

Abstract
Type-II kinase inhibitors represent a class of chemicals that trap their target kinases in an inactive, so-called DFG-out, state, occupying a hydrophobic pocket adjacent to the ATP binding site. These compounds are often more specific than those targeting active, DFG-in, kinase conformations. Unfortunately, the discovery of novel type-II scaffolds presents a considerable challenge, partly because the lack of compatible kinase structures makes structure-based methods inapplicable. We present a computational protocol for converting multiple available DFG-in structures of various kinases (∼70% of mammalian structural kinome) into accurate and specific models of their type-II-bound state. The models, described as Deletion-Of-Loop asp-PHe-gly-IN (DOLPHIN) kinase models, demonstrate exceptional performance in various inhibitor discovery applications, including compound pose prediction, screening, and in silico activity profiling. Given the abundance of the DFG-in structures, the presented approach opens possibilities for kinome-wide discovery of specific molecules targeting inactive kinase states.
Keywords: kinase, DFG-in, DFG-out, type-II inhibitor, imatinib, structure-based inhibitor discovery, compound screening, compound profiling, phosphorylation, cancer therapeutics
Introduction
Protein kinases have been long recognized as important drug targets 1. Several dozens of small molecule kinase inhibitors are either approved or studied for various human diseases including cancer, cardio-vascular disorders, and inflammation 2, 3.
Functional states of a typical protein kinase can be characterized by the position of a conserved DFG (or, rarely, D[LWY]G) motif in its activation loop. The absolute majority of inhibitors target the ATP-site of the kinase in its active, DFG-in, state. In contrast, the so-called type-II inhibitors (e.g. imatinib and sorafenib) induce a distinct, DFG-out, conformation, and occupy an additional hydrophobic pocket created by this rearrangement 4-8 (Figure 1(a)). These inhibitors possess several advantages over ATP-site compounds, including improved kinase selectivity and slower off-rates 9.
Figure 1.

(a) Type II inhibitors induce, stabilize, and exploit a distinct “DFG-out” conformation of the target kinases. (b) Conformational statistics of mammalian structural kinome (based on PDB release of June 2008).
The phenomenon of type-II inhibition was initially thought to be specific to only a few protein kinases. A small residue at the so-called gatekeeper position in the kinase hinge region was considered a prerequisite for type-II inhibition. That view is challenged by recent advances in targeting kinases with medium-size gatekeepers, such as TIE and MET 10-12. Type-II inhibitor discovery for a wider range of kinases is therefore a topic of great interest and importance.
Unfortunately, the de novo identification of type-II inhibitors presents a considerable challenge. They are often overlooked in traditional enzymatic assays and high-throughput screening (HTS), because of low affinity to active, phosphorylated kinases. To overcome this obstacle, several phosphorylation state-independent binding assays have been developed, some involving competition binding to immobilized probes 13-16, and others based on temperature-dependent unfolding of the protein 17-19. These assays, however, address the problem only partially, as they not as cost-effective as biochemical assays, and are hard to apply in a high-throughput fashion. Not surprisingly, most known type-II inhibitors to date have been developed via QSAR-guided modifications of ATP-site ligands, rather than directly from HTS.
The QSAR strategies were generalized by Liu and Gray 20 and Okram et al 21, who presented a universal chemical modification protocol converting known ATP-site inhibitors into their type-II counterparts. This revolutionary work demonstrated that type-II inhibition is a relatively common phenomenon, for which general methods can be successfully developed and applied. Their approach, however, was restricted to only a small fraction of chemical space, and, being completely chemistry-based, yielded compounds with unpredictable kinase specificity.
Structure-based computational methods, including Virtual Ligand Screening (VLS) have a potential of both dramatically widening the chemical space and reducing the number of candidates for experimental validation. VLS techniques were found successful in a wide variety of applications (e.g. 22-24), especially combined with improved scoring functions 25, 26. However, the lack of relevant kinase structures limits the applicability of these techniques to type-II inhibitor discovery. The DFG-in structures, representing ∼70% of the mammalian structural kinome, are type-II-incompatible, as well as intermediate (∼22%) and even apo-DFG-out (∼3%) structures. Reliable methods for modeling the DFG-in / DFG-out transition have not been reported to date.
Here we propose a new approach to structure-based type-II inhibitor discovery and evaluation. We designed a general deterministic modeling protocol for converting the abundant DFG-in structures of various kinases into accurate and specific models of their type-II-bound state, the so-called DOLPHIN (Deletion-Of-Loop PHe-IN) kinase models. The models were validated on a comprehensive kinase-ligand benchmark and demonstrated exceptional performance in all three types of structure-based inhibitor discovery applications: (i) ligand docking (binding pose prediction); (ii) ligand screening (recognition of active type-II compounds in a large dataset); (iii) ligand activity profiling (evaluation of the relative ligand affinities to different kinases). Given the extensive representation of the DFG-in conformations in structural kinome, this approach opens new possibilities for discovery of novel type-II inhibitors for a wide range of kinases.
Results
DFG-in Conformations are Predominant in the Structural Kinome
The June 2008 release of the Protein Data Bank27 contained 1,216 structures of 122 mammalian protein kinase domains. Conformational analysis of this set showed that 95 kinases were represented at least once in the DFG-in state (840 structures) (Figure 1(b)). The set of type-II-compatible structures, on the contrary, was limited to only 9 kinases (ABL1, LCK, MET, KIT, SRC, BRAF1, VGFR2, TIE2, and MK14) that have already been co-crystallized with type-II inhibitors (69 structures). Neither 268 structures of intermediate conformations, nor even 39 apo-DFG-out structures represented reasonable models of type-II-bound states.
Conservation of Structural Features of Type-II-bound Conformations in the DFG-in State Suggests DOLPHIN Transformation
DFG-in / DFG-out transition is a dramatic conformational change induced by type-II kinase inhibitors, and their characterizing feature. We observed, however, that except for the DFG-out state, determinants of type-II ligand binding are preserved in most DFG-in structures. These determinants include (i) presence of the conserved lysine-glutamate salt bridge and (ii) sufficient pocket width (the distance between the carboxyl group of the conserved αC-helix glutamate and the backbone amide nitrogen of the DFG-motif aspartate).
With reasonable margins, both conserved salt bridge and sufficiently wide pocket (> 4.3 Å) were observed in as many as 600 mammalian DFG-in structures (71%). Some representative counterexamples included PDB 1pkg (activated KIT kinase, the pocket width of only 4.2 Å), 1fmk (SRC kinase, the conserved glutamate points away from the active site) and chain B of PDB 1yom (SRC, the conserved glutamate disordered). Fortunately, these cases were a minority.
Structural conservation of the two determinants of type-II inhibition suggested that DFG-motif excision might convert the DFG-in structures into accurate models of type-II-bound state of their respective kinases, which led to the development of DOLPHIN (Deletion-Of-Loop PHe-IN) protocol (Figure 2, Methods). To compensate for possible crystallographic errors and enhance the model performance, we also introduced a weak non-specific pharmacophore-like field in place of selected removed atoms.
Figure 2.
