

Abstract
Alcoholism involves compulsive behaviors of alcohol drinking, which is thought to be related at least initially to the rewarding effect of alcohol. It has been shown that mu-opioid receptors play an essential role in drug reward and dependence for many drugs of abuse including alcohol, but the function of delta-opioid receptors (DOR) in drug reward remains largely unknown at present. Previous animal studies using systemic approaches with DOR antagonists or DOR knockout animals have yielded inconsistent results, showing a decrease, an increase or no change in alcohol consumption and behaviors of alcohol reward after DOR inhibition or deletion. In the present study, we used ethanol-conditioned rats to investigate adaptive DOR function in neurons of the central nucleus of the amygdala (CeA), a key brain site for alcohol reward and addiction. We found that functional DOR was absent in glutamate synapses of CeA neurons from control rats, but it emerged and inhibited glutamate synaptic currents in CeA neurons from rats displaying ethanol-induced behavior of conditioned place preference (CPP). Analysis of paired-pulse ratios and miniature glutamate synaptic currents revealed that the recruited DOR was present on glutamatergic presynaptic terminals. Similar induction of functional DOR was also found on GABA synapses. Furthermore, microinjection of a DOR antagonist into the CeA reversed ethanol-induced CPP behavior in rats in vivo. These results suggest that repeated alcohol exposure recruits new functional DOR on CeA glutamate and GABA synapses, which may be involved in the expression or maintenance of ethanol-induced CPP behavior.
Keywords: Glutamate synaptic transmission, central amygdala, reward, addiction, rat
Alcoholism is a widespread disorder characterized by compulsive behaviors of alcohol consumption, leading to alcohol dependence and addiction (Tabakoff and Hoffman, 1996). Understanding the neural mechanisms underlying alcohol addiction is critical for the development of effective therapies for alcoholism. Previous studies have demonstrated that the central nucleus of the amygdala (CeA) is involved in stimulus-reward learning for many drugs of abuse, particularly alcohol (Koob et al., 1998, Baxter and Murray, 2002, McBride, 2002, Gottfried et al., 2003, See et al., 2003, Balleine and Killcross, 2006, Murray, 2007). Among many central targets of alcohol actions is the central glutamate synapse, which undergoes significant neuroadaptive changes in response to prolonged exposure to alcohol (Siggins et al., 2003, Hyman et al., 2006). In CeA neurons, while acute alcohol decreases glutamate synaptic activity through pre- and postsynaptic mechanisms, sustained or intermittent alcohol treatment increases presynaptic glutamate release (Roberto et al., 2004b, Roberto et al., 2006, Zhu et al., 2007). Furthermore, blockade of this increased glutamate synaptic activity abolishes the expression of ethanol-induced behavior of conditioned place preference (CPP) (Zhu et al., 2007). Thus, glutamate synaptic activity and its adaptive changes in CeA neurons appear to play an important role in a behavior related to alcohol reward. In addition, alcohol also induces adaptive changes in CeA GABA synaptic activity, contributing to the mechanisms of alcohol reinforcement (Koob, 2004, Siggins et al., 2005).
The endogenous opioid system is essential for the rewarding effects of many abused drugs including alcohol. Naltrexone, a nonselective antagonist of opioid receptors, is the most consistent medication for clinical treatment of alcohol dependence (Krystal et al., 2001, Assanangkornchai and Srisurapanont, 2007). Animal studies have shown that the CeA is the most effective brain site for opioid antagonists to inhibit alcohol self-administration (Koob et al., 1998). Opioids produce their actions primarily through three types of opioid receptors: mu, delta and kappa (Waldhoer et al., 2004). It has been well established that the mu-opioid receptor (MOR) is required for the rewarding effect and associated behavior induced by opioids and alcohol (Contet et al., 2004). However, the role of the delta-opioid receptor (DOR) in alcohol reward and dependence remains largely unknown at present and inconsistent results have been reported in previous studies (Kieffer and Gaveriaux-Ruff, 2002). While systemic blockade of DOR reduced alcohol consumption and reward (Matsuzawa et al., 1999, Ciccocioppo et al., 2002), DOR knockout increased alcohol self-administration (Roberts et al., 2001); other studies reported no effect of DOR antagonism on alcohol consumption or its discriminative effect (Stromberg et al., 1998, Mhatre et al., 2000, Ingman et al., 2003). Interestingly, recent studies including ours have identified a unique feature of DOR properties: in pain-related brainstem areas and spinal cord, DOR, predominately localized in intracellular compartments and normally not functional, is trafficked from cytoplasm to surface membrane and becomes functional after prolonged opioid exposure, resulting in enhanced opioid analgesia (Cahill et al., 2001, Guan et al., 2005, Hack et al., 2005, Ma et al., 2006). However, it is currently unknown how this adaptive DOR functions in the brain mechanisms for drug reward and dependence. Therefore, in the present study, we focused on CeA neurons to investigated DOR function in ethanol-conditioned rats with CPP behavior.
EXPERIMENTAL PROCEDURES
All procedures involving the use of animals conformed to the guidelines and approved by the University of Texas–MD Anderson Cancer Center Animal Care and Use Committee. The general experimental methods used have been reported previously (Zhu et al., 2007).
Brain slice preparations
Male Wistar rats (150–250 g) were used to make CeA slice preparations for whole-cell recording in vitro. After ethanol conditioning, a rat was anesthetized with inhalation of halothane and then euthanized by decapitation. The brain was removed and cut in a vibratome in cold (4 °C) physiological saline to obtain coronal slices (200-300 μm thick) containing the CeA. A single slice was submerged in a shallow recording chamber and perfused with preheated (35 °C) physiological saline (in mM: NaCl, 126; KCl, 2.5; NaH2PO4, 1.2; MgCl2, 1.2; CaCl2, 2.4; glucose, 11; NaHCO3, 25, saturated with 95% O2 and 5% CO2, pH 7.2–7.4). Slices were maintained at around 35 °C throughout a recording experiment.
Whole-cell recording
Visualized whole-cell voltage-clamp recordings were obtained from neurons in the visually estimated medial part of the CeA in a slice with a glass pipette (resistance 2-4 MΩ) containing (in mM): K-gluconate, 126; NaCl, 10; MgCl2, 1; EGTA, 11; HEPES, 10; ATP, 2; GTP, 0.25; pH adjusted to 7.3 with KOH; osmolarity 280–290 mOsmol/l. For recording of GABAergic inhibitory postsynaptic current (IPSC), the internal concentration of K-gluconate was reduced to 100 mM and 10 mM KCl was added to facilitate IPSC recording, resulting in an IPSC in a downward direction. An AxoPatch 1-D amplifier and AxoGraph software were used for data acquisition and on-line/off-line data analyses. A seal resistance of 2 GΩ or above and an access resistance of 15 MΩ or less were considered acceptable. Series resistance was optimally compensated. The access resistance was monitored throughout the experiment. Junction potential was not corrected.
Synaptic activity
Electrical stimuli of constant current (0.25 ms, 0.04–0.2 mA) were used to evoke synaptic currents with bipolar stimulating electrodes placed in the ventrolateral part of the CeA. Glutamate-mediated excitatory postsynaptic currents (EPSCs) were recorded in the presence of GABAA receptor antagonist bicuculline (30 μM). Under these conditions, the recorded EPSC was predominantly mediated by non-NMDA receptors (Zhu and Pan, 2004). A pair of EPSCs was evoked by two stimuli with an interval of 40 ms and a paired-pulse ratio (PPR) was calculated by dividing the second EPSC amplitude by the first one. Six PPRs were averaged to obtain a mean PPR before and during application of a drug in a given cell. The PPR has been widely used to determine the involvement of a presynaptic action site (Dobrunz and Stevens, 1997, Ungless et al., 2001, Bie et al., 2005). Miniature EPSCs were obtained in 60-s epochs in the presence of tetrodotoxin (1 μM). A sliding template of miniature EPSC, defined by predetermined parameters with the AxoGraph software, was used to detect and analyze the spontaneous events of miniature EPSCs and their distribution. All GABA-mediated inhibitory postsynaptic currents (IPSCs) were recorded in the presence of glutamate receptor antagonists AP5 (30 μM) and CNQX (10 μM). Holding potential was -70 mV for EPSCs and -60 mV for IPSCs. Recordings in slices from ethanol- or saline-conditioned rats were made in an ethanol-free solution 2–4 hours after making the slice preparation. Drugs were applied through the bath solution.
