

Abstract
Background
c-Met and EGFR receptors are widely expressed on cancer cells; they are implicated in the development and progression of cancer through a plethora of effects on cell cycle progression, apoptosis, motility and metastasis and are potential targets for combination therapy. EGFR receptor tyrosine kinases are currently being targeted in a number of malignancies.
Methods
Apoptosis was studied by FACS analysis using propidium iodide. EGF and HGF signaling intermediates were studied by western blotting. Cell proliferation was determined by MTT assays. Cell motility was done by time lapse confocal microscopy.
Results
c-Met and EGFR were both expressed in A549, H1838, H2170, SW900, SW1573, H358, SKLU-1, and H1993 non small cell lung cancer (NSCLC) cell lines. Both EGF and HGF at 100 ng/ml in medium showed a synergistic effect on cell proliferation at 48–72 h as seen by a proliferation assay in A549, H1838, and SKMES cells. In A549 and H1838 cell lines, HGF (40 ng/ml) and EGF (5 ng/ml) induced synergistic phosphorylation on c-Met (Tyr 1003/1230/1234/1235). Additionally, synergistic phosphorylation of Akt (Ser-473) and phospho-ERK1+ERK2 (Thr202/Tyr204) was also seen indicating that EGF and HGF could induce synergistic phosphorylation of important signaling intermediates. Treatment with EGF and HGF at 100 ng/ml for 2 h also leads to an additive effect in inducing cell motility (especially membrane ruffling) in H1993 cells. A novel c-Met small molecule tyrosine kinase inhibitor SU11274 and EGFR tyrosine kinase inhibitors Tyrphostin AG1478 and gefitinib (Iressa) were tested to study their effect in combination on proliferation and apoptosis in lung cancer cells. Interestingly, a synergistic effect on inhibition of cell proliferation was seen in the presence of SU11274 and Tyrphostin AG1478. 0.5 µM Tyrphostin AG1478 and 2 µM SU11274 inhibited growth by 21% and 25%, respectively; a combination of both tyrosine kinase inhibitors inhibited growth by 65%. Interestingly, EGFR inhibitor (gefitinib, Iressa) and c-Met inhibitor (SU11274) also had a synergistic effect on apoptosis in H358 cells.
Conclusion
There was a synergistic effect of EGF and HGF on proliferation, downstream activation of signal transduction and an additive effect seen on motility. These studies show that a combination of HGF and EGF tyrosine kinase inhibitors on NSCLC, could potentially be targeted in a synergistic fashion.
Keywords: c-Met inhibitor SU11274, cross-talk, EGF, lung cancer, signal transduction via HGF
Introduction
NSCLC is a devastating malignancy for which current therapies do not offer satisfactory results. It is imperative to develop novel therapeutic targets in order to achieve better outcomes for this disease. The molecular targeting of RTKs has emerged as a promising approach to the treatment of NSCLC. c-Met and EGFR are known to be expressed and also play a very important role in the progression of NSCLC.[1,2] EGFR, in particular, has been extensively investigated, and its inhibitors have undergone clinical trials with limited success.[3] We have previously shown that the c-Met pathway plays a crucial role in the progression of SCLC,[4,5] NSCLC, and other invasive tumors.[6]
The EGFR protein is a 170-kD transmembrane glycoprotein, consisting of an extracellular ligand binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain.[7] The receptor is part of a family of human epidermal growth factor receptors, which has four members (HER1/erbB1, HER2/erbB2, HER3/erbB3, and HER4/erbB4). EGFR has six members, currently known endogenous ligands: EGF, amphiregulin, TGF-α, betacellulin, heparin binding EGF, and epiregulin.[8] When the ligand binds the receptor, the receptor undergoes homodimerization, or heterodimerization (commonly with HER2), and causes autophosphorylation of the tyrosine kinase domain and activation of critical signal transduction events, ultimately leading to a biological response. The EGFR inhibitor, gefitinib has been used as a single agent in NSCLC, but overall, the efficacy has been modest at best. However, there are a select group of patients that respond very dramatically to this agent, and very recently, these responses were correlated to the presence of activating mutations in the tyrosine kinase domain of EGFR in these tumors.[9,10]
c-Met is also highly overexpressed in 60%–80% of NSCLC and overexpressed in many solid tumors.[11] The c-Met receptor is a disulfide linked α-β heterodimer with a molecular weight of 190 kD.[12] The 140-kD β chain spans the membrane and possesses cytoplasmic tyrosine kinase activity and can be detected in its precursor form at 170 kD. Unlike EGFR, the only known natural ligand for c-Met is HGF (also known as scatter factor).[13–15] HGF stimulation of c-Met can lead to proliferation, increased survival, altered motility, enhanced invasion into extracellular matrix, and more rapid formation of tubules.[15] c-Met overexpression as well as activating mutations in the various domains can lead to carcinogenesis in multiple tumors. There are multiple activating mutations in the c-Met gene identified in hereditary papillary renal carcinomas.[16] On activation by autophosphorylation, c-Met can associate with and activate multiple signal transduction intermediates.[15] Novel small molecule inhibitors of c-Met, SU11274,[17] and PHA-665752[18] have been utilized in an in vitro setting and have shown to inhibit the phosphorylation of c-Met and also inhibit the proliferation of cell lines.