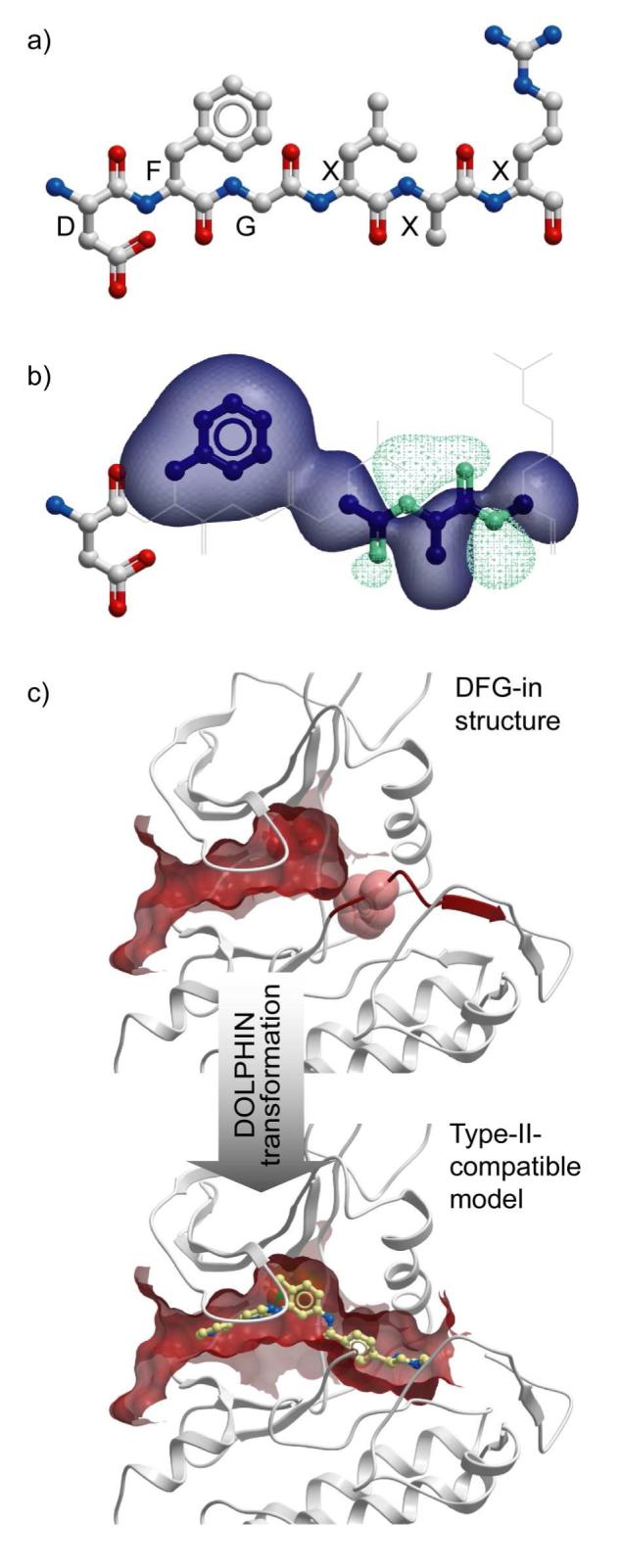
DOLPHIN transformation: (a) DFG Phe and the four next residues in the sequence are removed; (b) attractive density is generated from selected removed atoms. The transparent blobs represent equipotent contours of the lipophilic (dark-blue) and polar (sea-green) parts of the density; (c) this computational “surgery” dramatically affects the ligand binding pocket shape and size, producing a type-II compatible model.
The models were tested in docking, screening, and activity profiling of the known type-II inhibitors in two modes. In the so-called Single Receptor mode, the performance of each DOLPHIN was evaluated independently. In the Multiple Receptor Conformations (MRC) mode, all available DOLPHINs of a single kinase were combined with each compound represented by its best score in this ensemble.
The minority of DFG-in structures with narrow pocket and/or disrupted salt bridge was expected to demonstrate inferior performance in the above applications. We nevertheless included these structures in the experiment for the sake of exhaustiveness and to evaluate the relative roles of the two structural features.
Docking to DOLPHIN Models Correctly Predicts Type-II Ligand Binding Geometry
A comprehensive benchmark of publicly available DFG-in and type-II-bound kinase structures was used to test the DOLPHIN docking. We studied the performance of individual DOLPHINs and the effects of using MRC ensembles or the additional pharmacophore-like field.
Docking to a single rigid DOLPHIN model is successful in over 75% of the cases
In the single receptor docking, the near-native ligand binding geometry scored first for 142 of 184 DOLPHIN-ligand pairs (77%). For 153 cases (83%), it was found within three top-scoring ligand conformations (Table 1). The standard ligand heavy-atom RMSD cutoff of 2 Å was applied.
Table 1.
RMSD values for top-scoring docking poses of crystallographic type-II ligands in DOLPHIN models of their respective kinase targets. Table entries are in the format Rmsd (Rank 1st correct, if not 1); NS = Not Sampled. Correct poses ranking first are colored green, within top three yellow, otherwise grey. All compound chemical structures can be found in Supp. Figure 1.
| ABL1 DFG-in | 406 | 7MP | GIN | KIN | PRC | STI |
|---|---|---|---|---|---|---|
| 2f4jA | 1.91 | 0.46 | 0.87 | 0.62 | 1.73 | 0.52 |
| 2g2iA | 1.14 | 0.65 | 1.11 | 0.67 | 1.67 | 0.76 |
| 2g2iB | 2.12(2) | 0.53 | 3.76(2) | 0.71 | 2.62(5) | 0.98 |
| 2gqgAa | 2.09(4) | 0.83 | 2.06(2) | 0.98 | 12.62(16) | 1.69 |
| 2gqgB | 1.85 | 1.90 | 0.85 | 0.89 | 2.06(8) | 1.75 |
| 2hz4A | 0.92 | 1.98 | 0.99 | 0.84 | 12.72(9) | 6.65(3) |
| 2hz4B | 1.37 | 1.03 | 0.85 | 0.62 | 1.92 | 1.55 |
| 2hz4C | 7.44(12) | 1.70 | 11.66(4) | 12.35(5) | 14.99 (40) | 11.11(26) |
| 2qohA | 0.88 | 0.68 | 0.89 | 0.73 | 1.56 | 0.60 |
| 2qohB | 1.09 | 0.97 | 1.23 | 0.64 | 1.58 | 0.81 |
| 2v7aAb | 4.32(44) | 8.22(10) | 10.34(61) | 0.97 | 9.97(47) | 13.19(51) |
| 2v7aBb | 13.19(149) | 9.36(23) | 10.49(NS) | 1.12 | 10.85(48) | 9.23(62) |
| 2z60Ab | 1.04 | 1.93 | 1.92 | 0.74 | 12.20(3) | 1.06 |
| BRAF1 DFG-in | BAX | LCK DFG-in | 1N8 | 242 | 9NH | STI | SRC DFG-in | STI |
|---|---|---|---|---|---|---|---|---|
| 2fb8A | 1.59 | 1qpcA | 1.13 | 0.77 | 1.01 | 1.49 | 1fmkAc | 11.91(NS) |
| 2fb8B | 0.62 | 1qpdA | 1.02 | 0.71 | 1.36 | 1.44 | 1y57A | 1.33 |
| 1qpeA | 1.28 | 1.63 | 1.30 | 1.61 | 1yi6A | 16.33(3) | ||
| 1qpjA | 1.03 | 0.47 | 0.83 | 1.39 | 1yi6Ba | 2.80(4) | ||
| 2of2A | 1.21 | 0.76 | 1.41 | 1.55 | 1yomBd | 5.18(57) | ||
| KIT DFG-in | STI | 2of4A | 1.23 | 0.74 | 1.05 | 1.50 | 2bdfAa | 7.11(2) |
| 1pkgAa | 4.11(3) | 2ofuA | 1.28 | 0.54 | 1.01 | 1.40 | 2bdfBa | 7.02(3) |
| 1pkgBa | 3.73(3) | 3lckA | 1.43 | 0.76 | 2.06(2) | 1.41 | 2bdjAa | 1.12 |
| 2hwoAd | 13.77(18) | |||||||
| 2oiqBa | 1.12 | |||||||
| MK14 DFG-in | 1PP | AQZ | B96 | BMU | L09 | L10 | L11 | LI2 | LI3 | WBT |
|---|---|---|---|---|---|---|---|---|---|---|
| 1m7qA | 0.62 | 3.16e(5) | 0.77 | 1.07 | 1.58 | 0.45 | 1.03 | 0.73 | 1.40 | 0.68 |
| 1oukA | 0.67 | 3.17e(4) | 0.80 | 0.93 | 1.40 | 0.68 | 0.77 | 0.79 | 1.50 | 0.75 |
| 1ouyA | 0.73 | 3.61e(15) | 0.76 | 1.12 | 1.81 | 0.49 | 1.19 | 0.99 | 1.50 | 0.90 |
| 1oz1A | 0.65 | 1.29 | 1.17 | 0.97 | 1.14 | 0.44 | 0.57 | 0.82 | 0.85 | 0.62 |
| 2okrA | 0.75 | 1.49 | 4.36(4) | 1.08 | 10.99(11) | 0.67 | 1.32 | 1.35 | 1.43 | 1.30 |
| 2okrD | 0.80 | 4.56e(NS) | 4.30(82) | 1.27 | 1.85 | 0.68 | 1.45 | 1.57 | 1.56 | 1.46 |
Narrow pocket
T315I imatinib-resistant mutation
Conserved salt bridge disrupted
Conserved salt bridge disordered
The part of the ligand occupying the hydrophobic “selectivity” pocket docked correctly with partial RMSD < 1A.