Conditioned place preference
The conditioned place preference (CPP) paradigm has been widely used to measure the conditioned rewarding effect of many drugs of abuse including alcohol (Tzschentke, 2007). A standard rat CPP apparatus of three chambers (Med Associates Inc.) was used for analysis of ethanol conditioning-induced CPP behavior in rats. The apparatus’ two test chambers had distinct environmental cues: a black chamber with stainless steel rod grid floor and a white chamber with stainless steel mesh floor. A third center chamber in neutral gray connected the two test chambers with operational doors. Automated data were collected by fifteen infrared photobeam detectors on chamber floors and were automatically sent to a computer for storage and analysis.
The conditioning protocol used in this study was adopted from previous studies of ethanol-induced CPP behavior in rats (Bozarth, 1990, Bienkowski et al., 1996, Biala and Kotlinska, 1999, Matsuzawa et al., 1999) and has been reported previously (Zhu et al., 2007). The conditioning procedure consisted of four phases for a total of 18 consecutive days. Phase 1, habituation (days 1-2): after a saline injection (i.p.), a rat was placed in the center connecting chamber and allowed to move freely between the two test chambers for 30 minutes each day. Phase 2, pre-test (day 3): after a saline injection (i.p.), the rat was placed in the center chamber with free access to both test chambers and the time the rat spent in each of the two test chambers during a 30-min test period was recorded. This pre-test determined baseline preferences, i.e., whether the rat spent significantly more time in one test chamber (preferred) than in the other (non-preferred), regarded as equipment bias. Phase 3, ethanol conditioning (days 4-17): rats were randomly assigned to saline and ethanol groups; on day 4, the rat in the ethanol group was injected with ethanol (0.5 g/kg, 10% w/v, i.p.) and was immediately confined in the non-preferred chamber for 20 minutes. On day 5, the rat was injected with saline and confined in the other test chamber for 20 min. The same procedure of ethanol and saline conditioning on alternate days was repeated on days 6 & 7 through days 16 & 17. In the saline control group, rats received saline injection on all 14 conditioning days. Phase 4, post-test (day 18): after a saline injection (i.p.), the conditioned rat was placed in the center chamber with free access to both test chambers for 30 minutes and the time the rat spent in each test chamber was automatically recorded. For acute effect of ethanol, rats were conditioned with a single injection of ethanol (0.5 g/kg, 10% w/v, i.p.). On the following day, a saline injection (i.p.) was made and a CPP test (post-test) was performed before CeA slice preparations were prepared from the rats. The blood alcohol level, tested on day 18 after the ethanol conditioning, was not significant (<5 mg/dl), as shown in our previous study (Zhu et al., 2007).
Cannula implantation and microinjection
General methods for site-specific microinjection were similar to those used in our previous study (Ma and Pan, 2006). Before conditioning treatment, a rat was initially anesthetized with nembutal sodium solution (50 mg/Kg) and then maintained lightly anesthetized in a sterotaxic apparatus with a constant intravenous infusion of methohexital (10 mg/ml at 0.8 ml/hr). A 26-gauge single guide cannula was inserted on each side of the brain, aiming at the CeA (AP: -2.3 mm from the Bregma; L: ±4.0 mm; V: -8.0 mm from dura) (Paxinos and Watson, 1986). The guide cannula was then cemented in place to the skull and capped. After the implantation surgery, the rat was allowed to recover for one week before undergoing the conditioning procedure. After ethanol conditioning and demonstration of the ethanol-induced CPP behavior in a post-test on day 18, CeA microinjection was performed on day 19, 10-20 min before a CPP test of post-CeA injection. Drugs were delivered into the CeA through a 33-gauge single injector cannula with an infusion pump at a rate of 0.1 μl/min. A total of 0.5 μl was injected on each side and all cannula placements for bilateral CeA were histologically verified afterwards. The naltrindole dose (0.45 μg in 0.5 μl × 2, or 2 nmol total) was selected according to previously reported doses, ranging from 2-10 nmol, which selectively blocked DOR function in vivo (Schmidt et al., 2002, Cornish et al., 2005, Ward and Roberts, 2007).
Statistic analysis
General numerical data were statistically tested with a paired or unpaired Student’s t-test and presented as mean ± SEM. For some data of non-normal distribution, the non-parametric Wilcoxon Signed Rank test was used. Data of miniature EPSCs were tested by StatView software with the Kolmogorov–Smirnov test. CPP data were presented as actual time in seconds a rat spent in the ethanol-paired chamber of the 3-chamber apparatus during a 30-min test period (pre-test, post-test and post-microinjection test). CPP behavior was determined by comparing times spent in the ethanol-paired chamber between pre-test and post-test in the same rat. Paired t-tests were used to statistically analyze CPP data. p<0.05 was considered statistically significant. All drugs were purchased from Tocris Bioscience or from Sigma-Aldrich.
RESULTS
Ethanol conditioning induces DOR-mediated inhibition of glutamate transmission
Rats were randomly assigned to two groups and conditioned with either ethanol (ethanol group) or saline (control group). Consistent with our previous report (Zhu et al., 2007), ethanol conditioning induced the behavior of conditioned place preference (CPP) in the ethanol group (n=9) with significantly increased time spent in the ethanol-paired chamber after ethanol conditioning (pre-test, 295 ± 35 s, post test, 495 ± 56 s, n=9, p<0.05, Fig. 1). No CPP behavior was observed in the saline-conditioned control group (pre-test, 295 ± 26 s, post test, 314 ± 29 s, n=8, p>0.05).
Fig. 1.
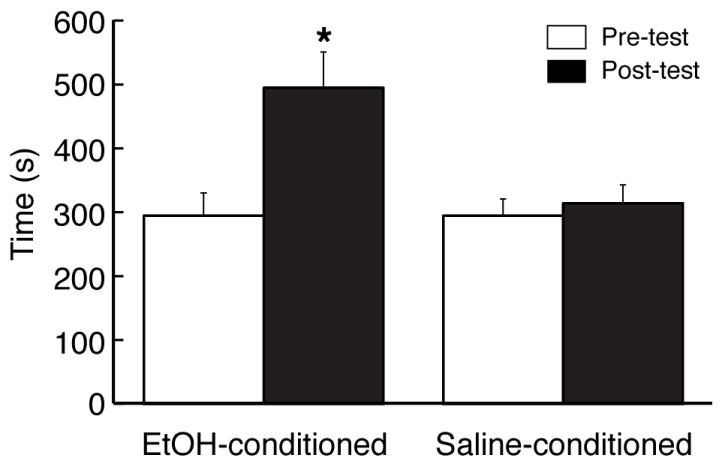
Ethanol conditioning induces behavior of conditioned place preference (CPP). Data are expressed as actual time in seconds a rat spent in the ethanol (EtOH)-paired chamber before (pre-test) and after (post-test) conditioning treatment with EtOH or saline. CPP was determined by comparing times between pre-test and post-test in the same animal. * p<0.05.
