaCl, 1% Triton X-100, 1× protease inhibitor pill (1 pill/10mL) (Roche Applied Science, Indianapolis, IN), 20 μl/mL Phosphatase Inhibitor Cocktail 1 (Sigma-Aldrich, St. Louis, MO), 20μl/mL Phosphatase Inhibitor Cocktail 2 (Sigma), 50μM NaVO4). Protein containing lysates were then cleared of cellular debris by centrifugation and protein concentration was determined using a Bradford Protein Assay (BioRad, Hercules, CA). 50 μg of each sample were resolved by SDS-PAGE electrophoresis on 12% polyacrylamide gels. Proteins were transferred to Immobilon P membrane (Millipore, Billerica, MA) and probed with primary antibody according to the manufacturer’s recommendations. Primary antibodies utilized included phospho-Akt (Ser 473), phospho-p38 (Thr180/Tyr182), phospho-p44/p42 (ERK) (Thr202/Tyr204), and native p44/p42 from Cell Signaling Technology (Danvers, MA). Native Akt antibody was obtained from BioVision (Mountain View, CA). Native p38 antibody and chicken anti-rabbit horseradish peroxidase conjugate were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Specific protein bands were visualized using Western Lightning Chemiluminescence reagent (Perkin Elmer, Waltham, MA). Blots were then developed on X-ray film and densitometric analysis performed using Image J (NIH, Bethesda, MD). For densitometric analysis, results from at least 3 separate experiments were collated.
2.3 Cytokine ELISAs
To measure cytokines secreted by BMDMϕ cultures under conditions discussed in the results section, cell culture supernatant fluid were removed from macrophage monolayers and cleared by centrifugation. IL-12 p70 concentrations were measured using OptEIA ELISA kits (BD Biosciences) according to manufacturer’s instructions. Supernatants were applied in triplicates to 96 well plates (Nalge Nunc International, Rochester, NY), which had been pre-coated with capture antibody (4.4 μg/ml). Plates were developed using HRP substrate solutions (BD Biosciences) and plates were read at 450nm on a Powerwave 200 spectrophotometer (Bio-Tek, Winooski, VT). The cytokine concentration in each sample was extrapolated from a standard curve generated from the measured absorbance obtained from recombinant standards supplied with the kit. The sensitivity of IL-12 p70 ELISA was 62.5 pg/ml.
2.4 Reverse Transcriptase Quantitative Polymerase Chain Reaction (rtPCR)
To determine the presence of mRNA transcripts of IL-12p40, rtPCR was performed using an OneStep rtPCR kit (Qiagen, Valencia, CA) and TaqMan FRET probes on an iCycler Thermal Cycler (Biorad). RNA was purified from cell lysates using RNeasy kit (Qiagen, Valencia, CA) and 2 μg of total RNA was used in each reaction. To generate cDNA from the RNA pool, each sample was first incubated for 30 minutes at 50°C. Forward and reverse primers for IL-12 p40 and the positive control, GAPDH (Biosource, Carlsbad, CA) were used at the manufacturer’s recommended dilutions. Additionally, the positive control was used to determine efficiency for each primer set. Quantitation of message was determined by an increase in fluorescence above a threshold as TaqMan probe was incorporated into the product. The threshold cycle (CT) obtained for each cytokine was normalized to the reference gene GAPDH message levels using the delta Ct method (Bustin et al, 2005). Briefly, this method approximates the fold increase in target gene expression and involves the comparison of the ratio of control gene’s expression with the target gene’s expression between treatment groups; this method also takes into account primer efficiency.
TaqMan probe sequence for IL-12 (Biosource) was 5’-FAM-TTC AAC ATC AAG AGC AGT AGC AGT TCC CCT-Black Hole Quencher-3’; GAPDH TaqMan probe sequence was 5’-FAM-CAT TCT CGG CCT TGA CTG TGC CGT-Black Hole Quencher-3’ and was synthesized by Sigma Genosys (The Woodlands, TX). Polymerase chain reaction conditions were as follows: 95°C for 15 minutes, and 40 cycles of 95°C (1 minute) and 60°C(1 minute) and 68°C (1 minute). rtPCR for each sample was run in triplicates. The results presented are the average values from at least three experiments.
2.5 Promoter Activity Assay
IL-12 p40 promoter activity was determined by nucleofecting RAW 264.7 macrophages with an IL-12 promoter firefly luciferase construct. The pDM4300 plasmid has been previously characterized (Murphy et al, 1995) and was the generous gift of Dr. Kenneth Murphy (Washington University School of Medicine, St. Louis, MO). This plasmid contains 4300 bp upstream of the transcriptional start site of the murine IL-12 p40 gene ligated into pBlueScript-Luc (Stratagene, La Jolla, CA). Additionally, to normalize data, macrophages were co-nucleofected with phRL-TK, a plasmid containing the Renilla luciferase gene (Promega, Madison, WI) under control of the herpes virus thymidine kinase promoter. pDM4300 (10μg) and phRL-TK (500ng) were nucleofected into 1 × 107 macrophages using the Nucleofector System (Amaxa Biosystems, Gaithersburg, MD). After 24 hours, cells were infected with L. amazonensis promastigotes at an MOI of 10 along with the appropriate inhibitors. After an additional 24 hours, cells were harvested and luciferase activity determined utilizing the Dual Luciferase Reporter Assay System (Promega, Madison, WI) on a 20/20n luminometer (Turner Biosystems, Sunnyvale, CA). The activity of the plasmid was confirmed by its induction in response to LPS and, moreover, by its synergistic induction to LPS and IFN-γ (data not shown).
2.6 Statistics
Data are presented as the mean ± standard deviation, and data were considered significant with the student’s T test at a P value of ≤0.05. Significance of cytokine ELISA data were analyzed by analysis of variance.
3 Results
3.1 IL-12 is not produced after infection with Leishmania amazonensis promastigotes
Since Leishmania species and isolates differ in their interactions with macrophages (McDowell et al, 2002), it was important to establish that infection with the L. amazonensis parasites that was used in these experiments results in the absence of IL-12 production as well. Infections were evaluated with late stationary stage promastigotes of L. amazonensis. After a 24 hour infection of bone marrow derived macrophages obtained from BALB/c mice, IL-12 p70 was assayed in the supernatant fluid. The results of ELISA assays compiled from three experiments are shown in Figure 1a. Whereas a copious amount of IL-12 was produced when BMDMϕ were incubated with LPS and IFN-γ, incubation of BMDMϕ with promastigotes of L. amazonensis did not result in the production of IL-12 p70 above levels seen in supernatant fluid from resting macrophage cultures. These infected BMDMϕ produced significantly reduced levels of IL-12 upon incubation with LPS and IFN-γ (not shown). Similar results were obtained with cells derived from C57BL/6 mice (not shown).
Figure 1.