Docking performance of individual models varied significantly. For example, several models of the SRC kinase appeared incompatible with compound 1 (imatinib 4, PDB Het ID STI, see Figure 3) because of a disrupted salt bridge or other structural deviations. Interestingly, imatinib is an extremely weak binder to SRC kinase 28 (observable Kd > 10 μM), which might be due to low representation of appropriate SRC conformations in solution. Correct ligand placement also presented a challenge for both narrow-pocket DOLPHINs of the KIT kinase (1pkgA,B). Importantly, imatinib-resistant active site mutation, T315I, made 2v7aA,B the lowest-performing structures of ABL1. On the other hand, the most accurate models reproduced the binding geometry of all benchmark compounds.
Figure 3.

Chemical structures, names, and targets of type-II kinase inhibitors mentioned in the text.
In spite of sufficiently wide pocket and intact salt bridge, chain C in the 2hz4 structure of ABL1 kinase appeared to be a difficult target. We explain it by very high (>100) B-factors of the pocket atoms in this chain signaling of unstable crystal and leading to the unusual, incompatible with type-II ligands rotameric state of Met290.
Among the ligands, the most difficult one was compound 2 (an anilinoquinazoline type-II inhibitor of MK14 29, PDB Het ID AQZ). In the co-crystal of the kinase with 2 (PDB 2bak), the DFG Phe169 is re-oriented towards the hinge region and provides a stabilizing ligand interaction. The absence of this residue in DOLPHINs complicated the ligand positioning, so that its hinge-region moiety docked correctly in only 2 of 6 cases. However, the part of the ligand occupying the hydrophobic “selectivity” pocket docked correctly in all cases with partial RMSD below 1 Å.
The contribution of the pharmacophore-like field
As described above, a weak attractive field was introduced as a part of the DOLPHIN models in place of their removed DFG-motif atoms. Interestingly, most models were sufficiently accurate to achieve similar docking performance even in the absence of the field. Without it, the correct ligand binding geometry scored first in 112 of 184 cases (61%), and within top three poses in 141 cases (77%).
Multiple Receptor Conformation mode further improves pose prediction
When only the best-scoring ligand poses were selected for kinases with multiple DFG-in structures, the correct binding geometry ranked first for as many as 21 of 23 kinase/ligand pairs (91%) (Figure 4). For the 22nd pair (MK14 and compound 2), it ranked third, yielding to two poses with correctly docked hydrophobic pocket moiety. The only unsuccessful pair consisted of the narrow-pocket KIT ensemble and imatinib, with the near-native geometry ranking only ninth. For 18 of 23 kinase/ligand pairs, pose prediction was also successful in the absence of the pharmacophore-like field.
Figure 4.

Comparison of the top-scoring poses of the crystallographic type-II ligands in DOLPHIN MRC ensembles of their target kinases (red) with the crystallographic complexes (green). *For MK14 and compound 2 (PDB Het AQZ), the correct pose was ranked first after refinement and full-atom scoring.
Refinement and rescoring of the top three ligand poses raised the success rate to 22 of 23 cases (95%), with the pair of MK14 and compound 2 included. The improvement was apparently due to the use of full-atom scoring procedure that is more sensitive to hydrogen bonding and therefore rewards ligand poses with optimal hinge-region contacts.
DOLPHIN Models Recognize Type-II Inhibitors of Their Respective Kinases in Virtual Screening
We further studied the ability of DOLPHIN models to distinguish the active type-II inhibitors from other compounds in VLS. A dataset of 391 crystallographic kinase ligands was used for this purpose, a fraction of active type-II inhibitors ranging from 0.7% for BRAF1 and SRC to 3.6% for MK14. Docking and scoring the dataset compounds in each DOLPHIN model produced an ordered hit list, with the challenge of bringing the scarce active inhibitors to its top. The model screening performance was evaluated numerically as the Area Under ROC-Curve, or AUC.
VLS with a single DOLPHIN model
As Table 2 illustrates, 31 of the 41 DOLPHIN models proved highly selective towards their type-II ligands with the AUC exceeding 0.9. As expected, poor selectivity was observed for the imatinib-resistant mutant structures of ABL1 (2z60A and 2v7aA,B), the three salt bridge deficient SRC structures (1fmkA, 2hwoA, and 1yomB), and high B-factor ABL1 structure (2hz4C). Notably, the models that performed best in docking (e.g. 2g2iA, 2qohA,B and 1oz1A), also demonstrated the highest screening selectivity.
Table 2.
VLS performance of individual DOLPHIN models evaluated as the area under their ROC curves. Three to fourteen active type-II compounds per kinase were recognized in a set of 391 kinase ligands.
| ABL1 | AUC | BRAF1 | AUC |
|---|---|---|---|
| 2qohA | 0.95 | 2fb8B | 0.98 |
| 2qohB | 0.95 | 2fb8A | 0.97 |
| 2g2iA | 0.95 | ||
| 2g2iB | 0.94 | ||
| 2f4jA | 0.93 | LCK | AUC |
| 2hz4B | 0.91 | 2ofuA | 0.97 |
| 2gqgB | 0.91 | 3lckA | 0.97 |
| 2hz4A | 0.89 | 1qpcA | 0.96 |
| 2z60Ab | 0.88 | 1qpeA | 0.96 |
| 2gqgAa | 0.83 | 2of2A | 0.95 |
| 2v7aAb | 0.81 | 2of4A | 0.95 |
| 2hz4C | 0.73 | 1qpjA | 0.92 |
| 2v7aBb | 0.66 | 1qpdA | 0.92 |
| KIT | AUC | SRC | AUC |
|---|---|---|---|
| 1pkgBa | 0.96 | 2oiqBa | 0.96 |
| 1pkgAa | 0.92 | 2bdjAa | 0.96 |
| 1yi6Ba | 0.94 | ||
| MK14 | AUC | 1y57A | 0.93 |
| 1oz1A | 0.97 | 2bdfBa | 0.91 |
| 1oukA | 0.97 | 2bdfAa | 0.90 |
| 1ouyA | 0.95 | 1yi6A | 0.82 |
| 1m7qA | 0.95 | 1fmkAc | 0.77 |
| 2okrD | 0.94 | 2hwoAd | 0.73 |
| 2okrA | 0.93 | 1yomBd | 0.50 |
Narrow pocket
T315I imatinib-resistant mutation
Conserved salt bridge disrupted
Conserved salt bridge disordered.
Clearly, the quality of the source X-ray structures may have a dramatic effect on the results of DOLPHIN docking and screening. This quality, however, is not exhausted by simple characteristics such as the average resolution of the structure or even atomic B-factor values. For example, the 3.12 Å resolution structure 2g2iA of ABL1 (maximal pocket B-factor of 147) and the 2.9 Å resolution structures 2fb8A,B of BRAF1 nevertheless demonstrated very high docking and screening performance. On the other hand, high-resolution, low B-factors, or even good electron density in the vicinity of the binding pocket can sometimes be observed in a poorly performing model. For example, the excellently resolved SRC structure, 1fmkA (resolution 1.5A, pocket atom B-factors not exceeding 54), appeared type-II incompatible due to a disrupted salt bridge. While it has been previously shown that large-scale virtual screening efforts provide better results for high-resolution structures, in DOLPHIN docking and screening, the influence of structure quality is watered down by other factors. For this study, we used structures with resolution below 3.5 Å. Where available, local pocket electron density was checked using Uppsala Electron Density Server 30 and found acceptable in all cases.