We then made slice preparations containing the central nucleus of the amygdala (CeA) from these rats of both ethanol and control groups, and excitatory post-synaptic currents (EPSCs) were evoked in CeA neurons under whole-cell voltage-clamp recording in these CeA slices in the presence of GABAA receptor antagonist bicuculline (30 μM). These glutamatergic EPSCs were predominantly mediated by non-NMDA receptors as our previous study showed that under these recording conditions, the EPSC was nearly completely blocked by non-NMDA receptor antagonist CNQX (10 μM) with no detectable glycine or GABAB receptor-mediated synaptic currents in CeA neurons (Zhu and Pan, 2004). In CeA slices from saline-conditioned control rats, bath application of the selective DOR agonist [D-Pen2,D-Pen5]-enkephalin (DPDPE) (1 μM) failed to affect the amplitude of evoked EPSCs in all neurons tested (control, 225.1 ± 12.2 pA, DPDPE, 224.3 ± 21.6 pA, n=16 from 8 rats, p>0.05, Fig. 2A, C). This observation is consistent with our previous report (Zhu and Pan, 2005) indicating the absence of functional DOR on glutamatergic synapses in CeA neurons from normal rats.
Fig. 2.

Functional delta-opioid receptors (DOR) emerge on CeA glutamate synapses in rats with ethanol-induced CPP. (A) Representative glutamate-mediated excitatory post-synaptic currents (EPSCs) in the absence (control) and presence of the DOR agonist [D-Pen2,D-Pen5]-enkephalin (DPDPE, 1 μM) in a neuron of the central nucleus of the amygdala (CeA) from a saline-conditioned control rat. (B) Representative glutamate EPSCs in control, in DPDPE and in DPDPE plus the DOR antagonist naltrindole (1 μM) in a CeA neuron from an ethanol-conditioned rats displaying CPP behavior. (C) Summarized data of the DPDPE effects on EPSCs in CeA neurons of the two rat groups. (D) Summarized data of naltrindole blockade of the DPDPE effect. (E) A dose-response curve for DOR inhibition of EPSCs in CeA neurons (n=5-10 for each dose) from ethanol-conditioned rats. The estimated EC50 was 57.9 nM. ** p<0.01.
In slices from ethanol-conditioned rats displaying the CPP behavior, the averaged EPSC amplitude is not significantly different from that of control rats (248.9 ± 19.2 pA vs. 225.1 ± 12.2 pA, p>0.05), largely due to significant variations in EPSC amplitude from cell to cell in this between-group comparison. However, in contrast to the lack of DPDPE effect in control neurons, application of DPDPE at the same dose (1 μM) significantly attenuated the amplitude of EPSCs by an average of 29.0 ± 2.2% in 18 of 25 (72%) CeA neurons surveyed in slices from ethanol-conditioned rats (control, 248.9 ± 19.2 pA, DPDPE, 174.4 ± 13.9 pA, n=18 from 9 rats, p<0.01, Fig. 2B, C). This DPDPE-mediated inhibition was completely abolished by addition of the selective DOR antagonist naltrindole (1 μM) (control, 234.3 ± 40.4 pA, DPDPE, 171.2 ± 34.8 pA, p<0.01, DPDPE + naltrindole, 234.5 ± 44.1 pA, n=5 cells, p>0.05 when compared with control, Fig. 2D), suggesting that the EPSC inhibition was mediated by DOR. Furthermore, the DPDPE inhibition of EPSCs was dose-dependent, with a near maximum inhibition of 29% of control and an estimated EC50 of 57.9 nM (Fig. 2E). Together, these results indicate that the conditioning treatment with ethanol, which induces the CPP behavior, recruits new functional DOR on glutamate synapses, and activation of this newly emerged DOR inhibits glutamate synaptic transmission in CeA neurons.
DOR decreases presynaptic release of glutamate
To determine the synaptic site of this DOR action, we first used the paradigm of paired-pulse ratio (PPR), which is relatively much less variable among individual cells and has an inverse relationship with the probability of presynaptic transmitter release so that an increased PPR indicates a reduced transmitter release, and vice versa (Dobrunz and Stevens, 1997, Ungless et al., 2001, Zhu et al., 2007). In CeA slices from saline-conditioned control rats, the averaged PPR was 1.62 ± 0.06 (n=23 cells), and the DOR agonist DPDPE (1 μM) did not significantly alter the PPR in these control neurons (control, 1.61 ± 0.15, DPDPE, 1.57 ± 0.09, n=6 cells/4 rats, p>0.05, Fig. 3A). In slices from ethanol-conditioned rats, however, the averaged PPR by comparison was significantly smaller than that from control rats (1.38 ± 0.08, n=10 vs. 1.62 ± 0.06, n=23, p<0.05), indicating that the ethanol conditioning treatment significantly increased synaptic release of glutamate in CeA neurons, an effect originally reported in our previous study (Zhu et al., 2007). Moreover, DPDPE (1 μM) significantly increased the PPR of EPSCs in CeA neurons from ethanol-conditioned rats (control, 1.38 ± 0.08, DPDPE, 1.90 ± 0.16, n=10 cells/6 rats, p<0.01 with the non-parametric Wilcoxon Signed Rank test, Fig. 3B). The increased PPR indicates that the DPDPE-mediated inhibition of EPSCs involves a presynaptic action site with a decreased probability of synaptic release of glutamate.
Fig. 3.
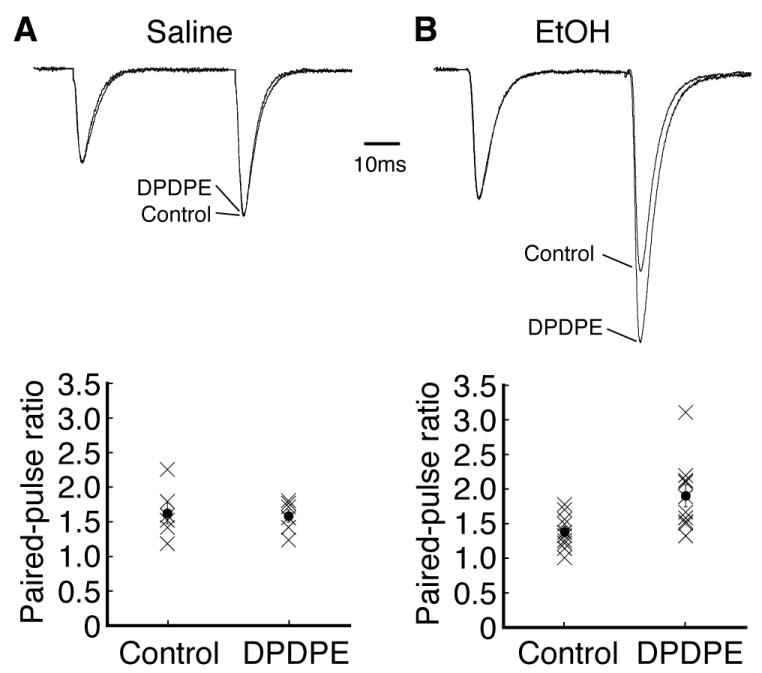
DOR activation increases the paired-pulse ratio (PPR) of EPSCs. (A, B) Representative pairs of EPSCs (upper) and PPR distributions (lower) before (control) and after addition of DPDPE (1 μM) in CeA neurons from control rats (A) and from ethanol-conditioned rats (B). The EPSC pairs were scaled to the first EPSC amplitude to show a changed ratio. Filled circles and associated vertical bars are means ± standard errors superimposed on the PPR of each cells (cross).