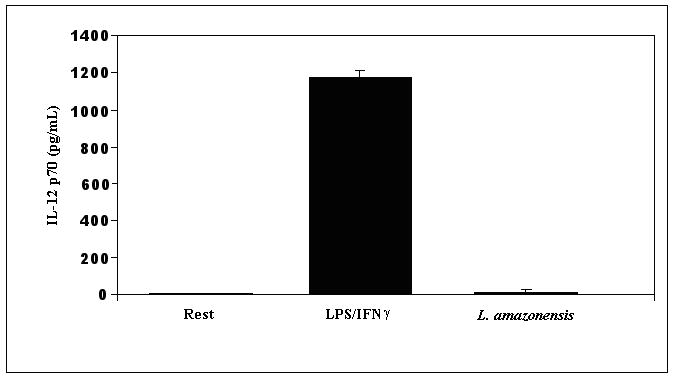
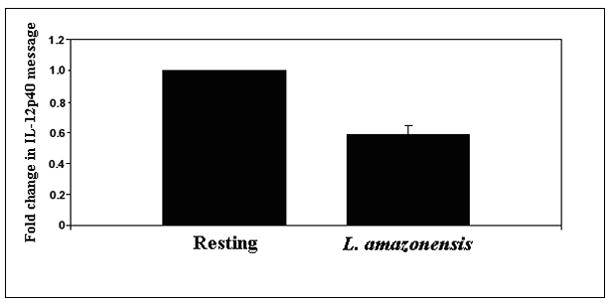
IL-12 is not produced when BMDMϕ are infected with L. amazonensis promastigote forms. (A) BMDMϕs were incubated with late stationary L. amazonensis promastigotes. Culture supernatant fluid was recovered after 24 hours and the presence of IL-12p70 was determined by ELISA. IL-12 concentration in these cultures was compared to supernatant fluid obtained from resting cultures or from cultures of BMDMϕ that were pre-incubated with IFN-γ before stimulation with LPS. (B) mRNA levels of IL-12p40 were determined by real time PCR in infected macrophage samples obtained after 12 hours infection. The threshold cycle (CT) of IL-12p40 for each treatment was normalized to the CT of the reference gene GAPDH for each treatment run in parallel. The fold change in IL-12p40 message compared to uninfected resting macrophages was determined using the delta CT method described in Materials and Methods. For each experiment, the resting BMDMϕ IL-12 message level was arbitrarily set to 100% (i.e. 1) and the fold change from this value for L. amazonensis infected cells was calculated. This allowed comparisons to be made between separate experiments. These results are compiled from 3 experiments.
To further establish that Leishmania promastigotes do not induce IL-12 expression, analysis of IL-12 mRNA levels by real time RT-PCR using Taqman probe was performed. IL-12 p40 mRNA levels were measured after infection and then normalized against mRNA levels of GAPDH. Figure 1b shows the peak levels of IL-12 p40 at 12 hrs post infection. Even though the level of IL-12 p40 mRNA was very low in resting cells and these cells did not produce IL-12, we consistently found that IL-12 p40 mRNA levels in L. amazonensis-infected cells were lower than the levels in uninfected cells (Figure 1b). Taken together, these observations confirm that L. amazonensis infection of macrophages does not result in IL-12 p40 production and consequently, no IL-12p70.
To commence evaluating the mechanisms that underlie the apparent prevention of IL-12 production upon infection with Leishmania promastigotes, we considered the role of IL-10. Infection with complement-opsonized parasites resulted in IL-10 production, and only minimal, if any, IL-10 was produced upon infection with unopsonized parasites (not shown). Furthermore, prevention of IL-12 production by L. amazonensis promastigotes was not reversed in experiments in which antibodies to IL-10 were added to BMDMϕs infections (data not shown).
3.2 Inhibition of PI3K/Akt signaling restores IL-12 production by Leishmania-infected cells
We have previously shown that infection of RAW 264.7 macrophages with Leishmania promastigotes activates PI3K/Akt signaling, which confers on the infected cell the capacity to resist apoptosis (Ruhland et al, 2007). Here we show that in bone marrow derived macrophages as well, there is sustained activation of Akt in response to infection with L. amazonensis (Figure 2a). The figure shows that Akt is phosphorylated within 15 minutes of infection and reaches a plateau at 4 hours which is maintained throughout the first 24 hours of infection.
Figure 2.
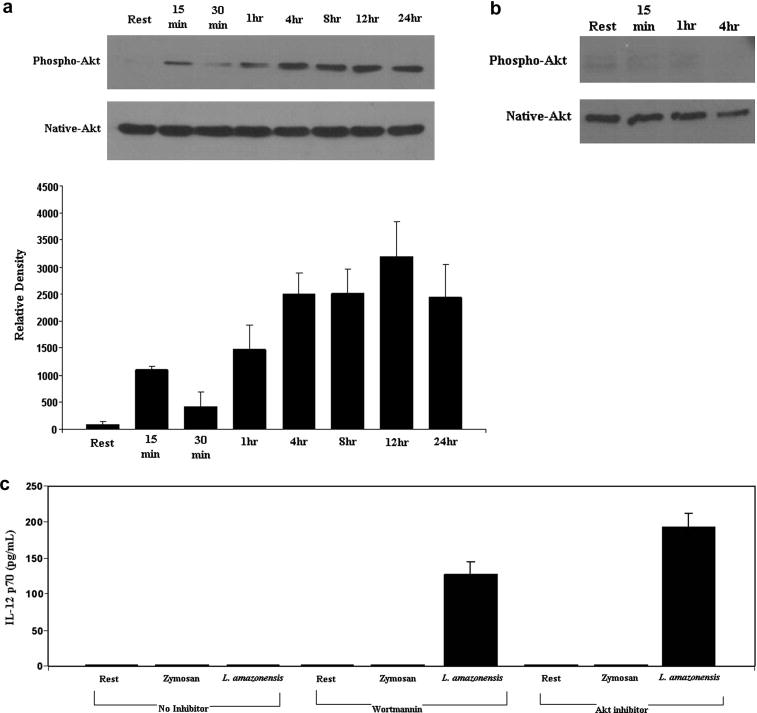
L. amazonensis infection activates PI3K/Akt signaling and inhibition of components in this pathway restores IL-12p70 production in L. amazonensis infected BMDMϕ. A) Macrophage lysates were prepared at the indicated times after infection and then analyzed in Wesern blots for the presence of activated Akt with anti-phospho-Akt (Ser 473). The blots were stripped and re-probed with antibodies to native Akt. Densitometric analysis of blots from several experiments was performed with ImageJ software and results were normalized to native protein levels. B) Akt activation was inhibited when infection was performed in the presence of the PI3K inhibitor wortmannin (100nM). Parallel infections were performed with vehicle and infected cells with results similar to the data presented in Figure 2A. C) Supernatant fluid was obtained after 24 hours from macrophage cultures that were incubated with Zymosan or infected with L. amazonensis parasites at an MOI of 10. IL-12p70 concentrations in these culture supernatants were determined by ELISA. Resting cells yielded similar results to vehicle treated resting and vehicle treated infected cells. These data are compiled from three experiments
We next determined whether signaling through the PI3KAkt pathway is important for the prevention of IL-12 by determining the effect of the PI3K inhibitor, wortmannin on IL-12 production by infected cells. The activity of wortmannin was confirmed by Western blot analyses (Figure 2b), which showed that infections that were performed in the presence of wortmannin did not result in the activation of Akt. We should acknowledge that fewer macrophages (~10% as compared to ~30%) were infected in the presence of wortmannin; but the inhibition of Akt phosphorylation was likely due to the inhibition of PI3K by wortmannin and not lower numbers of infected macrophages. It is intriguing that treating cultures with Akt inhibitor did not have a similar inhibition of infection.