VLS with multiple models of the same kinase
The ligand hit lists from all DOLPHIN models of a single kinase were combined, with each compound represented by its best score in the ensemble. This approach provided exceptionally high selectivity for all six kinases, with the AUC values of 0.96, 0.97, 0.95, 0.96, 0.98, and 0.96 for ABL1, BRAF1, KIT, LCK, MK14, and SRC respectively (Figure 5). Most (>50%) of the known type-II ligands were ranked in the top 3.32% of the list for ABL1 and BRAF1, and in the top 1.28%, 2.56%, 2.30% and 1.79% for KIT, LCK, MK14, and SRC. Even the narrow-pocket KIT ensemble recognized three of five type-II inhibitors of KIT (compounds 3, 4, and 5, Figure 3) as ranks two, four, and five, respectively.
Figure 5.
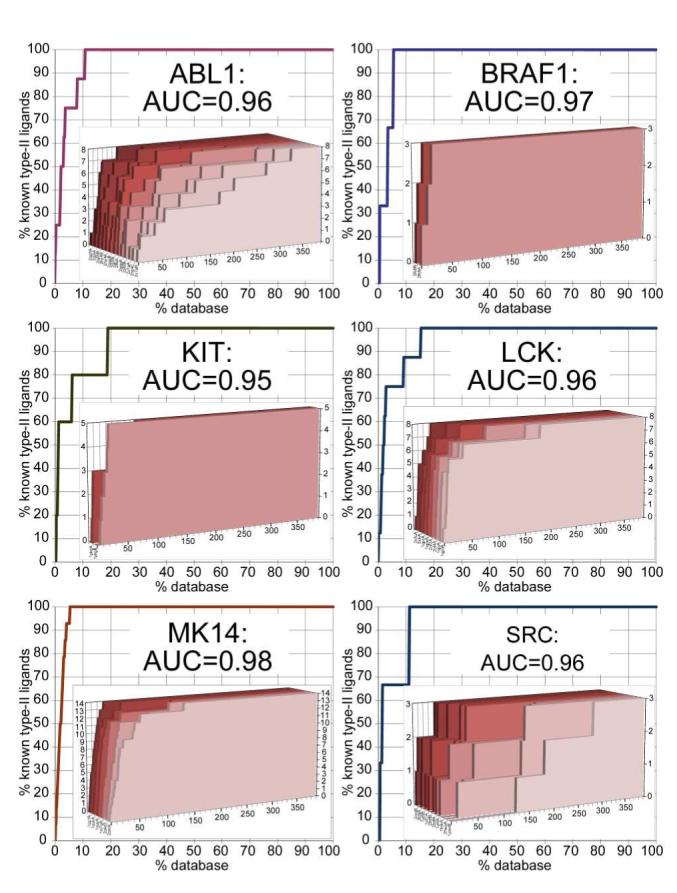
VLS performance of DOLPHIN MRC ensembles, evaluated as the area under their ROC curves. Three to fourteen active type-II compounds per kinase were recognized in a set of 391 kinase ligands. Red 3D inserts represent the ROC curves for individual DOLPHINs.
Application of the DOLPHIN screening: identification of compound off-target activities
We further analyzed top ∼5% of each hit lists looking specifically at the hypothetical false positives, i.e. high scoring compounds with no reported activity against the respective kinases. This exercise resulted in identification of the following secondary activities of crystallographic type-II inhibitors (Figure 6):
Compound 6, a dual TIE2 and VGFR2 tyrosine kinase inhibitor 10 (PDB Het ID GIG), scored well in the DOLPHIN ensembles of SRC and MK14 kinases (ranks 3 and 18 of 391, respectively). Its inhibitory activity against these kinases was confirmed experimentally (Masaichi Hasegawa, personal communication).
Compound 7 (INNO-406), a potent inhibitor of ABL1 and Lyn kinases 31 (PDB Het ID 406), was among the top-scoring compounds in the LCK ensemble (rank 33 of 391). That prediction was also confirmed, as INNO-406 inhibits LCK with IC50 ∼ 120 nM 32.
Compound 8, a VGFR2 inhibitor 33 (PDB Het ID 276), ranked fourth in the DOLPHIN ensemble of BRAF1. It was also confirmed to inhibit this kinase with IC50 ∼ 400 nM (Michele Potashman, personal communication).
Figure 6.
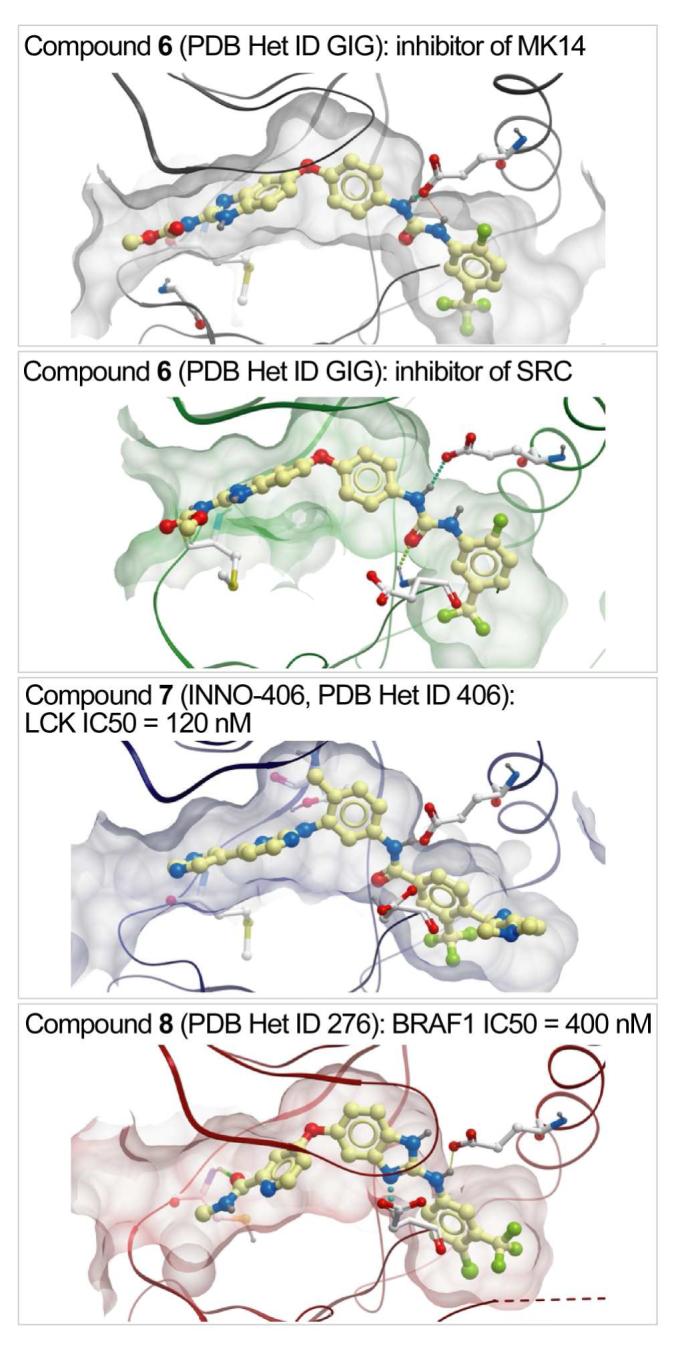
Examples of compound secondary activity prediction using DOLPHIN screening. Top-scoring poses of the inhibitors in DOLPHIN models of the off-target kinases are shown. Inhibition of these kinases was confirmed by the authors of the original studies.
Identification of a potent and selective inhibitor of CSK kinase
Our approach was further applied to a kinase with no available DFG-out structure. We chose human C-terminal Src kinase, CSK, for this case study, because it had a DFG-in structure in the public domain (PDB 1k9a 34), and a known type-II inhibitor 20. We built the DOLPHIN models from all six chains of the structure and screened all “second generation” type-II inhibitors 20 against the obtained MRC ensemble. Compound 9, the only annotated type-II CSK inhibitor in the set 20, ranked first in the resulting hit list.
In silico Compound Profiling Using DOLPHIN Kinase Models
Computational prediction of the relative binding affinity of a compound to different proteins remains an unsolved problem despite the significant progress towards better force fields and scoring functions. A major difficulty is presented by the fact that binding energies estimated from protein-ligand complex structures appear to be shifted relatively to the observed binding energies in a systematic, protein-specific fashion. Apart from inevitable fluctuations of model quality and energy functions, these systematic binding energy offsets are caused by thermodynamic reasons, namely, variations of the protein conformational equilibrium.