This notion was further confirmed by the following experiments examining the effect of DPDPE on spontaneous miniature EPSCs in CeA neurons in the presence of tetrodotoxin (1 μM). We found that, whereas DPDPE (1 μM) was ineffective on either the frequency or the amplitude of miniature EPSCs in CeA slices from control rats (frequency: control, 8.71 ± 0.48 Hz, DPDPE, 8.93 ± 0.84 Hz, p>0.05; amplitude: control, 18.33 ± 1.33 pA, DPDPE, 18.86 ± 1.62 pA, p>0.05, n=5 cell/4 rats, Fig. 4A-C), it significantly reduced the frequency of miniature EPSCs without altering the amplitude in neurons from ethanol-conditioned rats (frequency: control, 10.12 ± 1.20 Hz, DPDPE, 7.14 ± 0.84 Hz, p<0.05; amplitude: control, 16.12 ± 1.12 pA, DPDPE, 14.34 ± 1.02 pA, p>0.05, n=5 cells/5 rats, Fig. 4D-F). Taken together, these findings suggest that conditioning with alcohol induces functional DOR on presynaptic glutamatergic terminals and its activation inhibits presynaptic release of glutamate in CeA neurons.
Fig. 4.

DOR activation inhibits presynaptic release of glutamate. (A-C) Current traces (A) and cumulative distributions of miniature EPSC frequency (B) and amplitude (C) in control and in DPDPE in a representative CeA neuron from a saline-conditioned rat. (D-F) Corresponding data in a CeA neuron from an ethanol-conditioned rat.
Blockade of CeA DOR attenuates ethanol-induced CPP behavior
Next we determined the potential role of this newly emerged DOR in the ethanol-induced CPP behavior in behavioral studies with site-specific microinjections in vivo. Rats were implanted with bilateral cannula aiming at the CeA on both sides of the brain and allowed to recover from the surgery before ethanol conditioning for CPP induction. In the first experiment, we examined whether CeA opioid receptors were generally involved in the ethanol-induced CPP behavior. In a group of rats with implanted CeA cannula, ethanol conditioning induced a similar CPP behavior with increased time spent in the ethanol-paired chamber (pre-test, 232 ± 38 s, post-test, 491 ± 39 s, n=5 rats, p<0.05, Fig. 5A). One day after the post-test, a CPP test was performed again in the same rat following bilateral microinjection of saline into the CeA. The CPP behavior in those rats largely remained without significant decline (481 ± 14 s, n=5, p>0.05 when compared with the post-test), consistent with previous reports of long-lasting CPP behavior (Tzschentke, 2007). By contrast, in a separate group of ethanol-conditioned rats with CPP (n=8), bilateral CeA microinjection of the non-selective opioid receptor antagonist naltrexone (0.38 μg in 0.5 μl each side) completely abolished ethanol-induced CPP behavior to a level below the pre-conditioning one (pre-test, 208 ± 32 s, post-test, 360 ± 55 s, n=8 rats, t=4.83, p<0.05; CeA naltrexone, 140 ± 23 s, n=8 rats, t=4.94, p<0.01 when compared with the post-test and t= 2.50, p<0.05 when compared with the pre-test, Fig. 5A). The same CeA microinjection of naltrexone did not significantly affect the baseline preference behavior in saline-conditioned rats (pre-test, 171 ± 29 s, post-test, 204 ± 25 s, n = 4 rats, t=1.22, p>0.05; CeA naltrexone, 166 ± 25 s, n = 4 rats, t=1.20, p>0.05 when compared with the post-test, Fig. 5A). These results indicate that activation of opioid receptors in the CeA is required for the expression or maintenance of ethanol-induced CPP behavior.
Fig. 5.

Antagonism of CeA opioid receptors and DOR inhibits ethanol-induced CPP behavior. CPP behavior was first elicited by conditioning with ethanol and comparing pre-test and post-test. One day after the post-test, saline or opioid antagonist naltrexone (A) or DOR antagonist naltrindole (B) was bilaterally microinjected into the CeA, followed by a CPP test (post-CeA injection) in both ethanol-conditioned rats and saline-conditioned rats. * p<0.05. ** p<0.01. Sal, saline, Nltx, naltrexone, Nltd, naltrindole. (C) A photomicrograph of the CeA slice showing a dye-marked microinjection site (arrow) in the CeA. The scale bar is 1 mm.
We then examined whether the newly recruited DOR had a contributing role in the ethanol-induced CPP behavior. In an additional group of rats, ethanol conditioning similarly induced CPP behavior that persisted one day after the post-test following CeA microinjection of saline (pre-test, 182 ± 34 s, post-test, 343 ± 35 s, n=4 rats, t=4.73, p<0.05; CeA saline, 314 ± 33 s, n=4 rats, t=0.53, p>0.05 when compared with the post-test, Fig. 5B). In contrast, in another group of rats similarly conditioned with ethanol, bilateral microinjections of the DOR antagonist naltrindole (0.45 μg in 0.5 μl each side) into the CeA largely inhibited the CPP behavior (pre-test, 169 ± 10 s, post-test, 342 ± 20 s, n=5 rats, t=10.26, p<0.01; CeA naltrindole, 199 ± 39 s, n=5 rats, t=5.81, p<0.01, Fig. 5B). In saline-conditioned rats, as controls, the same CeA microinjection of naltrindole at the same dose had no significant effect on the baseline preference behavior (pre-test, 172 ± 20 s, post-test, 160 ± 10 s, n=4 rats, t=0.41, p>0.05; CeA naltrindole, 160 ± 35 s, n=4 rats, t=0.001, p>0.05, Fig. 5B). Figure 5C shows the microinjection site in the CeA. This lack of naltrindole effect argues that acute blockade of CeA DOR, if present under these control conditions, lacks an apparent effect on baseline preference behavior.
Acute ethanol fails to induce functional DOR
Next, we examined whether the functional DOR would be similarly induced by application of acute ethanol that should not induce the CPP behavior of alcohol reward. The effect of DOR agonist was tested on evoked EPSCs in CeA neurons treated with acute ethanol either in vitro or in vivo. For in vitro ethanol treatment, CeA slices taken from control rats were incubated in ethanol (20 mM) for 1 to 4 hours before recording. In these acute ethanol-treated slices, the DOR agonist DPDPE (1 μM) induced no apparent effect on the EPSC amplitude (control, 187.3 ± 21.2 pA, DPDPE, 184.4 ± 24.9 pA, n=6 cells/4 rats, p>0.05). For in vivo treatment of acute ethanol, rats (n=6) were conditioned with a single dose of ethanol and on the following day, a CPP test was performed after a saline injection and then CeA slice preparations were obtained. These rats conditioned with a single dose of ethanol did not display CPP behavior, but showed a tendency of conditioned place aversion (pre-test, 241 ± 32 s, post-test, 149 ± 18 s, n=6, p=0.057, Fig. 6A). In CeA slices from those single ethanol-treated rats, DPDPE (1 μM) failed to produce any inhibition on the EPSC amplitude (control, 217.9 ± 18.9 pA, DPDPE, 213.4 ± 19.3 pA, n=6, p>0.05, Fig. 6B). Thus, it appears that repeated ethanol exposure, but not acute ethanol, is essential for the induction of functional DOR in glutamate synapses of CeA neurons.
Fig. 6.