Whereas infection of BMDMϕ with Leishmania did not result in the production of IL-12, infections that were performed in the presence of wortmannin resulted in the production of IL-12 (Figure 2C). Even with fewer infected cells, significant levels of IL-12 were produced. Our previous experiments had shown that suppressing Akt levels by transfection of siRNA to all three Akt isoforms yielded comparable results to using the Akt inhibitor IV (Ruhland et al, 2007). So in these studies, Akt activity was abolished with the Akt inhibitor IV. Even greater levels of IL-12 were produced when macrophages were infected with Leishmania promastigotes in the presence of Akt inhibitor IV (Figure 2C). It is noteworthy that neither wortmannin nor Akt inhibitor IV induced resting cells to produce IL-12.
Experiments were performed to determine if inhibition of PI3K/Akt signaling during the phagocytosis of Zymosan particles would also result in IL-12 production. As Figure 2C shows, no IL-12 was produced in response to inhibition of PI3K/Akt signaling during the phagocytosis of Zymosan particles. Experiments with opsonized Zymosan particles yielded similar results (not shown). The results of these later experiments suggest that signaling during Leishmania infection is distinct from that which occurs in generic phagocytosis since internalization of either opsonized or non-opsonized Zymosan in the presence of wortmannin or Akt inhibitor did not lead to IL-12 production. Taken together, these studies show that the activation of PI3K/Akt signaling upon parasite contact with and entry into macrophages inhibits signaling that can result in IL-12 production by infected cells. Stated conversely, inhibition of PI3K/Akt signaling relieves suppression of IL-12 production by Leishmania infected macrophages, which results in IL-12 production by infected cells without addition of exogenous stimuli.
3.3 Inhibition of Akt activity relieves IL-12 p40 production at the level of transcription
Quantitative reverse transcriptase mediated real time PCR was utilized to determine IL-12p40 mRNA levels in resting and infected cells treated with Akt inhibitor. A Taqman FRET probe specific for an internal sequence within the IL-12p40 gene was utilized as described in Materials and Methods. Infections were carried out at an MOI of 10, with and without Akt inhibitor. As Figure 3a shows, the inhibition of Akt in infected cell cultures leads to an approximate 6 fold increase in IL-12p40 mRNA, which is statistically significant. This effect was consistently seen at the 12 hour time point and not at earlier times (data not shown). Addition of Akt inhibitor to resting cells has only a minimal effect on IL-12p40 mRNA levels.
Figure 3.
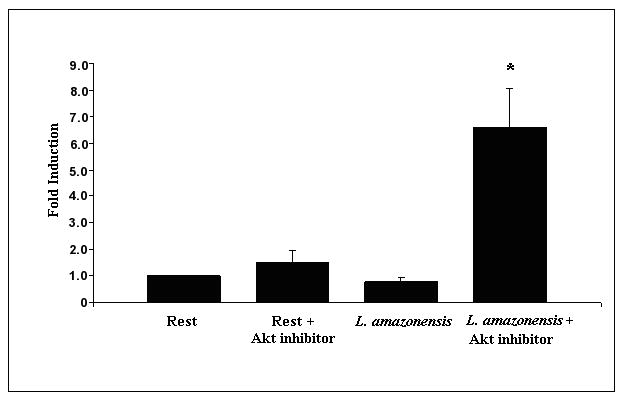
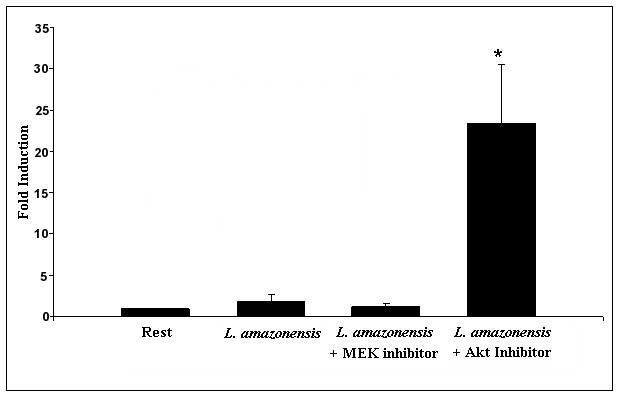
Inhibition of Akt activation during infection with L. amazonensis relieves suppression of IL-12p40 at the transcriptional level. (A) BMDMϕ were infected with L. amazonensis in the presence or absence of Akt inhibitor IV and RNA was extracted at 12 hours. IL-12p40 mRNA transcript levels were determined in macrophages by quantitative rtPCR. IL-12p40 CT values were normalized to GAPDH CT values obtained for each treatment. Fold-induction of IL-12p40 was determined by comparing this value to that obtained for uninfected resting BMDMϕ (i.e. resting BMDMϕ fold change = 1). (B) Activity of IL-12 promoter driven luciferase plasmid (pDM4300) in L. amazonensis infected cells. Cells were co-nucleofected with a constitutively expressed Renilla luciferase plasmid. Values are normalized and represented as fold induction over resting cells and are the collation of 3 sets of experimental data. Resting cells were treated with DMSO vehicle. (*) denotes statistical significance, which was determined by the Student’s T test (p<0.05).
Further experiments were performed with a plasmid construct containing the 4300 bp sequence upstream of the IL-12p40 transcriptional start site that drives a firefly luciferase reporter (Murphy et al, 1995). This promoter construct contains multiple enhancer elements, and, in response to inflammatory stimuli such as LPS/IFN-γ, leads to significant luciferase production (data not shown). In order to normalize results, firefly luciferase values obtained were normalized to a constitutively expressed Renilla luciferase construct, which was co-transfected into the cells. In these experiments, we utilized the RAW 264.7 macrophage cell line since nucleofection of BMDMϕ resulted in very low transfection efficiencies when compared to the cell line. As Figure 3b shows, in transfected macrophages infected with L. amazonensis there is little change in luciferase levels, which indicates that infection results in minimal induction of the IL-12 p40 promoter. However, treatment of infected cells with Akt inhibitor resulted in an approximate 20 fold induction over resting levels. In comparison addition of the MEK inhibitor PD98059 had no effect on the induction of IL-12 p40. This data taken together with the rtPCR results, confirms that Akt acts by suppressing IL-12 p40 at the transcriptional level.