The latter considerations are of particular importance to the present study. Our ligands of interest bind exclusively to the DFG-out kinase species, so their observed affinity depends on the relative concentrations of DFG-in and DFG-out molecules. Equilibrium variations between kinases, mutants of the same kinase, and experimental conditions introduce different offsets to the observed binding energies. For example, experimental data analysis gives the estimated offset difference of 3.15 kcal/mol to the observed type-II compound binding energy to phosphorylated vs. unphosphorylated ABL1, 0.7 kcal/mol to LCK vs. ABL1, and 4 kcal/mol to SRC vs. ABL1 (see Supplementary Materials). Here we demonstrate that adding the offsets to the binding energy estimates from DOLPHIN complexes makes them suitable for ligand activity profiling.
Computational determination of kinase-specific binding energy offsets
For most kinases, the offsets cannot be directly derived from experimental data, and must be found by fitting experimental to calculated binding energies. Using this approach, we obtained the following offset differences for the five kinases with respect to unphosphorylated ABL1:
bBRAF1 — bABL1 = 1.17 kcal/mol
bKIT — bABL1 = - 4.48 kcal/mol
bLCK — bABL1 = 0.20 kcal/mol
bMK14 — bABL1 = - 0.17 kcal/mol
bSRC — bABL1 = 3.55 kcal/mol
For the two kinases whose relative offsets w.r.t. ABL1 could be estimated from the experimental binding data (LCK and SRC), these values are in a good agreement with the experiment (0.20 vs. 0.7 kcal/mol for LCK and 3.55 vs. 4 kcal/mol for SRC). The unusually low predicted offset for KIT (-4.48 kcal/mol) is due to the properties of the source DFG-in structures rather than the equilibrium considerations: the calculated binding energies for the correctly docked ligands were consistently higher in the narrow pocket KIT ensemble than in other kinases.
Ligand activity/selectivity profiling
The kinase-specific binding energy offsets were combined with previously calculated ligand binding scores in the DOLPHIN MRC ensembles to give the estimates of their observed binding affinities. Comparison of the obtained values with the experimental data showed a strong correlation (Figure 7). For example, the DOLPHIN models correctly characterized compound imatinib (PDB Het ID STI) as a potent inhibitor of ABL1 and LCK kinases, but not BRAF1, MK14, or SRC. BIRB-796 (PDB Het ID B96) was found to be more active against MK14 than against the other five kinases. Sorafenib (PDB Het ID BAX) was confirmed to be a relatively non-specific compound inhibiting all six kinases, SRC to a lesser extent than others. The plot features only two false negatives: INNO-406 and imatinib were not identified as the inhibitors of KIT due to their poor scoring in the available KIT DFG-in structures.
Figure 7.

Comparison of experimental activities of known type-II kinase inhibitors with their binding affinities predicted by the DOLPHIN kinase models.
Several false positives on the plot are likely due to the mixed nature of the experimental data: our calculated binding affinities approximated Kd, but were fitted to mixed IC50 / Kd / Ki data. The experimental IC50 / Kd ratios are known to vary from 1 to >100 due to unnatural peptide substrates, dependence on the ATP concentration relative to the KM,ATP, and other factors 35, 36, which most likely explains the observed deviations.
Discussion
Recent advances in medicinal chemistry demonstrated that type-II inhibition phenomenon might extend to a wide range of kinases10, 12 and ligand chemotypes (e.g. 33, 37). Unfortunately, the efficient structure-based discovery of type-II inhibitors is hindered by the lack of compatible kinase structures. Better stability of the DFG-in state makes it a primary material of both experimental ligand screening and X-ray crystallography, introducing a strong bias towards DFG-in conformations in the structural kinome. We demonstrated that despite their obvious type-II-incompatibility, the DFG-in structures often preserve the determinants of type-II ligand binding and can be converted into accurate and specific models of type-II-bound kinases.
Building and testing the DOLPHIN models on a comprehensive kinase benchmark revealed their potential for predicting type-II ligand binding poses (95% success). The top-ranking ligand poses also reproduced the detailed inter-atomic contacts of the complex. Figure 8 provides a comparison of DOLPHIN docking and crystallographic complexes of compound 10 (BIRB-796) with MK14 kinase: the predicted complex reconstitutes all essential intermolecular hydrogen bonds and non-polar contacts identified by X-ray crystallography 9. Similar results were achieved with other ligand/kinase pairs. The accurate prediction of inter-atomic contacts makes DOLPHIN docking complexes good starting points for structure-based ligand optimization.
Figure 8.

Side-by-side comparison of (a) the best scoring pose of compound 10 (BIRB-796) in the DOLPHIN ensemble of the MK14 kinase with (b) the crystallographic complex: DOLPHIN docking reproduces the detailed inter-atomic contacts. Hydrogen bonds are shown as colored dots; the receptor pocket surface as transparent mesh.
Most DOLPHIN models demonstrated high screening selectivity, even as single rigid receptors. Taking into account the kinase conformational flexibility (MRC approach) further improved the results, providing recognition of most known type-II inhibitors in the top 1.5%-3.5% of the hit lists. We therefore recommend DOLPHIN MRC ensembles as the most efficient virtual screening tool. We were especially pleased to observe that several cases initially classified as false positives were later confirmed as secondary (off-target) activities of the benchmark compounds. This validated the use of DOLPHIN approach for compound off-target activity prediction, a task of critical practical importance.
The DOLPHIN protocol proved to be sensitive to kinase active site mutations. The three ABL1 structures carrying T315I imatinib-resistance mutation (2v7aA,B and 2z60A) clearly behaved differently in docking and screening, down-scoring the inhibitors of the wild-type kinase. Instead, high scores and ranks were assigned to different compounds that now await experimental validation as type-II inhibitors of T315I ABL1.
Current understanding of the phenomenon of type-II inhibition gives two major reasons for higher affinity (or exclusive binding) of a type-II inhibitor to one of two different kinases. First, and most obvious reason lies in variations of the binding site residue composition (it explains, for example, why imatinib binds to ABL1 but not to MK14). In particular, a single change in the gatekeeper residue may exhibit most profound effect on the inhibitor binding due to a steric clash. Another, more subtle reason is the varying energetic penalty of adopting the DFG-out conformation (this, for example, explains low observed affinity of imatinib to SRC28). The two reasons, though not entirely independent 38, do not appear to directly correlate across the kinome: small gatekeeper does not result in high DFG-out propensity (e,g. SRC has a small gatekeeper and low DFG-out propensity) and vice versa (e,g, IGF1R has a bulky gatekeeper but high DFG-out propensity). While the calculated binding energies for DOLPHIN docking complexes capture the residue composition aspect of the binding affinity, the predefined kinase-specific systematic binding energy offsets introduced in this study represent the numerical expression of the DFG-out propensities. By combining these values, we showed that the DOLPHIN approach could be used to predict the affinity of a single inhibitor to different kinases, evaluate its cross-reactivity, and determine its selectivity profile. That puts our computational technique in line with advanced in vitro activity profiling assays 39 addressing a particularly challenging problem of targeting inactive kinase conformations.
It is important to note that high selectivity may or may not be a desirable feature for a kinase inhibitor. In the recent years, several compounds annotated as “multi-targeted kinase inhibitors” entered clinical trials and were proved to exhibit therapeutic effects by simultaneously shutting down more than one kinase in the same or related pathways. Rational development of such compounds requires both extensive screening and profiling. Combined with experimentally-derived protein-specific offsets, the methodology proposed in this paper may be of great help in designing inhibitors with desired activity profiles.