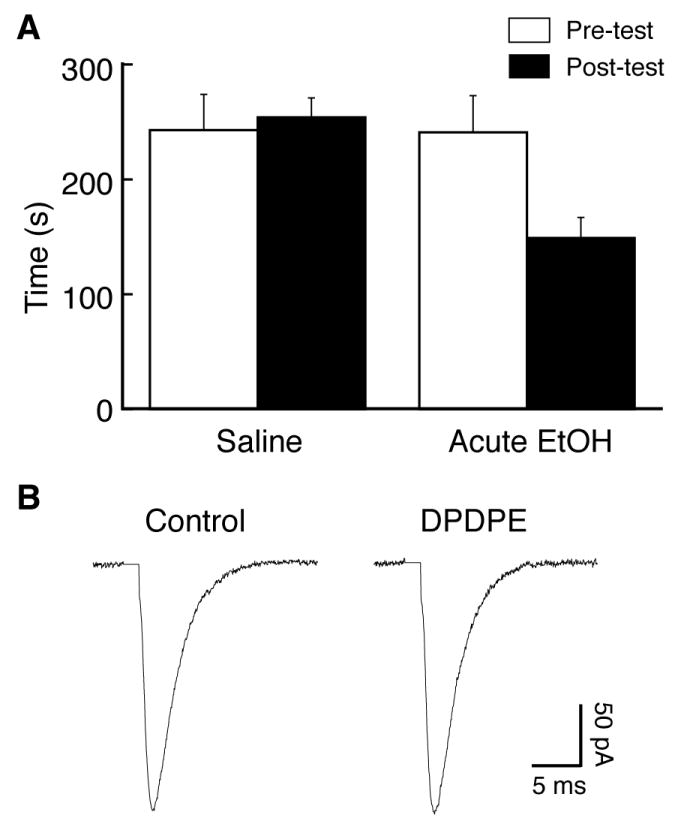
Acute ethanol treatment fails to induce CPP or functional DOR. (A) Data of CPP tests from rats conditioned with saline or with a single dose of ethanol. (B) Representative EPSCs in control and in DPDPE in a CeA neuron from a rat conditioned with a single dose of ethanol.
Ethanol conditioning also induces functional DOR on GABA synapses
We have shown previously that blocking CeA glutamate synaptic transmission inhibits ethanol-induced CPP behavior (Zhu et al., 2007). However, our current results showed that reversing DOR inhibition of CeA glutamate EPSCs with DOR antagonist, which would promote glutamate synaptic activity, also attenuated ethanol-induced CPP behavior. Although the reasons for this discrepancy between the synaptic DOR action and CPP behavior in vivo are presently unknown, one of the possibilities is that the DOR antagonist administered in vivo blocked DOR actions on multiple sites in the CeA, particularly on GABA synapses whose involvement in alcohol reinforcement and addiction has been well documented (Koob, 2004). Therefore, we performed additional experiments examining potential ethanol-induced DOR function on CeA GABA synapses. In slices from saline-conditioned control rats (n=10), DPDPE (1 μM) was ineffective on the amplitude of GABAergic inhibitory postsynaptic current (IPSC) in CeA neurons (control, 210.4 ± 24.4 pA, DPDPE, 204.3 ± 23.7 pA, n=16, p>0.05, Fig. 7A, C). By contrast, in slices from ethanol-conditioned rats (n=12), DPDPE (1 μM) produced a significant inhibition of IPSC in a total of 19 cells tested (control, 253.1 ± 25.1 pA, DPDPE, 185.6 ± 21.7 pA, n=19, p<0.01 with the paired t-test or non-parametric Wilcoxon Signed Rank test, Fig. 7B, C). This inhibition was completely reversed by addition of the selective DOR antagonist naltrindole (1 μM, n=6, Fig. 7B, D), indicating a specific DOR action on GABA IPSCs.
Fig. 7.

Functional DOR emerges on CeA GABA synapses in rats with ethanol-induced CPP. (A, B) Representative GABAergic inhibitory post-synaptic currents (IPSCs) in control and in DPDPE (1 μM) without or with naltrindole in CeA neurons from a saline-conditioned control rat (A) and from an ethanol-conditioned rat (B). (C, D) Summarized data of DPDPE effects on IPSCs and of naltrindole blockade of the DPDPE inhibition in CeA neurons from the two rat groups. ** p<0.01.
DISCUSSION
The present study has demonstrated that conditioning with intermittent ethanol that induces CPP behavior recruits new functional DOR on glutamate and GABA synapses in the CeA, an important brain structure for alcohol addiction. The ethanol-recruited DOR appears on presynaptic glutamatergic terminals and its activation inhibits presynaptic glutamate release and synaptic activity. Furthermore, pharmacological blockade of DOR function in the CeA, having no apparent effect on baseline preference behavior, reverses the ethanol-induced CPP behavior. These findings provide original evidence that intermittent alcohol exposure that induces drug-reward-related CPP behavior is able to bring out new functional DOR into play in CeA modulation of preference behavioral induced by alcohol conditioning. This may represent an important adaptive function of DOR that might be involved in behavioral effects of repeated alcohol such as alcohol reinforcement and addiction.
Functions of opioid receptors in alcohol reward
Extensive evidence shows that the endogenous opioid system plays a key role in alcohol reward and addiction. For example, systemic application of opioid antagonist naltrexone or naloxone significantly reduces the rewarding effect of alcohol and alcohol consumption both in animals and in humans (Krystal et al., 2001). Although the specific type of opioid receptors mediating the alcohol reward and alcohol consumption was not identified in early studies, mice lacking MOR displayed significantly decreased rewarding effect of alcohol and reduced alcohol consumption, suggesting an essential role of MOR in alcohol reward and addiction as well as in addiction to other drugs of abuse (Hall et al., 2001, Contet et al., 2004). Further extending those previous studies of the systemic opioid system, our results on the reversal of ethanol-induced CPP behavior by CeA-applied naltrexone suggest that the opioid receptors in the CeA are important for the maintenance of an acquired CPP behavior potentially related to alcohol reward.
In contrast, the function of DOR in addiction to alcohol, and to other drugs of abuse in general, is largely unknown at present. Previous studies using a systemic approach targeting DOR in the whole CNS have reported inconsistent findings. While some studies showed that systemically blocking DOR decreased alcohol reward or alcohol consumption (Matsuzawa et al., 1999, Ciccocioppo et al., 2002), DOR knockout animals exhibited instead increased alcohol self-administration (Roberts et al., 2001). Yet other studies reported that systemic DOR antagonism had no effect on alcohol consumption or alcohol-mediated discriminative effect (Stromberg et al., 1998, Mhatre et al., 2000, Ingman et al., 2003). Although the reasons for the discrepancy are unclear, contributing factors may include differential alcohol modulation of DOR functions in different brain areas as well as different animal models and training protocols used. Given the current finding of ethanol-induced recruitment of new DOR function in CeA neurons and its involvement in ethanol-induced CPP behavior, it is likely that the DOR function in alcohol reward may also depend on the ability of prolonged alcohol treatment to recruit new functional DOR in a specific brain area.
CPP as a behavioral measure of alcohol reward
Alcohol-induced CPP behavior has been mostly reported in mice, with relatively more alcohol-induced place aversion reported in rats (Cunningham et al., 2003, Tzschentke, 2007). The reasons for the species difference in rodents are currently unknown. Besides routes of alcohol administration and genetic variables of animals used, it has been suggested that varying timing of ethanol administration and ethanol-pairing cues can induce both CPP and place aversion in the same animal (Cunningham et al., 2002, Green and Grahame, 2008), underscoring the effect of experimental conditions on the outcome of alcohol conditioning. In fact, there are numerous previous reports of ethanol-induced CPP in rats with either biased or unbiased procedures (Bozarth, 1990, Bienkowski et al., 1996, Biala and Kotlinska, 1999, Ciccocioppo et al., 2003, Cole et al., 2003, Kotlinska et al., 2007, Walker and Ettenberg, 2007, Peana et al., 2008). Under our experimental conditions, both ethanol and morphine, a strong rewarding opioid, induce similar CPP, supporting the involvement of a rewarding effect of alcohol in the CPP behavior in rats (Zhu et al., 2007). Since a biased CPP apparatus was used and ethanol conditioning was paired with the non-preferred chamber in this study, some anti-aversive or anti-anxiolytic effect of ethanol cannot be ruled out in the CPP behavior we observed. However, we have shown previously that the same ethanol conditioning paired with the preferred chamber induces similar CPP behavior, further arguing that the ethanol-induced CPP behavior involves a rewarding effect of ethanol (Zhu et al., 2007).