3.4 MAPK signaling does not mediate the inhibitory effect of Leishmania amazonensis infection on IL-12 production
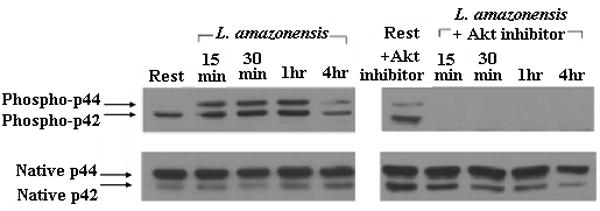
We assessed the activation of MAPKs in L. amazonensis-infected cells. As Figure 4a and 4b show, p38 and ERK are phosphorylated during infection by L. amazonensis. Although p38 phosphorylation levels fluctuate over the 24 hour infection course, the overall levels of this signaling intermediate remain above resting levels. In contrast, the phosphorylation of ERK seems transient: activated forms peak 1 hour post infection and then drop to resting levels. Phophorylation of ERK was apparently mediated by the upstream kinase MEK since use of PD98059, a MEK inhibitor, abrogated phosphorylation of ERK (Figure 4c).
Figure 4.
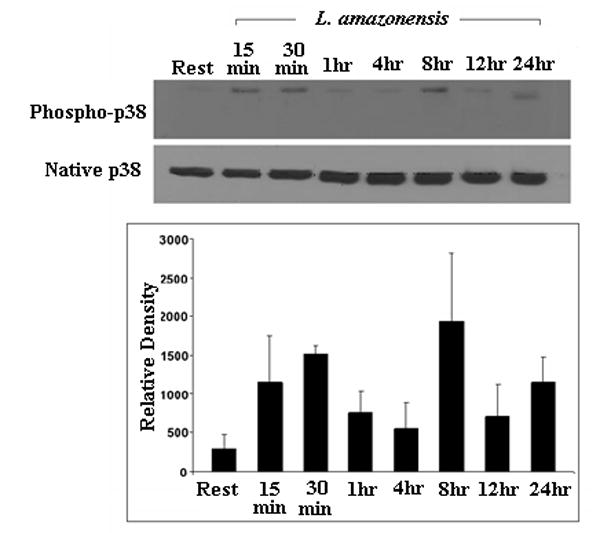
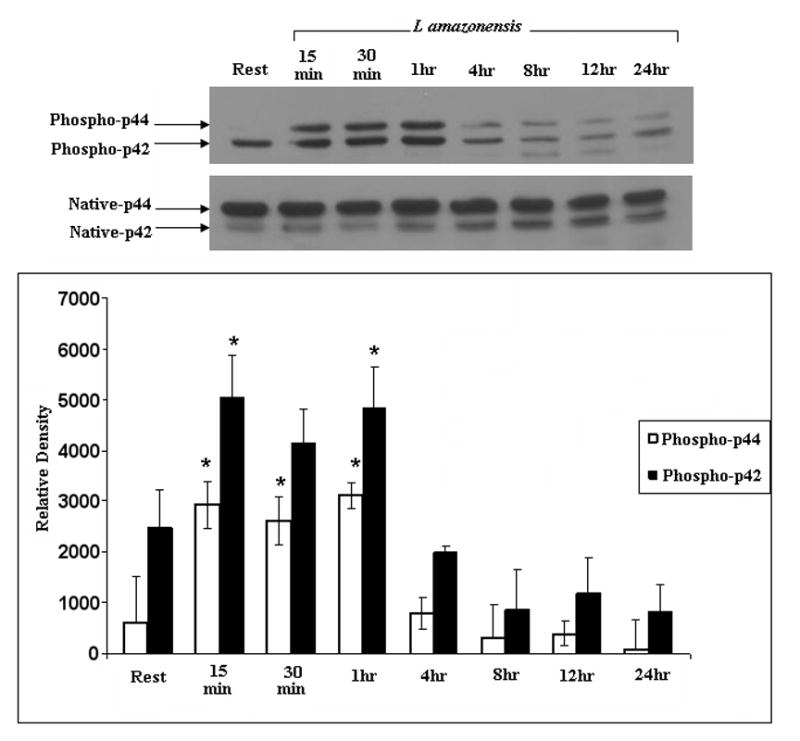
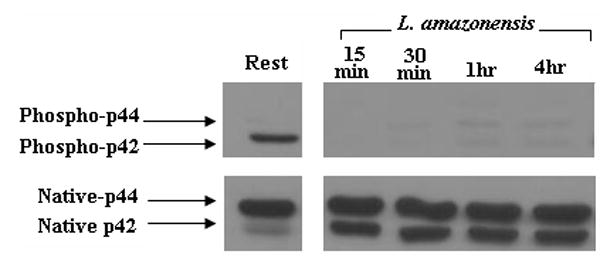
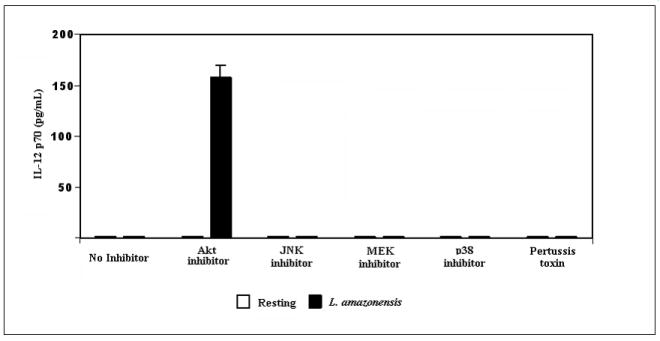
ERK and p38 MAP kinases are phosphorylated during L. amazonensis infection but inhibition of MAPK pathways or GCPRs, does not result in IL-12p70 production by infected BMDMϕs. Infections were performed as described in Materials and Methods and activation of MAP kinases was determined by western blot analysis. (A) Blots were probed for phosphorylated p38, then stripped and probed with antibodies to native p38. Densitometric scans from 3 experiments were normalized to native p38 levels and show the profile of p38 activation.(*) denotes statistically significant differences. (B) Samples were similarly analyzed for ERK activation. (C) Specificity of p42/p44 phosphorylation was determined when infections were performed in the presence of PD98059, a MEK inhibitor. This treatment completely abolishes ERK phosphorylation in response to infection. (D) BMDMϕ were infected with L. amazonensis promastigotes in the presence of the MAP kinase pathway inhibitors JNK inhibitor II (20μM), p38 MAPK inhibitor (SB203580, 20μM), MEK inhibitor (PD98059, 50μM) as well as the inhibitor of GCPRs (Pertussis toxin, 200ng/ml) and Akt inhibitor IV(10μM). Resting cells were treated with DMSO vehicle. Culture supernatants were collected after 24 hours and assayed for IL-12p70 content by ELISA as described in Materials and Methods.