Ab initio prediction of the absolute DFG-out propensity values and related binding energy offsets for different kinases is an unsolved problem and is out of the scope of this paper. The propensity can be, however, deduced from experimental binding data on at least one known type-II inhibitor. Recent large-scale kinase inhibitor profiling efforts (e.g. 39 and 40) provide data for indirect evaluation of the fraction of kinases with significant DFG-out propensity and estimating selected kinase-specific offsets. For example, at least 108 of 281 protein kinases tested by Ambit 39 (mutants excluded) bind five known type-II inhibitors — 1 (imatinib 6), 3 (sorafenib, 41), 10 (BIRB-796 9), 11 (ABT-869 42), and 12 (AST-487 43, 44), — with affinity of ≤ 1 μM. Similar tendency is observed within the Ser/Thr kinase domain extensively screened by Fedorov et al 40. It is highly unlikely that the inhibitors achieve this high potency while binding to the kinases in DFG-in conformations, therefore, 40% (i.e. 108/281) represents a reasonable estimate of the fraction of kinases readily adopting a DFG-out conformation in solution. Interestingly, 44 of the 108 kinases have crystal structures in PDB; among them, only 8 are crystallized with a type-II inhibitor, but 26 (60%) are found in the classical DFG-in conformation thus representing immediate candidates for DOLPHIN transformation and DOLPHIN-based compound cross-reactivity studies.
In summary, the proposed DOLPHIN methodology provides reliable structure-based identification of novel type-II ligands, their binding geometry, and kinase selectivity profiles. The abundance of DFG-in conformations in the structural kinome makes this approach applicable to a wide range of kinases, opening new possibilities for discovery of novel specific kinase-targeting therapeutics in cancer and other diseases.
Methods
Identification of Protein Kinase Domains
Protein kinase domain sequence annotations were taken from SwissProt 45, 46 (release 55.5 of June 2008). The sequences were searched against a non-redundant subset of PDB sequences with common protein tags removed. The identified kinase domain structures were clustered to 95% sequence identity. The procedure yielded 122 mammalian kinases with available X-ray 3D information, (856 PDB entries, 1,216 structures when distinct chains within a single PDB entry are counted separately).
Automated Conformational Classification of the Entire Structural Kinome
Each kinase domain was superimposed onto a template DFG-in structure of ABL1 kinase (PDB ID 2gqg, chain A) using only backbone heavy atoms in the 5A vicinity of the imatinib binding site, with activation loop excluded. Residue matching for the superimposition was established from a sequence alignment. Superimposition algorithm iteratively optimized a weighted RMSD with lower weights assigned to the minority of the most deviating atoms.
DFG-in / DFG-out classification of the superimposed structure was performed based on the position and the orientation of the middle residue in its DFG motif. Orientation of the residue was determined as the sum of cosines of angles between the four covalent bonds formed by its Cα, Cβ, Cγ , Cδ1,2 atoms and the corresponding bonds in the template structure. The resulting “Phe orientation index” ranged from 0 to 4, with larger values indicating similar orientations. Position of the residue was determined as the distance between its Cα atom and the Phe382 Cα of template structure. The so-called DFG-in score (SDFG-in) was calculated for each kinase domain as follows:
Supp. Figure 3 presents the histogram of distribution of DFG-in scores for all X-ray structures of kinase domain in PDB, and examples of structures with different values of the DFG-in score.
For the purpose of this study, a kinase domain structure was classified as DFG-in if its DFG-in score was below 3. It was classified as DFG-out if no more than 1 heavy atom was present in the 2 A vicinity of the DFG phenyl ring of the template structure after superimposition. The structures in which the DFG motif was disordered or overlapped with the template DFG with SDFG-in ≥ 3 were considered intermediate (neither DFG-in nor DFG-out).
A small molecule ligand was classified as a type-II ligand if it had mode than 4 atoms in the 2A vicinity of the DFG phenyl ring of the template after kinase domain superimposition. The corresponding kinase domain structure was called a type-II-bound structure.
Benchmark for Ligand Binding Geometry Prediction by DOLPHIN Models
The DOLPHIN docking set consisted of all kinases for which both DFG-in and type-II-bound X-ray structures were found in PDB release of March 2008. It included 41 DFG-in structures of six kinases and 20 crystallographic type-II inhibitors (Table 1, Supp. Table 2). Each ligand was found in a co-crystal with one kinase, except imatinib co-crystallized with ABL1, SRC, KIT, and LCK. This produced the total of 23 kinase/inhibitor pairs and 184 DFG-in structure/inhibitor docking pairs.
Benchmark for Testing DOLPHIN Model Screening and Selectivity Properties
A comprehensive set of crystallographic kinase inhibitors was collected from PDB release of March 2008 (391 compounds). Among them, 28 compounds (Supp. Table 1, Supp. Figures 1 and 2) have been co-crystallized in the type-II binding mode with one or more kinases. Each of these compounds was classified as a binder to a kinase if it had been found in a co-crystal with that kinase in PDB, or if one of experimentally determined IC50, Ki, Kd was below 10 μM; otherwise it was considered a non-binder. The type-II binders made the positive part of the set for the kinase; all other compounds, including ATP-site inhibitors and type-II ligands with no available activity data, were considered negative. The positive parts of the set for ABL1, BRAF1, KIT, LCK, MK14, and SRC consisted of 8, 3, 5, 8, 14, and 3 type-II compounds, respectively.
DOLPHIN Model Preparation from DFG-in Kinase Structures
Protocol of DOLPHIN model preparation consisted of the two fully automatic steps: (i) removal of all atoms of DFG Phe and the next four residues in the sequence; (ii) generation of the pharmacophore-like field from side-chain atoms of DFG Phe and backbone atoms of the following residues, with DFG Gly skipped (Figure 2(a)). The pharmacophore-like density was generated in the spirit of Atomic Property Fields 47, using a single property (lipophilicity). The density slightly rewarded ligand docking poses occupying the hydrophobic “selectivity” pocket. No ligand-related information was employed by the algorithm.
ICM Grid Docking
ICM molecular modeling software 48, 49 was used for ligand docking and scoring. ICM ligand docking is based on biased probability Monte Carlo optimization of the ligand internal coordinates in the set of grid potential maps of the receptor. A diverse set of conformers was first generated from PDB ligand coordinates by ligand sampling in vacuo. Each conformer was locally minimized with relaxed bond lengths and bond angles using the MMFF-94 force field in order to remove any bias towards receptor-bound covalent geometry. The generated conformers were then placed into the binding pocket in four principal orientations and used as starting points for Monte Carlo optimization. The optimized energy function included the ligand internal strain and a weighted sum of the grid map values in ligand atom centers. The attractive DOLPHIN density map was added to the standard set of ICM receptor maps. The number of sampling steps was limited to 50,000 per ligand per receptor (20-40 seconds on Intel® Core™ 2 Duo E6600 @ 2.40 GHz).
ICM Full-Atom Ligand-Receptor Complex Refinement and Scoring
The top-scoring ligand poses were merged with their DOLPHIN receptors to obtain full-atom models of the complexes. Each complex was refined by local gradient minimization of the ligand and surrounding pocket side-chains and global Monte Carlo optimization of rotatable hydrogens. During the refinement, the ligand heavy atoms were tethered to their docking positions with a harmonic restraint whose weight was iteratively decreased.
The complexes were evaluated with full-atom ICM ligand binding score 26 that has been previously derived from a multi-receptor screening benchmark as a compromise between approximated Gibbs free energy of binding and numerical errors. The score was calculated by:
where Evw, Eel, Ehb, Ehp, and Esf are Van der Waals, electrostatic, hydrogen bonding, non-polar and polar atom solvation energy differences between bound and unbound states, Eint is the ligand internal strain, ΔSTor is its conformational entropy loss upon binding, T = 300 K, and αi are ligand- and receptor-independent constants. The score was artificially raised for irrelevant ligand poses outside the hydrophobic/selectivity pocket, i.e. more than 2 Å from the sidechain of the removed DFG Phe or more than 5 Å from the Cβ atom of either conserved Lys or the gatekeeper.
Virtual Ligand Screening
The 391 crystallographic kinase ligands were docked into each DOLPHIN model. Top three poses per ligand were refined and rescored. An ordered ligand hit list was built from the best scoring ligand poses. The ROC-curves were obtained by plotting the number of top compounds in the list against the number of correct type-II ligands among them.