Behavioral driving forces for alcohol-induced CPP behavior
Our results showed that prolonged ethanol treatment, but not acute ethanol, concomitantly induced a new DOR function and CPP behavior that was blocked by a locally applied DOR antagonist, suggesting the potential importance of this adaptive DOR function in the CPP behavior, a commonly used behavioral measure of drug reward in rodents. Alcohol-induced CPP or alcohol-seeking behavior could be driven by both positive reinforcement of alcohol and by negative reinforcement of alcohol withdrawal (Koob and Kreek, 2007), and it remains unknown that by blocking which component the DOR antagonist inhibited the CPP behavior. Given the observations that naltrexone, which primarily blocks MOR, abolished the CPP behavior (Fig. 5) and that MOR activation also inhibits CeA glutamate synaptic transmission (Zhu and Pan, 2005), it is likely that DOR antagonism, similar to MOR antagonism, attenuates ethanol-induced CPP by blocking a DOR-mediated component of positive reinforcement (rewarding effect) of alcohol in the CeA. This notion is consistent with previous reports showing that systemically applied DOR antagonists decrease alcohol consumption and promote alcohol-induced conditioned taste aversion, suggesting a DOR contribution to the positive rewarding effect of alcohol (Froehlich et al., 1998, June et al., 1999, Matsuzawa et al., 1999, Ciccocioppo et al., 2002). In addition, since CeA neurons both in vitro and in vivo in our experimental conditions could be under a condition of alcohol withdrawal (if present), the CPP attenuation by DOR antagonist might be related to reduction of withdrawal-induced negative reinforcement. It is also possible that the DOR antagonist-produced CPP attenuation is due to its inhibition of alcohol- or alcohol withdrawal-induced anxiolytic effect. Further studies are needed to determine the specific psychophysiological mechanisms.
Synaptic mechanisms for alcohol-induced CPP behavior
The receptor mechanisms for alcohol-induced DOR recruitment in central synapses are unknown. Similar DOR recruitment induced by prolonged opioids, resulting from exocytotic membrane trafficking of intracellular DOR, has been reported on postsynaptic membrane and presynaptic GABA terminals in brainstem and spinal cord neurons related to pain modulation, leading to enhanced DOR-mediated analgesia (Hack et al., 2005, Ma et al., 2006, Zhang et al., 2006, Cahill et al., 2007). A recent study shows that prolonged morphine treatment also induces DOR trafficking in basal ganglia (Lucido et al., 2005). Although alcohol may increase DOR gene expression as shown in cultured cell lines (Charness et al., 1993), it is possible that similar receptor trafficking mechanisms are involved in alcohol recruitment of functional DOR on glutamate and GABA synapses in CeA neurons observed in this study. Detailed molecular determinants and signaling pathways regulating this DOR membrane trafficking in central neurons have yet to be demonstrated.
The synaptic mechanisms underlying the roles of recruited CeA DOR as well as normally existing MOR in ethanol-induced CPP behavior remain an interesting topic of current research. Glutamate synaptic transmission and its adaptive changes induced by prolonged exposure to alcohol have been implicated in the mechanisms for reward- and addiction-related behaviors induced by many abused drugs including alcohol (Siggins et al., 2003, Hyman et al., 2006). Chronic alcohol increases synaptic release of glutamate in alcohol-dependent animals and augments NMDA receptor function through subunit-specific upregulation of surface proteins (Roberto et al., 2006, Qiang et al., 2007); and general blockade by glutamate receptor antagonists of ethanol-induced increase in glutamate synaptic activity in CeA neurons reverses ethanol-induced CPP (Zhu et al., 2007). These studies indicate that the enhanced glutamate synaptic activity in the CeA may underlie part of the mechanisms for the acquirement of alcohol-induced rewarding effect and/or for the maintenance of alcohol-induced CPP behavior in an alcohol-dependent or alcohol-withdrawn state. Nevertheless, both DOR (current finding) and MOR (Zhu and Pan, 2005) agonists inhibit glutamate synaptic transmission in the CeA, and antagonism of CeA DOR or CeA MOR by naltrexone, which would promote glutamate synaptic activity, attenuates alcohol-induced CPP. Apparently, DOR actions on glutamate synapses alone cannot account for the behavioral effect of its blockade on CPP behavior and other potential actions of DOR as well as MOR in the CeA and related mechanisms must also be considered.
While many such mechanisms may exist for the integrated behavioral outcome, another important target of alcohol and opioids is the CeA GABA synapse, which also has been implicated in alcohol-induced CPP behavior and alcohol addiction in general (Koob, 2004, Siggins et al., 2005). Acute ethanol augments GABA synaptic transmission through CRF1 receptors and chronic ethanol increases GABA synaptic release in the CeA (Nie et al., 2004, Roberto et al., 2004a). Acute blockade of systemic GABAA receptor function reduces operant alcohol self-administration in rats (Koob, 2004). Our current findings showed that an adaptive DOR function also occurred on CeA GABA synapses, indicating its contribution to the ethanol-induced CPP behavior and its blockade by DOR antagonism. Thus, the current study provides important evidence for the adaptive DOR modulation of CeA glutamate and GABA synaptic activity and its involvement in the behavior of alcohol reward. However, it remains to be investigated what role this synaptic DOR function play in the mechanisms that drive the CPP behavior largely because the contribution of increased glutamate and GABA synaptic activity in the CeA induced by alcohol to the driving mechanisms for CPP behavior is still unknown. Anatomically, DOR staining has been shown in CeA cells of naïve animals, probably on axon terminals (Mansour et al., 1988, Wilson et al., 2002). We did not observe a significant effect of DOR agonist on GABA IPSCs in control CeA neurons, which is at odds with a previous report indicating that some functional DOR is present on GABA synapses in CeA neurons of wild-type mice (Kang-Park et al., 2007). This may reflect an important difference between species besides other potential differences in experimental conditions. Nonetheless, our observation that antagonism of CeA DOR had no effect on basal preference behavior in control animals indicates the lack of a behavioral effect of DOR, if present on normal CeA GABA or glutamate synapses, on baseline preference behavior. Ultimately, because MOR is responsible for the main positive reinforcement of compulsive alcohol consumption in alcohol addiction and its abrupt blockade or inactivation inevitably induces aversive symptoms of alcohol withdrawal, pharmacological antagonism of DOR might be a useful therapeutic strategy for the treatment of alcohol addiction.
Acknowledgments
This work was supported by the National Institute on Drug Abuse grants DA014524 and DA023069, and by an Institution Research Grant of UT-MD Anderson Cancer Center.