c-Met co-immunoprecipitates with EGFR in protein extracts from tumor cells but not in extracts from normal hepatocytes, suggesting a new tumor-specific cross-talk for the activation of HGF/MET signaling by the TGFα/EGFR axis.[19] Recently, several papers suggest an interaction between HGF/MET and EGFR signaling pathways. In a glioma cell line, c-Met activation results in a wave of transcription-dependent EGFR activation, which contributes to HGF-induced cell proliferation.[20] In mammary carcinoma cells, EGFR inhibition significantly blocked HGF activation of c-Met and EGFR and inhibition of these pathways mitigated HGF-induced proliferation and motility.[21] HGF can induce transactivation of EGFR in corneal epithelial cells through amphiregulin and heparin-binding epidermal growth factor-like growth factor, and this is a prerequisite for induction of full motility.[22] It has also been found that cross-talk between EGFR and c-Met may play a key role in regulating retinal pigment epithelium cell migration, proliferation, and wound healing.[23] In addition, a recent study showed that c-Met amplification leads to gefitinib secondary resistance and could be an explanation for this resistance in some patients. It was found in NSCLC that amplification of c-Met causes gefitinib resistance by driving ERBB3 (HER3)-dependent activation of PI3K, a pathway thought to be specific to EGFR/ERBB family receptors.[24] Recently, second-site point mutations (T790M) associated with 50% of the cases with acquired resistance to EGFR tyrosine kinase inhibitors have been found in lung adenocarcinomas harboring EGFR mutations in exons encoding the tyrosine kinase domain. It has also been found that c-Met amplification occurs independent of EGFRT790M mutations and that c-Met may be a clinically relevant therapeutic target for some patients with acquired resistance to gefitinib or erlotinib.[25] Rikova et al. have suggested using a global survey of phosphotyrosine signaling based on activated kinases identified that in a given tumor, there are opportunities to therapeutically intervene using multiple kinase inhibitors.[26]
Our rationale behind the investigation of the interaction between c-Met and EGFR are multifold. Both receptors are overexpressed in NSCLC and both have been implicated in cell motility, generation of reactive oxygen species,[5,27] angiogenesis[4,28] and several other critical biological phenomena. Since there are many common signaling pathways between both receptors, we sought to determine if EGF and HGF could synergistically/cooperatively lead to increased cellular proliferation, motility, as well as downstream signaling. EGFR inhibitors have been used and continue to be used clinically, and have shown only a modest benefit in the treatment of NSCLC.[3] With the advent of a novel small molecule inhibitor of c-Met, SU11274, we thought that in respect to the interactions between EGFR and c-Met, it would be crucial to demonstrate a synergistic inhibition of cell growth and apoptosis utilizing the respective inhibitors. Our demonstration of the functional and biochemical interaction between c-Met and EGFR and the synergism between their inhibitors will rationalize the future combination of these drugs in the management of NSCLC.