We then assessed the contribution of MAPK signaling to L. amazonensis-induced suppression of IL-12 production during infection of macrophages. The effect of widely used inhibitors of these signaling intermediates was determined. For these studies we used: JNK inhibitor II, a specific inhibitor of JNK; SB203580, which inhibits p38 MAPK; and PD98059 which inhibits MEK1/2, the MAP kinase kinase that acts just upstream of ERK. Western blot analyses show that ERK (p42 and p44) are not phosphorylated in infections performed in the presence of PD98059 (Figure 4c). As Figure 4d shows, inhibition of p38 MAPK, MEK1/2 or JNK did not reverse the inability of Leishmania-infected macrophages to produce IL-12.
3.5 Inhibition of IL-12 production by Leishmania-infected macrophages is not dependent on the engagement of G protein coupled receptors (GPCRs)
We assessed whether the effect of Leishmania infection on macrophage IL-12 production was the result of interactions with GPCRs. To address this issue, infections were performed in the presence of varying amounts of pertussis toxin, a known inhibitor of GPCRs (Bokoch and Gilman, 1984). Figure 4d shows that treatment with 200ng/ml pertussis toxin had little to no effect on IL-12 production by infected cells. Although pertussis toxin treatment did not affect IL-12 production, it reproducibly led to a small enhancement in the production of IL-10 under these infection conditions (data not shown).
3.6 Interplay between MAP kinase and PI3K/Akt signaling in L. amazonensis infected cells
We assessed the effect of PI3K/Akt activation on MAP kinase signaling during L. amazonensis infection. Figure 5a and 5b show the effect of Akt inhibition on the ERK and p38 pathways induced upon infection with L. amazonensis. Inhibition of Akt completely blocked ERK phosphorylation in response to infection with L. amazonensis. Conversely, p38 phosphorylation is greatly enhanced between 4 and 8 hours after infection when Akt is blocked. This data suggests a central role for Akt in relation to the activation of other cascades induced upon infection with L. amazonensis.
Figure 5.
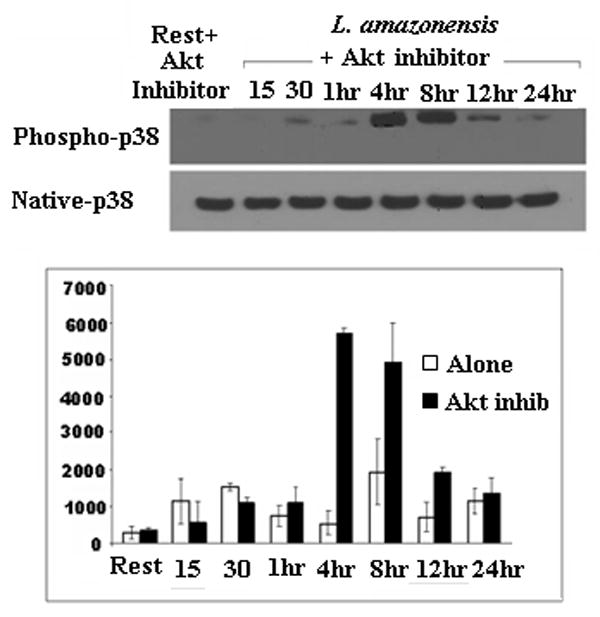
Inhibition of Akt has opposing effects on ERK and p38 phosphorylation. Resting BMDMϕ or those infected with L. amazonensis were incubated with or without Akt inhibitor IV and lysates recovered at various times. Western blot analysis for (A) ERK p44/p42 phosphorylation and (B) p38 phosphorylation are shown. Phospho-p38 levels in the presence of Akt inhibitor is compared to the phospho-p38 without inhibitor blot shown in Figure 4a., which was run in the same experiment (C) Quantitative densitometric analysis for phospho p38 levels was performed with ImageJ software.
4.0 Discussion
The decisive role of IL-12 in the development of a TH1 type immune response, which is necessary for host control of leishmaniasis is well established (Reiner and Locksley, 1995, Jones et al, 1998). There is however, recognition in some experimental models of leishmaniasis that the role of this cytokine is limited (Mattner et al, 1997, Buxbaum et al, 2002, Torrentera et al, 2002). This notwithstanding, it has been shown previously that infection with promastigotes of most Leishmania species does not result in the production of IL-12 by macrophages and some dendritic cells (Reiner et al, 1994, Carrera et al, 1996, Bennet et al, 2001). In light of recent studies that have implicated PI3K signaling as a negative regulator of IL-12 production (Fukao and Koyasu, 2003, Martin et al, 2003, Waggoner et al, 2005), we assessed whether activation of the PI3K pathway in macrophages by L. amazonensis infection contributes to the underlying mechanism that limits IL-12 production by infected macrophages. Activation of PI3K results in the production of lipid second messengers that in turn interact with several downstream kinases. One of these downstream kinases is Protein Kinase B, also known as Akt. Here, we show that L. amazonensis infection engages PI3K/Akt signaling in macrophages, which mediates the prevention of IL-12 production. Blocking PI3K signaling during infection results in IL-12 production in the absence of any additional stimulus, which would suggest that infection with these parasites engages both positive and negative IL-12 signaling pathways. Our observation that cells infected with L. amazonensis express even lower levels of IL-12 p40 than resting cells is consistent with the observations of others (Carrera et al, 1996). In those studies, reduction in IL-12 p40 levels was shown to be selective since mRNA levels of other cytokines were not similarly affected by Leishmania infection. Although this study was not designed to address the role of PI3K signaling in parasite induced suppression of IL-12 in response to LPS stimulation, a recent study by Kuroda et al (2008) showed that in the absence of PTEN, a negative regulator of PI3K signaling, even greater suppression of IL-12 production is achieved.
IL-12 suppression in Leishmania infections can be mediated by IL-10 (Kane and Mosser, 2001, Thomas and Buxbaum, 2008). However, it has been reported that IL-12 production is absent in IL-10-/- mice infected with Leishmania (Carrera et al, 1996). In addition, we observed that treatment of infected cultures with anti-IL-10 antibodies does not relieve the prevention of IL-12 production by Leishmania infection, which is consistent with observations by others (McDowell et al, 2002, Thomas and Buxbaum, 2008). This suggested that there is an IL-10 independent mechanism by which Leishmania parasites can prevent IL-12 production during infection.
Evidence that infection engages the PI3K signaling pathway was demonstrated by showing an increase in phospho-Akt levels soon after exposure of macrophages to parasites (Figure 1, and Ruhland et al, 2007). Inhibition of PI3K signaling with a PI3K inhibitor resulted in relief of IL-12 suppression in response to infection. Interestingly, higher levels of IL-12 were produced when activation of Akt, a downstream kinase in the PI3K signaling pathway was blocked (Figures 2). We interpret the finding that inhibition of Akt results in greater production of IL-12 to imply that even though parasite interactions with molecules on the macrophage surface are sufficient to initiate PI3K signaling that can block IL-12 production, sustained Akt activation by internalized parasites more effectively blocks processes that are modulated by PI3K/Akt signaling.