ABBREVIATIONS
- CeA
central nucleus of the amygdala
- CPP
conditioned place preference
- DOR
delta-opioid receptor
- DPDPE
[D-Pen2,D-Pen5]-enkephalin
- EPSC
excitatory post-synaptic current
- IPSC
inhibitory postsynaptic current
- MOR
mu-opioid receptor
- PPR
paired-pulse ratio
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Assanangkornchai S, Srisurapanont M. The treatment of alcohol dependence. Curr Opin Psychiatry. 2007;20:222–227. doi: 10.1097/YCO.0b013e3280fa837d. [DOI] [PubMed] [Google Scholar]
- Balleine BW, Killcross S. Parallel incentive processing: an integrated view of amygdala function. Trends Neurosci. 2006;29:272–279. doi: 10.1016/j.tins.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Baxter MG, Murray EA. The amygdala and reward. Nat Rev Neurosci. 2002;3:563–573. doi: 10.1038/nrn875. [DOI] [PubMed] [Google Scholar]
- Biala G, Kotlinska J. Blockade of the acquisition of ethanol-induced conditioned place preference by N-methyl-D-aspartate receptor antagonists. Alcohol Alcohol. 1999;34:175–182. doi: 10.1093/alcalc/34.2.175. [DOI] [PubMed] [Google Scholar]
- Bie B, Peng Y, Zhang Y, Pan ZZ. cAMP-mediated mechanisms for pain sensitization during opioid withdrawal. J Neurosci. 2005;25:3824–3832. doi: 10.1523/JNEUROSCI.5010-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienkowski P, Kuca P, Piasecki J, Kostowski W. Low dose of ethanol induces conditioned place preference in rats after repeated exposures to ethanol or saline injections. Alcohol Alcohol. 1996;31:547–553. doi: 10.1093/oxfordjournals.alcalc.a008190. [DOI] [PubMed] [Google Scholar]
- Bozarth MA. Evidence for the rewarding effects of ethanol using the conditioned place preference method. Pharmacol Biochem Behav. 1990;35:485–487. doi: 10.1016/0091-3057(90)90191-j. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Holdridge SV, Morinville A. Trafficking of delta-opioid receptors and other G-protein-coupled receptors: implications for pain and analgesia. Trends Pharmacol Sci. 2007;28:23–31. doi: 10.1016/j.tips.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Lee MC, Vincent JP, Collier B, Beaudet A. Prolonged morphine treatment targets delta opioid receptors to neuronal plasma membranes and enhances delta-mediated antinociception. J Neurosci. 2001;21:7598–7607. doi: 10.1523/JNEUROSCI.21-19-07598.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charness ME, Hu G, Edwards RH, Querimit LA. Ethanol increases delta-opioid receptor gene expression in neuronal cell lines. Mol Pharmacol. 1993;44:1119–1127. [PubMed] [Google Scholar]
- Ciccocioppo R, Economidou D, Fedeli A, Massi M. The nociceptin/orphanin FQ/NOP receptor system as a target for treatment of alcohol abuse: a review of recent work in alcohol-preferring rats. Physiol Behav. 2003;79:121–128. doi: 10.1016/s0031-9384(03)00112-4. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Martin-Fardon R, Weiss F. Effect of selective blockade of mu(1) or delta opioid receptors on reinstatement of alcohol-seeking behavior by drug-associated stimuli in rats. Neuropsychopharmacology. 2002;27:391–399. doi: 10.1016/S0893-133X(02)00302-0. [DOI] [PubMed] [Google Scholar]
- Cole JC, Sumnall HR, O’Shea E, Marsden CA. Effects of MDMA exposure on the conditioned place preference produced by other drugs of abuse. Psychopharmacology (Berl) 2003;166:383–390. doi: 10.1007/s00213-002-1374-x. [DOI] [PubMed] [Google Scholar]
- Contet C, Kieffer BL, Befort K. Mu opioid receptor: a gateway to drug addiction. Curr Opin Neurobiol. 2004;14:370–378. doi: 10.1016/j.conb.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Cornish JL, Lontos JM, Clemens KJ, McGregor IS. Cocaine and heroin (’speedball’) self-administration: the involvement of nucleus accumbens dopamine and mu-opiate, but not delta-opiate receptors. Psychopharmacology (Berl) 2005;180:21–32. doi: 10.1007/s00213-004-2135-9. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Clemans JM, Fidler TL. Injection timing determines whether intragastric ethanol produces conditioned place preference or aversion in mice. Pharmacol Biochem Behav. 2002;72:659–668. doi: 10.1016/s0091-3057(02)00734-7. [DOI] [PubMed] [Google Scholar]
- Cunningham CL, Ferree NK, Howard MA. Apparatus bias and place conditioning with ethanol in mice. Psychopharmacology (Berl) 2003;170:409–422. doi: 10.1007/s00213-003-1559-y. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Froehlich JC, Badia-Elder NE, Zink RW, McCullough DE, Portoghese PS. Contribution of the opioid system to alcohol aversion and alcohol drinking behavior. J Pharmacol Exp Ther. 1998;287:284–292. [PubMed] [Google Scholar]
- Gottfried JA, O’Doherty J, Dolan RJ. Encoding predictive reward value in human amygdala and orbitofrontal cortex. Science. 2003;301:1104–1107. doi: 10.1126/science.1087919. [DOI] [PubMed] [Google Scholar]
- Green AS, Grahame NJ. Ethanol drinking in rodents: is free-choice drinking related to the reinforcing effects of ethanol? Alcohol. 2008;42:1–11. doi: 10.1016/j.alcohol.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Xu ZZ, Gao H, He SQ, Ma GQ, Sun T, Wang LH, Zhang ZN, Lena I, Kitchen I, Elde R, Zimmer A, He C, Pei G, Bao L, Zhang X. Interaction with vesicle luminal protachykinin regulates surface expression of delta-opioid receptors and opioid analgesia. Cell. 2005;122:619–631. doi: 10.1016/j.cell.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Hack SP, Bagley EE, Chieng BC, Christie MJ. Induction of delta-opioid receptor function in the midbrain after chronic morphine treatment. J Neurosci. 2005;25:3192–3198. doi: 10.1523/JNEUROSCI.4585-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall FS, Sora I, Uhl GR. Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology (Berl) 2001;154:43–49. doi: 10.1007/s002130000622. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Ingman K, Salvadori S, Lazarus L, Korpi ER, Honkanen A. Selective delta-opioid receptor antagonist N,N(CH3)2-Dmt-Tic-OH does not reduce ethanol intake in alcohol-preferring AA rats. Addict Biol. 2003;8:173–179. doi: 10.1080/1355621031000117400. [DOI] [PubMed] [Google Scholar]
- June HL, McCane SR, Zink RW, Portoghese PS, Li TK, Froehlich JC. The delta 2-opioid receptor antagonist naltriben reduces motivated responding for ethanol. Psychopharmacology (Berl) 1999;147:81–89. doi: 10.1007/s002130051145. [DOI] [PubMed] [Google Scholar]
- Kang-Park MH, Kieffer BL, Roberts AJ, Siggins GR, Moore SD. Presynaptic delta opioid receptors regulate ethanol actions in central amygdala. J Pharmacol Exp Ther. 2007;320:917–925. doi: 10.1124/jpet.106.112722. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Gaveriaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66:285–306. doi: 10.1016/s0301-0082(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Koob G, Kreek MJ. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry. 2007;164:1149–1159. doi: 10.1176/appi.ajp.2007.05030503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. A role for GABA mechanisms in the motivational effects of alcohol. Biochem Pharmacol. 2004;68:1515–1525. doi: 10.1016/j.bcp.2004.07.031. [DOI] [PubMed] [Google Scholar]
- Koob GF, Sanna PP, Bloom FE. Neuroscience of addiction. Neuron. 1998;21:467–476. doi: 10.1016/s0896-6273(00)80557-7. [DOI] [PubMed] [Google Scholar]