Materials and Methods
Reagents and antibodies: SU11274: [(3Z)-N-(3-chlorophenyl)-3-({3,5-dimethyl-4-[(4-methylpiperazin-1-yl)carbonyl]-1H-pyrrol-2-yl}methylene)-N-methyl-2-oxo-2,3-dihydro-1H-indole-5-sulfonamide] and gefitinib were obtained from American Customs Chemical Corporation (San Diego, CA). Tyrphostin AG1478 was obtained from Sigma-Aldrich (St. Louis, MO). All inhibitors were suspended in DMSO and kept in small aliquots at −20°C. HGF was obtained from EMD chemical (Gibbstown, NJ), and EGF was obtained from Invitrogen (Carlsbad, CA). Phosphospecific antibodies for pS473 on AKT were obtained from Cell Signaling Technology (Beverly, MA). Total c-Met was obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and β-actin from Sigma-Aldrich (St. Louis, MO). Phospho c-Met (Tyr 1230/1234/1235), phospho c-Met (Tyr 1003), and phospho-ERK1+ERK2 (Thr202/Tyr204) were obtained from Invitrogen (Carlsbad, CA). All antibodies were used as described in the manufacturer's protocol.
Cell lines and apoptosis analysis
All NSCLC cell lines A549, H1838, H2170, SW900, SW1573, H358, SKLU-1, and H1993 were obtained from the American Type Culture Collection (Rockville, MD, USA), and were cultured according to the instructions (http://www.ATCC.org).
Cell lines were incubated at 37°C and 5% CO2. For studying the effect of Iressa (gefitinib) and SU11274 alone and in combination, H358 cells were plated in 6 well plates and treated after 24 h with different concentrations of Iressa and SU11274. After 72 h of treatment, cells were collected by trypsinization, stained with propidium iodide and the sub-G0 DNA content was determined by FACS analysis and analyzed by Cell-Quest software.
Kinetics of HGF- and EGF-induced phosphorylation of c-Met and downstream mediators
Cells were starved overnight in media containing 0.5% BSA (w/v) (Sigma, St. Louis, MO, USA) for 18 h. The cells were then stimulated with 40 ng/ml HGF or 5 ng/ml of EGF or both for various intervals of time and lysed in lysis buffer containing 20 mmol/L Tris (pH, 8.0), 150 mmol/L NaCl, 10% glycerol, 1% NP40, 0.42% NaF, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L Na3 VO4, and 10 µL/mL protease inhibitor cocktail (Sigma-Aldrich) as described previously.[5] Cell lysates were separated by 7.5% or 10% SDS-PAGE under reducing conditions. Proteins were transferred to an immobilization membrane (Bio-Rad Laboratories, Hercules, CA) and immunoblotted using the enhanced chemiluminescence technique (PerkinElmer Life and Analytical Sciences, Torrence, CA). The membrane was then probed with the antibodies against p-Akt (Serine 473), phospho c-Met (Tyr 1230/1234/1235), phospho c-Met (Tyr 1003), phospho-ERK1+ERK2 (Thr202/Tyr204), and β-actin.
MTT assays
To study the synergistic effects of HGF and EGF and the effect of inhibitors of EGFR and c-Met on cell growth, cell viability was measured by a MTT colorimetric dye reduction assay (Sigma, St. Louis, MO) using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT) according to the manufacturer's instructions. Each experiment was done in four replicate wells for each drug concentration. The IC50 value is defined as the concentration needed for a 50% reduction in absorbance calculated from the growth curves. Each data point is representative of three independent experiments, and the standard deviations were calculated accordingly. To study the synergistic effect of HGF and EGF, cells (5000 cells/well) were plated in a 96 -well plate. After 24 h, wells were treated with HGF 100 ng/ml, EGF 100 ng/ml or both in medium with 10% FBS. At given intervals of time, cell viability was measured by adding MTT reagent and solubilization solution, and the plates were read at 570 nm. Percentage of cell viability was determined relative to the control which had no additional growth factors. To determine the inhibitory effects of EGFR inhibitor Tyrphostin AG1478 and c-Met inhibitor, H2170 cells were plated as described above and treated after 24 h with various concentrations of Tyrphostin and SU11274 for 72 h after which an MTT was performed. All drug inhibition studies to study proliferation were performed in media containing 10% FBS.
Motility studies using time lapse video microscopy
For the motility studies, H2170 cells were plated in 35 mm glass bottom dishes (MatTek Co., Ashland, MA) coated with collagen. Cells were allowed 12 h to adhere to the plate and were subsequently starved in serum-free medium containing 0.5% BSA for 24 h. Cells were pre-incubated for 2 h prior to imaging with HGF alone (100 ng/ml), EGF alone (100 ng/ml), and HGF + EGF at the same doses. Images were taken by using a Zeiss time lapse video microscope. Data was analyzed and quantified using Metamorph and Image J (NIH).