Akt is a serine threonine kinase in the PI3K signaling pathway that is recruited to membrane sites of phosphoinositide generation. At those membrane sites, Akt becomes anchored through its pleckstrin homology domain and undergoes phosphorylation on Ser473 and Thr308. Following phosphorylation, Akt is redistributed in the cytoplasm or nucleus where it can in turn phosphorylate other substrates (Brazil et al, 2004). Some of the well characterized substrates of Akt include: the pro-apoptotic molecule Bad that upon phosphorylation becomes unable to promote the release of cytochrome C from the mitochondria, which therefore results in resistance to apoptosis (Datta et al, 1997) and the nuclear transcription factor FoxO that controls phosphorylation of p27, a regulator of progression through the cell cycle (Medema et al, 2000). Akt has recently been shown to be activated in several tumors and as a result has become the target of several therapeutic strategies aimed at targeting these tumors (Chen et al, 2005). With the availability of drugs against Akt for in vivo use, the observations presented here suggest that therapeutic strategies that block Akt activation in infected subjects might be useful in the modulation of the immune response to Leishmania. It should be noted that a previous study of L. major infection of mice in which the p85 gene that encodes the regulatory subunit of PI3K was deleted resulted in a lower parasite burden in infected mice and a more resistant phenotype (Fukao et al, 2002).
At present, we do not know which molecules are involved in the initiation of PI3K signaling during infection. PI3K signaling can be initiated in response to the engagement of a broad range of cell surface molecules (Deane and Fruman, 2004). Although Leishmania parasites can be internalized by opsonin and non-opsonin dependent receptors, it is not known which of these receptors plays a dominant role in initiating signal transduction during infection. Recent studies have shown that some G protein coupled receptors (GPCRs) trigger PI3K signaling, through their β and γ subunits (He et al, 2000). We assessed whether the effect of the parasite on IL-12 production was mediated by interactions with GPCRs. Pertussis toxin, which is an accepted blocking agent of GPCRs had no effect on the prevention of IL-12 production in Leishmania infected cells. As we noted earlier, pertussis toxin treatment had some effect on IL-10 production in infected cells. This is consistent with the findings of others (He et al, 2000). This might be an indication that there is a differential threshold in signaling that regulates IL-10 and IL-12 secretion during a parasite infection.
Studies in other infection systems as well as in Leishmania have considered the role of MAP kinases in the generation of IL-12 (Feng et al, 1999, Kim et al, 2005, Mason et al, 2004). There isn’t a clear consensus on the role of MAPK signaling in the regulation of IL-12 production. In infections with the intracellular pathogen, Toxoplasma gondii, induction of p38 MAPK has been shown to drive IL-12 production (Kim et al, 2005, Mason et al, 2004). This is consistent with the observation that MAP kinase kinase 3 knockout mice are defective in IL-12 production (Lu et al, 1999). Mice deficient in another MAP kinase, JNK, were found to have normal capacity to produce IL-12 (Constant et al, 2000). However, it was recently shown that JNK mediates IL-12 suppression upon engagement of GPCRs (la Sala et al, 2005). It is of interest that in the latter study, it was shown that inhibition of ERK signaling had no effect on GPCR inhibition of IL-12 production.
Differential activation of MAP kinase signaling, in particular ERK and p38, has been implicated in the suppression of IL-12 production by lpg derived from L. donovani parasites (Feng et al, 1999, Piedrafita et al, 1999). A noteworthy observation in those studies and in studies by others was the demonstration that the MAP kinases have competing effects and that timing of the activation of either p38 or ERK could affect the outcome of the host cell response. We found that ERK and p38 MAP kinases are activated in macrophages soon after incubation with L. amazonensis promastigotes. However, inhibition of MAP kinase activation with specific inhibitors of these signaling molecules had no effect on IL-12 production by bone marrow derived macrophages incubated with L. amazonensis promastigotes (Figure 4). One explanation for why our studies could not establish a role for MAP kinase signaling in the prevention of IL-12 production during L. amazonensis infection might simply be that L. mexicana complex parasites as compared to L. donovani parasites elicit quantitatively and qualitatively different host cell responses (McMahon-Pratt and Alexander, 2004).
We found that inhibition of Akt can profoundly affect the amplitude of MAPKs, which were activated upon infection with L. amazonensis promastigotes. A recent report has similarly linked activation of PI3K/Akt signaling to the suppression of p38 phosphorylation, which results in the negative regulation of IL-12 (Yang et al, 2006). Taken together, the data from the current study can be summarized in a putative pathway shown in Figure 6. Whereas Akt activation promotes the downstream phosphorylation of ERK, this event has little effect on the suppression of IL-12 p40 since blocking of ERK did not yield similar results to blocking Akt upstream. The fact that inhibition of PI3K/Akt signaling results in IL-12 production implies that L. amazonensis promastigote infection induces positive signaling as well, that mediates IL-12 production. Although our studies are inconclusive on whether this positive signaling pathway is mediated by p38, we showed that inhibition of Akt led to hyper-phosphorylated levels of p38. Therefore, it can be said that infection initiates divergent pathways; activation of PI3K/Akt signaling suppresses signaling that initiates IL-12 p40 transcription.
Figure 6.
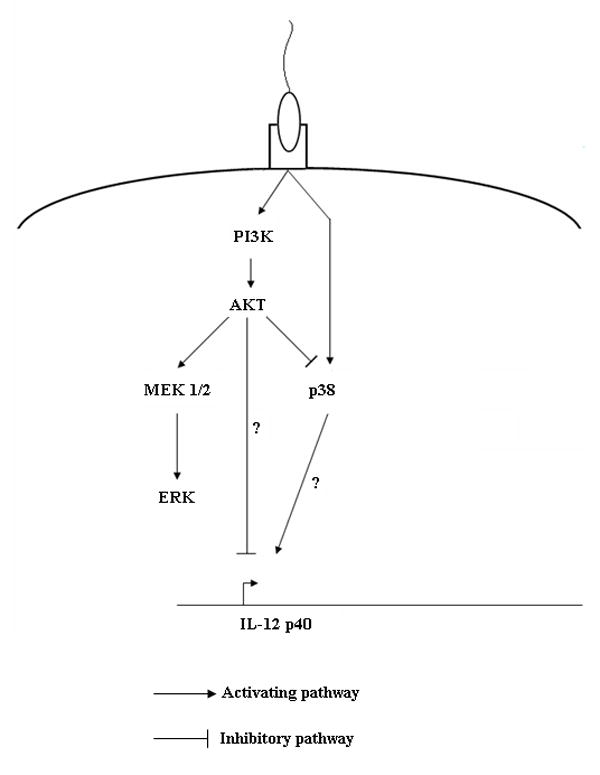
Hypothetical signal transduction scheme involving PI3K/Akt and MAP kinase signaling that controls IL-12 production in L. amazonensis-infected macrophages. Akt activation differentially modulates ERK and p38 activation; whereas inhibition of PI3K/Akt results in the induction of p38, Akt inhibition suppresses ERK activation. Given that IL-12 suppression is not relieved upon ERK inhibition, it is unlikely that ERK is directly downstream of Akt. Although p38 is negatively regulated by Akt, its induction is independent of PI3K activation.