Determination of Kinase-Specific Binding Energy Offsets and Compound Profiling
The observed Gibbs energy of binding of a ligand to a kinase, ΔGbobs, was calculated as RT ln k, T = 300 K. Where available, we used the binding constant Kd for k; in all other cases, IC50 or Ki values were used. We assumed a linear relation between the experimental ΔGbobs and the calculated binding score:
where m is a universal constant and b is a kinase-dependent binding energy offset. The set of 53 kinase-inhibitor pairs with available experimental binding data was described by a vector Sbind of the calculated binding scores and six kinase characterization vectors, σj (j ∈ {ABL1, BRAF1, KIT, LCK, MK14, SRC}, σj = 1 for all data points with j as the target kinase, 0 for the rest). Partial Least Squares regression was trained to predict experimental ΔGbobs as the function of Sbind and σj:
The obtained values of bj gave the kinase-specific binding energy offsets. They were used to predict the binding energy for each kinase-inhibitor pair as ΔGbpred = m × Sbind + bj, where bj is the offset for the corresponding kinase.
Supplementary Material
Acknowledgements
Authors thank Ketan S. Gajiwala, Pfizer, and Mazen W. Karaman, Ambit, for valuable discussions. We would also like to thank Michelle Potashman, Amgen, Tomoko Niwa, Nippon Shinyaku, and Masaichi Hasegawa, GlaxoSmithKline, for providing activity data on PDB Het ID’s 276, 406, and GIG, respectively. All molecular images were prepared using ICM (http://www.molsoft.com/icm/). This work was supported by NIH grant 1-R01-GM074832-01A1.
Abbreviations
- AUC
Area Under Curve
- DOLPHIN
Deletion-Of-Loop asp-PHe-gly-IN models
- DFG
conserved Asp-Phe-Gly motif
- ICM
Internal Coordinate Mechanics
- HTS
High Throughput Screening
- MRC
Multiple Receptor Conformations
- QSAR
Quantitative Structure Activity Relationship
- RMSD
Root Mean Square Deviation
- ROC
Receiver Operating Characteristic
- VLS
Virtual Ligand Screening
Footnotes
Supporting Information Available: Compound structures and experimental activities; DFG-in score distribution in the publicly available structural kinome; Benchmark for DOLPHIN ligand binding geometry prediction; Derivation of kinase-specific binding energy offsets. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 2.Cohen P. Protein kinases - the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1(4):309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 3.Bogoyevitch MA, Fairlie DP. A new paradigm for protein kinase inhibition: blocking phosphorylation without directly targeting ATP binding. Drug Discovery Today. 2007;12(1516):622–633. doi: 10.1016/j.drudis.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural Mechanism for STI-571 Inhibition of Abelson Tyrosine Kinase. Science. 2000;289(5486):1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 5.Lowinger TB, Riedl B, Dumas J, Smith RA. Design and discovery of small molecules targeting raf-1 kinase. Curr Pharm Des. 2002;8(25):2269–78. doi: 10.2174/1381612023393125. [DOI] [PubMed] [Google Scholar]
- 6.Cowan-Jacob SW, Fendrich G, Floersheimer A, Furet P, Liebetanz J, Rummel G, Rheinberger P, Centeleghe M, Fabbro D, Manley PW. Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukaemia. Acta Crystallographica Section D. 2007;63(1):80–93. doi: 10.1107/S0907444906047287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, Kuriyan J. Crystal Structures of the Kinase Domain of c-Abl in Complex with the Small Molecule Inhibitors PD173955 and Imatinib (STI-571) Cancer Res. 2002;62(15):4236–4243. [PubMed] [Google Scholar]
- 8.Weisberg E, Manley PW, Breitenstein W, Brüggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C, Mohammed A, Neuberg D, Wright RD, Gilliland DG, Griffin JD. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7(2):129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N, Pav S, Regan J. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Mol Biol. 2002;9(4):268–272. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- 10.Hasegawa M, Nishigaki N, Washio Y, Kano K, Harris PA, Sato H, Mori I, West RI, Shibahara M, Toyoda H, Wang L, Nolte RT, Veal JM, Cheung M. Discovery of Novel Benzimidazoles as Potent Inhibitors of TIE-2 and VEGFR-2 Tyrosine Kinase Receptors. J. Med. Chem. 2007;50(18):4453–4470. doi: 10.1021/jm0611051. [DOI] [PubMed] [Google Scholar]
- 11.Hodous BL, Geuns-Meyer SD, Hughes PE, Albrecht BK, Bellon S, Caenepeel S, Cee VJ, Chaffee SC, Emery M, Fretland J, Gallant P, Gu Y, Johnson RE, Kim JL, Long AM, Morrison M, Olivieri PR, Patel VF, Polverino A, Rose P, Wang L, Zhao H. Synthesis, structural analysis, and SAR studies of triazine derivatives as potent, selective Tie-2 inhibitors. Bioorganic & Medicinal Chemistry Letters. 2007;17(10):2886–2889. doi: 10.1016/j.bmcl.2007.02.067. [DOI] [PubMed] [Google Scholar]
- 12.Cai Z-W, Wei D, Schroeder GM, Cornelius LAM, Kim K, Chen X-T, Schmidt RJ, Williams DK, Tokarski JS, An Y, Sack JS, Manne V, Kamath A, Zhang Y, Marathe P, Hunt JT, Lombardo LJ, Fargnoli J, Borzilleri RM. Discovery of orally active pyrrolopyridine- and aminopyridine-based Met kinase inhibitors. Bioorganic & Medicinal Chemistry Letters. 2008;18(11):3224–3229. doi: 10.1016/j.bmcl.2008.04.047. [DOI] [PubMed] [Google Scholar]
- 13.Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias J-M, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23(3):329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 14.Nordin H, Jungnelius M, Karlsson R, Karlsson OP. Kinetic studies of small molecule interactions with protein kinases using biosensor technology. Anal Biochem. 2005;340(2):359–368. doi: 10.1016/j.ab.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 15.Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW. Functional Interrogation of the Kinome Using Nucleotide Acyl Phosphates. Biochemistry. 2007;46(2):350–358. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- 16.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, Drewes G. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotech. 2007;25(9):1035–1044. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 17.Mayhood TW, Windsor WT. Ligand binding affinity determined by temperature-dependent circular dichroism: cyclin-dependent kinase 2 inhibitors. Anal Biochem. 2005;345(2):187–197. doi: 10.1016/j.ab.2005.07.032. [DOI] [PubMed] [Google Scholar]
- 18.Vedadi M, Niesen FH, Allali-Hassani A, Fedorov OY, Finerty PJ, Wasney GA, Yeung R, Arrowsmith C, Ball LJ, Berglund H, Hui R, Marsden BD, Nordlund P, Sundstrom M, Weigelt J, Edwards AM. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci U S A. 2006;103(43):15835–15840. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson PA, Wang S, Howett LJ, Wang M-M, Patel R, Averill A, Showalter RE, Li B, Appleman JR. Identification of ligand binding by protein stabilization: comparison of ATLAS with biophysical and enzymatic methods. Assay Drug Dev Technol. 2008;6(1):69–81. doi: 10.1089/adt.2007.100. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2(7):358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 21.Okram B, Nagle A, Adrian FJ, Lee C, Ren P, Wang X, Sim T, Xie Y, Wang X, Xia G, Spraggon G, Warmuth M, Liu Y, Gray NS. A General Strategy for Creating “Inactive-Conformation” Abl Inhibitors. Chemistry & Biology. 2006;13(7):779–786. doi: 10.1016/j.chembiol.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 22.Totrov M, Abagyan R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins: Structure, Function, and Genetics. 1997;29(S1):215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 23.Chen H, Lyne PD, Giordanetto F, Lovell T, Li J. On Evaluating Molecular-Docking Methods for Pose Prediction and Enrichment Factors. J. Chem. Inf. Model. 2006;46(1):401–415. doi: 10.1021/ci0503255. [DOI] [PubMed] [Google Scholar]