Results
EGFR and c-Met expression in NSCLC cell lines
The expression of EGFR and c-Met were determined by immunoblotting of whole cell lysates prepared from cells cultured in regular media. EGFR and c-Met were both expressed at variable levels in the entire group of 8 cell lines tested [Figure 1]. EGFR expression was the highest in H2170, and c-Met expression was the highest in H1993 [Figure 1]. H2170, A549, and H1838 had robust expression of both c-Met and EGFR and were used for several investigations on signal transduction, cell viability, cell proliferation, and cell motility.
Figure 1.
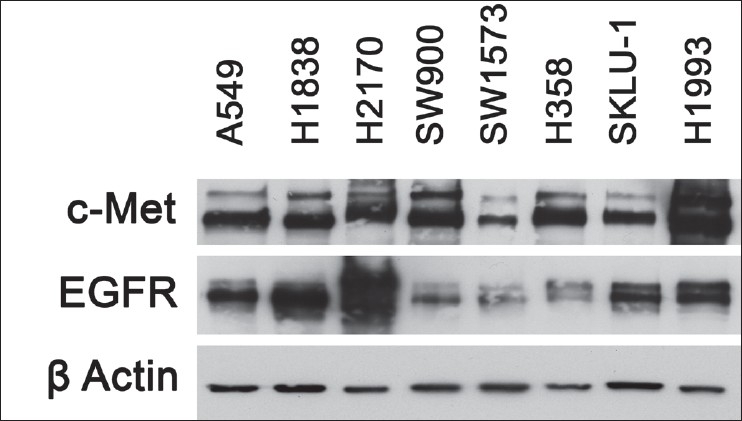
Expression of EGFR and c-Met in NSCLC cell lines. Whole cell lysates were prepared from 8 NSCLC cell lines, grown in media containing 10% FBS. Cell lines include A549, H1838, H2170, SW900, SW1573, H358, SKLU-1, and H1993. Expression of EGFR and c-Met was examined by immunoblotting as described in the Material and Methods section using polyclonal EGFR and c-Met antibodies. The c-Met immunoblot shows an upper 170-kDa protein band which represents the precursor form of the glycosylated c-Met and the lower 145-kDa band which is the biologically active transmembrane β-subunit of c-Met. β-actin was used as an internal loading control
EGF and HGF effect on the growth of NSCLC cell lines
The effect of HGF and EGF on cell growth was studied by treating cells with 100 ng of EGF or HGF either alone or in combination, and cell proliferation was determined by MTT as described in the section Materials and Methods [Figure 2A–C]. In A549, H1838, and SKMES cells, a combination of HGF and EGF had a synergistic effect (90%, 230% and 80%, respectively) [Figure 2A–C]. A synergistic effect can be defined as an effect that is more than the additive effect of HGF and EGF alone. These results indicate that there was a synergistic proliferative effect of both cytokines combined. These results could be due to potential cross-talks of HGF and EGF signaling pathways with each other and with other protumorigenic factors that have been shown to promote tumor cell growth. Functions of distinct signaling pathways such as proliferation can be amplified since cytoplasmic downstream components of most growth factor ligand/receptor complexes (e.g., adaptor proteins, kinases, and transcription factors) are frequently utilized by more than one signaling pathway.[29]
Figure 2.
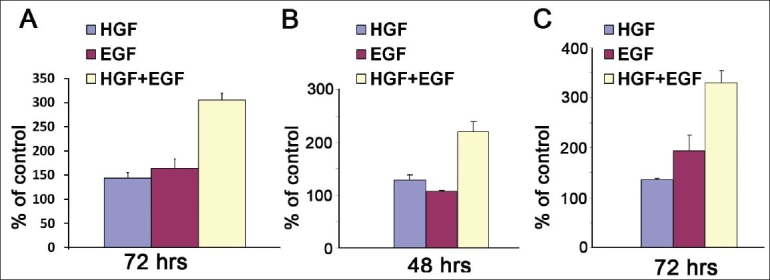
HGF and EGF stimulate growth in a synergistic manner in the NSCLC cell lines. A549 (A), H1838 (B), and SKMES (C) cells (5000/well) were plated in 96-well plates. After 24 h, wells were treated with HGF 100 ng/ml, EGF 100 ng/ml or both in medium with 10% FBS. At given intervals of time, viable cells were detected by MTT assays and cell viability was plotted as a mean ± SD of three independent experiments. Percentage of cell viability was determined relative to the control that had no additional growth factors. HGF and EGF both induced cell proliferation, and a combination of HGF and EGF had a synergistic effect on cell proliferation in A549 (A), H1838 (B), and SKMES (C)
HGF and EGF induce synergistic phosphorylation of c-Met receptor and of downstream signaling intermediates in a synergistic fashion
In the NSCLC cell lines, H1838 and A549, we studied the phosphorylation of HGF receptors and several of their downstream signaling intermediates [Figure 3A–C]. After densiometric analysis using NIH image we determined that HGF and EGF induced synergistic phosphorylation of phospho-c-Met (Tyr 1003) [Figure 3A, B] and phospho-c-Met (Tyr 1230/1234/1235) [Figure 3B]. This synergistic phosphorylation of HGF receptor could occur due to the association of c-Met with EGFR in tumor cells[19] or due to transcription-dependent cross-talk between c-Met and EGFR,[20] which could facilitate synergistic phosphorylation of these growth factor receptors.