Acknowledgments
We thank Delbert Abi Abdallah for help with some experiments. We also thank Dr. Howard Johnson for critically reading this manuscript. This work was supported by a grant from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Buxbaum LU, Uzonna JE, Goldschmidt MH, Scott P. Control of New World cutaneous leishmaniasis is interleukin-12 independent but STAT4 dependent. European Journal of Immunology. 2002;32:3206–3215. doi: 10.1002/1521-4141(200211)32:11<3206::AID-IMMU3206>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Belkaid YB, Butcher B, Sacks DL. Analysis of cytokine production by inflammatory mouse macrophages at the single-cell level: selective impairment of IL-12 induction in Leishmania-infected cells. European Journal of Immunology. 1998;28:1389–1400. doi: 10.1002/(SICI)1521-4141(199804)28:04<1389::AID-IMMU1389>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Misslitz A, Colledge L, Aebischer T, Blackburn C. Silent infection of bone marrow-derived dendritic cells by Leishmania mexicana amastigotes. European Journal of Immunology. 2001;31:876–83. doi: 10.1002/1521-4141(200103)31:3<876::aid-immu876>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Nolan T, Pfaffl MW. Quantitative real-time RT-PCR--a perspective. Journal of Molecular Endocrinology. 2005;34:597–601. doi: 10.1677/jme.1.01755. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signaling: AKTion on multiple front. Trends in Biochemical Science. 2004;29:233–242. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Braun MC, Kelsall BL. Regulation of interleukin-12 production by G-protein-coupled receptors. Microbes and Infection. 2001;3:99–107. doi: 10.1016/s1286-4579(00)01357-5. [DOI] [PubMed] [Google Scholar]
- Cameron P, McGachy A, Anderson M, Paul A, Coombs GH, Mottram JC, Alexander J, Plevin R. Inhibition of lipopolysaccharide-induced macrophage IL-12 production by Leishmania mexicana amastigotes: the role of cysteine peptidases and the NF-kappaB signaling pathway. Journal of Immunology. 2004;173:3297–3304. doi: 10.4049/jimmunol.173.5.3297. [DOI] [PubMed] [Google Scholar]
- Carrera L, Gazzinelli RT, Badolato R, Hieny S, Muller W, Kuhn R, Sacks DL. Leishmania promastigotes selectively inhibit interleukin 12 induction in bone marrow-derived macrophages from susceptible and resistant mice. Journal of Experimental Medicine. 1996;183:515–526. doi: 10.1084/jem.183.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YL, Law PY, Loh HH. Inhibition of PI3K/Akt signaling: an emerging paradigm for targeted cancer therapy. Current Medicinal Chemistry-Anticancer Agents. 2005;5:575–89. doi: 10.2174/156801105774574649. [DOI] [PubMed] [Google Scholar]
- Constant SL, Dong C, Yang DD, Wysk M, Davis RJ, Flavell RA. JNK1 is required for T cell-mediated immunity against Leishmania major infection. Journal of Immunology. 2000;165:2671–2676. doi: 10.4049/jimmunol.165.5.2671. [DOI] [PubMed] [Google Scholar]
- D’Andrea A, Aste-Amezaga M, Valiante NM, Ma X, Kubin M, Trinchieri G. Interleukin 10 (IL-10) inhibits human lymphocyte interferon γ-production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. Journal of Experimental Medicine. 1993;178:1041–1048. doi: 10.1084/jem.178.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Deane JA, Fruman DA. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annual Review of Immunology. 2004;22:563–598. doi: 10.1146/annurev.immunol.22.012703.104721. [DOI] [PubMed] [Google Scholar]
- Feng GJ, Goodridge HS, Harnett MM, Wei XQ, Nikolaev AV, Higson AP, Liew FY. Extracellular signal-related kinase (ERK) and p38 mitogen-activated protein (MAP) kinases differentially regulate the lipopolysaccharide-mediated induction of inducible nitric oxide synthase and IL-12 in macrophages: Leishmania phosphoglycans subvert macrophage IL-12 production by targeting ERK MAP kinase. Journal of Immunology. 1999;163:6403–6412. [PubMed] [Google Scholar]
- Fukao T, Tanabe M, Terauchi Y, Ota T, Matsuda S, Asano T, Kadowaki T, Takeuchi T, Koyasu S. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nature Immunology. 2002;3:875–881. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- He J, Gurunathan S, Iwasaki A, Ash-Shaheed B, Kelsall BL. Primary role for Gi protein signaling in the regulation of interleukin 12 production and the induction of T helper cell type 1 responses Journal of Experimental Medicine. 2000;191:1605–1610. doi: 10.1084/jem.191.9.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- Jones DE, Elloso MM, Scott P. Host susceptibility factors to cutaneous leishmaniasis. Frontiers in Bioscience. 1998;3:D1171–1180. doi: 10.2741/a353. [DOI] [PubMed] [Google Scholar]
- Kane MM, Mosser DM. The role of IL-10 in promoting disease progression in leishmaniasis. Journal of Immunology. 2001;166:1141–1147. doi: 10.4049/jimmunol.166.2.1141. [DOI] [PubMed] [Google Scholar]
- Kim L, Del Rio L, Butcher BA, Mogensen TH, Paludan SR, Flavell RA, Denkers EY. p38 MAPK autophosphorylation drives macrophage IL-12 production during intracellular infection. Journal of Immunology. 2005;174:4178–4184. doi: 10.4049/jimmunol.174.7.4178. [DOI] [PubMed] [Google Scholar]
- Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21:643–653. doi: 10.1016/j.immuni.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Kitani A, Fuss I, Nakamura K, Kumaki F, Usui T, Strober W. Transforming growth factor (TGF)-β1-producing regulatory T cells induce Smad-mediated interleukin 10 secretion that facilitates coordinated immunoregulatory activity and amelioration of TGF-β1-mediated fibrosis. Journal of Experimental Medicine. 2003;198:1179–1188. doi: 10.1084/jem.20030917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda S, Nishio M, Sasaki T, Horie Y, Kawahara K, Sasaki M, Natsui M, Matozaki T, Tezuka H, Ohteki T, Förster I, Mak TW, Nakano T, Suzuki A. Effective clearance of intracellular Leishmania major in vivo requires Pten in macrophages. European Journal of Immunology. 2008;38:1331–1340. doi: 10.1002/eji.200737302. [DOI] [PubMed] [Google Scholar]
- Lu HT, Yang DD, Wysk M, Gatti E, Mellman I, Davis RJ, Flavell RA. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO Journal. 1999;18:1845–1857. doi: 10.1093/emboj/18.7.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Sala A, Gadina M, Kelsall BL. G(i)-protein-dependent inhibition of IL-12 production is mediated by activation of the phosphatidylinositol 3-kinase-protein 3 kinase B/Akt pathway and JNK. Journal of Immunology. 2005;175:2994–2999. doi: 10.4049/jimmunol.175.5.2994. [DOI] [PubMed] [Google Scholar]