- 24.Lovell T, Chen H, Lyne PD, Giordanetto F, Li J. On Evaluating Molecular-Docking Methods for Pose Prediction and Enrichment Factors. [J. Chem. Inf. Model. 46, 401-415 (2006)] by. J. Chem. Inf. Model. 2008;48(1):246–246. doi: 10.1021/ci0503255. [DOI] [PubMed] [Google Scholar]
- 25.Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR. Towards the development of universal, fast and highly accurate docking//scoring methods: a long way to go. Br J Pharmacol. 2007;153(S1):S7–S26. doi: 10.1038/sj.bjp.0707515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schapira M, Totrov M, Abagyan R. Prediction of the binding energy for small molecules, peptides and proteins. Journal of Molecular Recognition. 1999;12(3):177–190. doi: 10.1002/(SICI)1099-1352(199905/06)12:3<177::AID-JMR451>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 27.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucl. Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seeliger MA, Nagar B, Frank F, Cao X, Henderson MN, Kuriyan J. c-Src Binds to the Cancer Drug Imatinib with an Inactive Abl/c-Kit Conformation and a Distributed Thermodynamic Penalty. Structure. 2007;15(3):299–311. doi: 10.1016/j.str.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan JE, Holdgate GA, Campbell D, Timms D, Gerhardt S, Breed J, Breeze AL, Bermingham A, Pauptit RA, Norman RA, Embrey KJ, Read J, VanScyoc WS, Ward WHJ. Prevention of MKK6-Dependent Activation by Binding to p38a MAP Kinase. Biochemistry. 2005;44(50):16475–16490. doi: 10.1021/bi051714v. [DOI] [PubMed] [Google Scholar]
- 30.Kleywegt GJ, Harris MR, Zou JY, Taylor TC, Wahlby A, Jones TA. The Uppsala Electron-Density Server. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2240–2249. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
- 31.Horio T, Hamasaki T, Inoue T, Wakayama T, Itou S, Naito H, Asaki T, Hayase H, Niwa T. Structural factors contributing to the Abl/Lyn dual inhibitory activity of 3-substituted benzamide derivatives. Bioorganic & Medicinal Chemistry Letters. 2007;17(10):2712–2717. doi: 10.1016/j.bmcl.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 32.Kimura S, Naito H, Segawa H, Kuroda J, Yuasa T, Sato K, Yokota A, Kamitsuji Y, Kawata E, Ashihara E, Nakaya Y, Naruoka H, Wakayama T, Nasu K, Asaki T, Niwa T, Hirabayashi K, Maekawa T. NS-187, a potent and selective dual Bcr-Abl/Lyn tyrosine kinase inhibitor, is a novel agent for imatinib-resistant leukemia. Blood. 2005;106(12):3948–3954. doi: 10.1182/blood-2005-06-2209. [DOI] [PubMed] [Google Scholar]
- 33.Potashman MH, Bready J, Coxon A, DeMelfi TM, DiPietro L, Doerr N, Elbaum D, Estrada J, Gallant P, Germain J, Gu Y, Harmange J-C, Kaufman SA, Kendall R, Kim JL, Kumar GN, Long AM, Neervannan S, Patel VF, Polverino A, Rose P, van der Plas S, Whittington D, Zanon R, Zhao H. Design, Synthesis, and Evaluation of Orally Active Benzimidazoles and Benzoxazoles as Vascular Endothelial Growth Factor-2 Receptor Tyrosine Kinase Inhibitors. J. Med. Chem. 2008;51(3):699–699. doi: 10.1021/jm070034i. [DOI] [PubMed] [Google Scholar]
- 34.Ogawa A, Takayama Y, Sakai H, Chong KT, Takeuchi S, Nakagawa A, Nada S, Okada M, Tsukihara T. Structure of the Carboxyl-terminal Src Kinase, Csk. J. Biol. Chem. 2002;277(17):14351–14354. doi: 10.1074/jbc.C200086200. [DOI] [PubMed] [Google Scholar]
- 35.O’Hare T, Druker BJ. BIRB-796 is not an effective ABL(T315I) inhibitor. Nat Biotech. 2005;23(10):1209–1210. doi: 10.1038/nbt1005-1209. [DOI] [PubMed] [Google Scholar]
- 36.Knight ZA, Shokat KM. Features of Selective Kinase Inhibitors. Chemistry & Biology. 2005;12(6):621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 37.Hu E, Tasker A, White RD, Kunz RK, Human J, Chen N, xfc, rli R, Hungate R, Novak P, Itano A, Zhang X, Yu V, Nguyen Y, Tudor Y, Plant M, Flynn S, Xu Y, Meagher KL, Whittington DA, Ng GY. Discovery of Aryl Aminoquinazoline Pyridones as Potent, Selective, and Orally Efficacious Inhibitors of Receptor Tyrosine Kinase c-Kit. J. Med. Chem. 2008;51(11):3065–3068. doi: 10.1021/jm800188g. [DOI] [PubMed] [Google Scholar]
- 38.Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 2008 doi: 10.1038/nsmb.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotech. 2008;26(1):127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 40.Fedorov O, Marsden B, Pogacic V, Rellos P, Muller S, Bullock AN, Schwaller J, Sundstrom M, Knapp S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc Natl Acad Sci U S A. 2007;104(51):20523–20528. doi: 10.1073/pnas.0708800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahmad T, Eisen T. Kinase Inhibition with BAY 43-9006 in Renal Cell Carcinoma. Clin Cancer Res. 2004;10(18):6388S–6392. doi: 10.1158/1078-0432.CCR-040028. [DOI] [PubMed] [Google Scholar]
- 42.Dai Y, Hartandi K, Ji Z, Ahmed AA, Albert DH, Bauch JL, Bouska JJ, Bousquet PF, Cunha GA, Glaser KB, Harris CM, Hickman D, Guo J, Li J, Marcotte PA, Marsh KC, Moskey MD, Martin RL, Olson AM, Osterling DJ, Pease LJ, Soni NB, Stewart KD, Stoll VS, Tapang P, Reuter DR, Davidsen SK, Michaelides MR. Discovery of N-(4-(3-amino-1H-indazol-4-yl)phenyl)-N’-(2-fluoro-5-methylphenyl)urea (ABT-869), a 3-aminoindazole-based orally active multitargeted receptor tyrosine kinase inhibitor. J Med Chem. 2007;50(7):1584–1597. doi: 10.1021/jm061280h. [DOI] [PubMed] [Google Scholar]
- 43.Weisberg E, Roesel J, Bold G, Furet P, Jiang J, Cools J, Wright RD, Nelson E, Barrett R, Ray A, Moreno D, Hall-Meyers E, Stone R, Galinsky I, Fox E, Gilliland G, Daley JF, Lazo-Kallanian S, Kung AL, Griffin JD. Anti-leukemic effects of the novel, mutant FLT3 inhibitor, NVP-AST487: Effects on PKC412-sensitive and -resistant FLT3-expressing cells. Blood. 2008 doi: 10.1182/blood-2008-02-138065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akeno-Stuart N, Croyle M, Knauf JA, Malaguarnera R, Vitagliano D, Santoro M, Stephan C, Grosios K, Wartmann M, Cozens R, Caravatti G, Fabbro D, Lane HA, Fagin JA. The RET kinase inhibitor NVP-AST487 blocks growth and calcitonin gene expression through distinct mechanisms in medullary thyroid cancer cells. Cancer Res. 2007;67(14):6956–6964. doi: 10.1158/0008-5472.CAN-06-4605. [DOI] [PubMed] [Google Scholar]
- 45.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh L-SL. The Universal Protein Resource (UniProt) Nucl. Acids Res. 2005;33(suppl1):D154–159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boeckmann B, Blatter M-C, Famiglietti L, Hinz U, Lane L, Roechert B, Bairoch A. Protein variety and functional diversity: Swiss-Prot annotation in its biological context. Comptes Rendus Biologies. 2005;328(1011):882–899. doi: 10.1016/j.crvi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 47.Totrov M. Atomic Property Fields: Generalized 3D Pharmacophoric Potential for Automated Ligand Superposition, Pharmacophore Elucidation and 3D QSAR. Chemical Biology & Drug Design. 2008;71(1):15–27. doi: 10.1111/j.1747-0285.2007.00605.x. [DOI] [PubMed] [Google Scholar]
- 48.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235(3):983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 49.Abagyan R, Totrov M, Kuznetsov DA. ICM: A New Method for Protein Modeling and Design: Applications to Docking and Structure Prediction from the Distorted Native Conformation. J Comp Chem. 1994;15:488–506. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.