- Kotlinska J, Pachuta A, Dylag T, Silberring J. Neuropeptide FF (NPFF) reduces the expression of morphine- but not of ethanol-induced conditioned place preference in rats. Peptides. 2007;28:2235–2242. doi: 10.1016/j.peptides.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Cramer JA, Krol WF, Kirk GF, Rosenheck RA. Naltrexone in the treatment of alcohol dependence. N Engl J Med. 2001;345:1734–1739. doi: 10.1056/NEJMoa011127. [DOI] [PubMed] [Google Scholar]
- Lucido AL, Morinville A, Gendron L, Stroh T, Beaudet A. Prolonged morphine treatment selectively increases membrane recruitment of delta-opioid receptors in mouse basal ganglia. J Mol Neurosci. 2005;25:207–214. doi: 10.1385/JMN:25:3:207. [DOI] [PubMed] [Google Scholar]
- Ma J, Pan ZZ. Contribution of brainstem GABA(A) synaptic transmission to morphine analgesic tolerance. Pain. 2006;122:163–173. doi: 10.1016/j.pain.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Ma J, Zhang Y, Kalyuzhny AE, Pan ZZ. Emergence of functional delta-opioid receptors induced by long-term treatment with morphine. Mol Pharmacol. 2006;69:1137–1145. doi: 10.1124/mol.105.019109. [DOI] [PubMed] [Google Scholar]
- Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Anatomy of CNS opioid receptors. Trends Neurosci. 1988;11:308–314. doi: 10.1016/0166-2236(88)90093-8. [DOI] [PubMed] [Google Scholar]
- Matsuzawa S, Suzuki T, Misawa M, Nagase H. Different roles of mu-, delta- and kappa-opioid receptors in ethanol-associated place preference in rats exposed to conditioned fear stress. Eur J Pharmacol. 1999;368:9–16. doi: 10.1016/s0014-2999(99)00008-4. [DOI] [PubMed] [Google Scholar]
- McBride WJ. Central nucleus of the amygdala and the effects of alcohol and alcohol-drinking behavior in rodents. Pharmacol Biochem Behav. 2002;71:509–515. doi: 10.1016/s0091-3057(01)00680-3. [DOI] [PubMed] [Google Scholar]
- Mhatre MC, Carl K, Garrett KM, Holloway FA. Opiate delta-2-receptor antagonist naltriben does not alter discriminative stimulus effects of ethanol. Pharmacol Biochem Behav. 2000;66:701–706. doi: 10.1016/s0091-3057(00)00272-0. [DOI] [PubMed] [Google Scholar]
- Murray EA. The amygdala, reward and emotion. Trends Cogn Sci. 2007;11:489–497. doi: 10.1016/j.tics.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Nie Z, Schweitzer P, Roberts AJ, Madamba SG, Moore SD, Siggins GR. Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science. 2004;303:1512–1514. doi: 10.1126/science.1092550. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Sydney: Academic Press; 1986. [DOI] [PubMed] [Google Scholar]
- Peana AT, Enrico P, Assaretti AR, Pulighe E, Muggironi G, Nieddu M, Piga A, Lintas A, Diana M. Key role of ethanol-derived acetaldehyde in the motivational properties induced by intragastric ethanol: a conditioned place preference study in the rat. Alcohol Clin Exp Res. 2008;32:249–258. doi: 10.1111/j.1530-0277.2007.00574.x. [DOI] [PubMed] [Google Scholar]
- Qiang M, Denny AD, Ticku MK. Chronic intermittent ethanol treatment selectively alters N-methyl-D-aspartate receptor subunit surface expression in cultured cortical neurons. Mol Pharmacol. 2007;72:95–102. doi: 10.1124/mol.106.033043. [DOI] [PubMed] [Google Scholar]
- Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR. Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology. 2006;31:988–996. doi: 10.1038/sj.npp.1300840. [DOI] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Increased GABA release in the central amygdala of ethanol-dependent rats. J Neurosci. 2004a;24:10159–10166. doi: 10.1523/JNEUROSCI.3004-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci. 2004b;24:1594–1603. doi: 10.1523/JNEUROSCI.5077-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Gold LH, Polis I, McDonald JS, Filliol D, Kieffer BL, Koob GF. Increased ethanol self-administration in delta-opioid receptor knockout mice. Alcohol Clin Exp Res. 2001;25:1249–1256. [PubMed] [Google Scholar]
- Schmidt BL, Tambeli CH, Barletta J, Luo L, Green P, Levine JD, Gear RW. Altered nucleus accumbens circuitry mediates pain-induced antinociception in morphine-tolerant rats. J Neurosci. 2002;22:6773–6780. doi: 10.1523/JNEUROSCI.22-15-06773.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See RE, Fuchs RA, Ledford CC, McLaughlin J. Drug addiction, relapse, and the amygdala. Ann N Y Acad Sci. 2003;985:294–307. doi: 10.1111/j.1749-6632.2003.tb07089.x. [DOI] [PubMed] [Google Scholar]
- Siggins GR, Martin G, Roberto M, Nie Z, Madamba S, De Lecea L. Glutamatergic transmission in opiate and alcohol dependence. Ann N Y Acad Sci. 2003;1003:196–211. doi: 10.1196/annals.1300.012. [DOI] [PubMed] [Google Scholar]
- Siggins GR, Roberto M, Nie Z. The tipsy terminal: presynaptic effects of ethanol. Pharmacol Ther. 2005;107:80–98. doi: 10.1016/j.pharmthera.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Stromberg MF, Casale M, Volpicelli L, Volpicelli JR, O’Brien CP. A comparison of the effects of the opioid antagonists naltrexone, naltrindole, and beta-funaltrexamine on ethanol consumption in the rat. Alcohol. 1998;15:281–289. doi: 10.1016/s0741-8329(97)00131-6. [DOI] [PubMed] [Google Scholar]
- Tabakoff B, Hoffman PL. Alcohol addiction: an enigma among us. Neuron. 1996;16:909–912. doi: 10.1016/s0896-6273(00)80113-0. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM. Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol. 2007;12:227–462. doi: 10.1111/j.1369-1600.2007.00070.x. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Waldhoer M, Bartlett SE, Whistler JL. Opioid receptors. Annu Rev Biochem. 2004;73:953–990. doi: 10.1146/annurev.biochem.73.011303.073940. [DOI] [PubMed] [Google Scholar]
- Walker BM, Ettenberg A. Intracerebroventricular ethanol-induced conditioned place preferences are prevented by fluphenazine infusions into the nucleus accumbens of rats. Behav Neurosci. 2007;121:401–410. doi: 10.1037/0735-7044.121.2.401. [DOI] [PubMed] [Google Scholar]
- Ward SJ, Roberts DC. Microinjection of the delta-opioid receptor selective antagonist naltrindole 5’-isothiocyanate site specifically affects cocaine self-administration in rats responding under a progressive ratio schedule of reinforcement. Behav Brain Res. 2007;182:140–144. doi: 10.1016/j.bbr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Mascagni F, McDonald AJ. Sex differences in delta opioid receptor immunoreactivity in rat medial amygdala. Neurosci Lett. 2002;328:160–164. doi: 10.1016/s0304-3940(02)00481-0. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bao L, Guan JS. Role of delivery and trafficking of delta-opioid peptide receptors in opioid analgesia and tolerance. Trends Pharmacol Sci. 2006;27:324–329. doi: 10.1016/j.tips.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Zhu W, Bie B, Pan ZZ. Involvement of non-NMDA glutamate receptors in central amygdala in synaptic actions of ethanol and ethanol-induced reward behavior. J Neurosci. 2007;27:289–298. doi: 10.1523/JNEUROSCI.3912-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Pan ZZ. Synaptic properties and postsynaptic opioid effects in rat central amygdala neurons. Neuroscience. 2004;127:871–879. doi: 10.1016/j.neuroscience.2004.05.043. [DOI] [PubMed] [Google Scholar]
- Zhu W, Pan ZZ. Mu-opioid-mediated inhibition of glutamate synaptic transmission in rat central amygdala neurons. Neuroscience. 2005;133:97–103. doi: 10.1016/j.neuroscience.2005.02.004. [DOI] [PubMed] [Google Scholar]