- Ilg T. Lipophosphoglycan is not required for infection of macrophages or mice by Leishmania mexicana. EMBO Journal. 2000;19:1953–1962. doi: 10.1093/emboj/19.9.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Trinchieri G. Regulation of interleukin-12 production in antigen-presenting cells. Advances in Immunology. 2001;79:55–92. doi: 10.1016/s0065-2776(01)79002-5. [DOI] [PubMed] [Google Scholar]
- Martin M, Schifferle RE, Cuesta N, Vogel SN, Katz J, Michalek SM. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. Journal of Immunology. 2003;171:717–725. doi: 10.4049/jimmunol.171.2.717. [DOI] [PubMed] [Google Scholar]
- Marth T, Kelsall BL. Regulation of interleukin-12 by complement receptor 3 signaling. Journal of Experimental Medicine. 1997;185:1987–1995. doi: 10.1084/jem.185.11.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason NJ, Fiore J, Kobayashi T, Masek KS, Choi Y, Hunter CA. TRAF6-dependent mitogen-activated protein kinase activation differentially regulates the production of interleukin-12 by macrophages in response to Toxoplasma gondii. Infection and Immunity. 2004;72:5662–5667. doi: 10.1128/IAI.72.10.5662-5667.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattner F, Di Padova K, Alber G. Interleukin-12 is indispensable for protective immunity against Leishmania major. Infection and Immunity. 1997;65:4378–83. doi: 10.1128/iai.65.11.4378-4383.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell MA, Marovich M, Lira R, Braun M, Sacks D. Leishmania priming of human dendritic cells for CD40 ligand-induced interleukin-12p70 secretion is strain and species dependent. Infection and Immunity. 2002;70:3994–4001. doi: 10.1128/IAI.70.8.3994-4001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon-Pratt D, Alexander J. Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunology Reviews. 2004;201:206–224. doi: 10.1111/j.0105-2896.2004.00190.x. [DOI] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NF-kappa B half-site. Molecular and Cellular Biology. 1995;15:5258–5267. doi: 10.1128/mcb.15.10.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedrafita D, Proudfoot L, Nikolaev AV, Xu D, Sands W, Feng GJ, Thomas E, Brewer J, Ferguson MA, Alexander J, Liew FY. Regulation of macrophage IL-12 synthesis by Leishmania phosphoglycans. European Journal of Immunology. 1999;29:235–244. doi: 10.1002/(SICI)1521-4141(199901)29:01<235::AID-IMMU235>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Reiner SL, Zheng S, Wang ZE, Stowring L, Locksley RM. Leishmania promastigotes evade interleukin 12 (IL-12) induction by macrophages and stimulate a broad range of cytokines from CD4+ T cells during initiation of infection. Journal of Experimental Medicine. 1994;179:447–456. doi: 10.1084/jem.179.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner SL, Locksley RM. The Regulation of immunity to Leishmania major. Annual Review of Immunology. 1995;13:151–177. doi: 10.1146/annurev.iy.13.040195.001055. [DOI] [PubMed] [Google Scholar]
- Ropert C, Gazzinelli RT. Signaling of immune system cells by glycosylphosphatidylinositol (GPI) anchor and related structures derived from parasitic protozoa. Current Opinion in Microbiology. 2000;3:395–403. doi: 10.1016/s1369-5274(00)00111-9. [DOI] [PubMed] [Google Scholar]
- Ruhland AM, Leal N, Kima PE. Leishmania promastigotes activate PI3K/Akt signaling to confer host cell resistance to apoptosis. Cellular Microbiology. 2007;9:84–96. doi: 10.1111/j.1462-5822.2006.00769.x. [DOI] [PubMed] [Google Scholar]
- Soong L, Xu JC, Grewal IS, Kima P, Sun J, Longley BJ, Jr, Ruddle NH, McMahon-Pratt D, Flavell RA. Disruption of CD40-CD40 ligand interactions results in an enhanced susceptibility to Leishmania amazonensis infection. Immunity. 1996;4:263–273. doi: 10.1016/s1074-7613(00)80434-3. [DOI] [PubMed] [Google Scholar]
- Sutterwala FS, Noel GJ, Clynes R, Mooser DM. Selective suppression of IL-12 induction after macrophage receptor ligation. Journal of Experimental Medicine. 1997;185:1977–1985. doi: 10.1084/jem.185.11.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BN, Buxbaum LU. FcgammaRIII mediates immunoglobulin G-induced interleukin-10 and is required for chronic Leishmania mexicana lesions. Infection and Immunity. 2008;76:623–631. doi: 10.1128/IAI.00316-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrentera AF, Laman JD, Van Meurs M, Adorini L, Muraille E, Carlier Y. Endogenous interleukin-12 is critical for controlling the late but not the early stage of Leishmania mexicana infection in C57BL/6 mice. Infection and Immunity. 2002;70:5075–5080. doi: 10.1128/IAI.70.9.5075-5080.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G, Scott P. Interleukin-12: basic principles and clinical applications Current Topics in Microbiology and Immunology. 1999;238:57–78. doi: 10.1007/978-3-662-09709-0_4. [DOI] [PubMed] [Google Scholar]
- Turco SJ. Adversarial relationship between the leishmania lipophosphoglycan and protein kinase C of host macrophages. Parasite Immunology. 1999;21:597–600. doi: 10.1046/j.1365-3024.1999.00266.x. [DOI] [PubMed] [Google Scholar]
- Waggoner SN, Cruise MW, Kassel R, Hahn YS. gC1q receptor ligation selectively down-regulates human IL-12 production through activation of the phosphoinositide 3-kinase pathway. Journal of Immunology. 2005;175:4706–4714. doi: 10.4049/jimmunol.175.7.4706. [DOI] [PubMed] [Google Scholar]
- Weinheber N, Wolfram M, Harbecke D, Aebischer T. Phagocytosis of Leishmania mexicana amastigotes by macrophages leads to a sustained suppression of IL-12 production. European Journal of Immunology. 1998;28:2467–77. doi: 10.1002/(SICI)1521-4141(199808)28:08<2467::AID-IMMU2467>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Wang R, Brattain MG. AKT can be activated in the nucleus. Cellular Signalling. 2006;10:1722–1731. doi: 10.1016/j.cellsig.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Yang CS, Song CH, Lee JS, Jung SB, Oh JH, Park J, Kim HJ, Park JK, Paik TH, Jo EK. Intracellular network of phosphatidylinositol 3-kinase, mammalian target of the rapamycin/70 kDa ribosomal S6 kinase 1, and mitogen-activated protein kinases pathways for regulating Mycobacteria-induced IL-23 expression in human macrophages. Cellular Microbiology. 2006;8:1158–1171. doi: 10.1111/j.1462-5822.2006.00699.x. [DOI] [PubMed] [Google Scholar]