Figure 3.
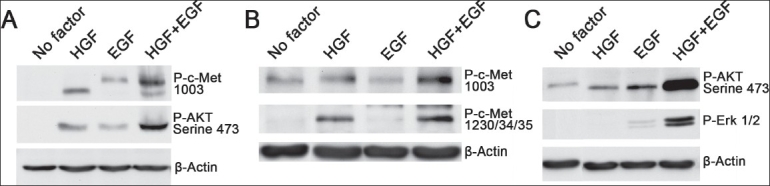
Kinetics of HGF- and EGF-induced phosphorylation, c-Met receptor, and its downstream signaling proteins. A549 and H1838 cells were starved overnight in media containing 0.5% BSA for 18 h and were stimulated with 40 ng/ml HGF or 5 ng/ml of EGF or both for various intervals of time. Cell lysates were prepared and western blotting performed as described in materials and methods section (A). In A549 cells, synergistic phosphorylation of c-Met (Tyr 1003) and downstream signaling protein Akt (Serine 473) was observed at 2.5 min after stimulation with a combination of EGF and HGF (B). In H1838 cells, HGF and EGF together induced synergistic phosphorylation of phospho c-Met (Tyr 1003) and phospho c-Met (Tyr 1230/1234/1235). (C) Synergistic phosphorylation of Akt (Serine 473) and ERK1+ERK2 (Thr202/Tyr204), which are downstream signaling intermediates
Synergistic phosphorylation of Akt (Serine 473) and phospho-ERK1+ERK2 (Thr202/Tyr204)[Figure 3A, C], which are downstream signaling intermediates from growth factor receptors, were also seen by densitometric analysis.
HGF and EGF cooperatively induce increased membrane ruffling
In order to study the changes in the cytoskeleton that occur via the c-Met/HGF axis and the EGFR/EGF axis, we studied cell motility by time lapse video microscopy. The images shown in the lower panel of Figure 4 are displayed in false color. An increase in membrane ruffling is observed with an increase in red color and a decrease is indicated by presence of blue color, as seen in the bar on the side of Figure 4. There was evidence of increased membrane ruffling that occurred when cells were stimulated with HGF and EGF independently and an additive effect when used together [Figure 4].
Figure 4.
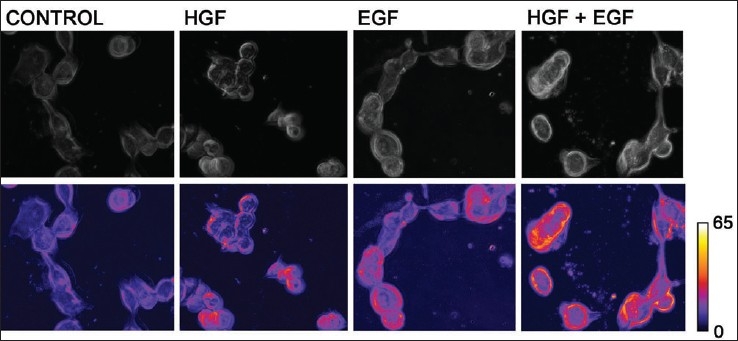
Increased membrane ruffling observed with HGF and EGF. H1993 were plated in glass bottom dishes coated with collagen (150,000 cells/plate). They were grown in serum-free medium containing 0.5% BSA for 24 h after which each plate was pretreated for 2 h with 100 ng/ml EGF, HGF or both. Cells were studied under TLVM for 4 h and photographs were taken at 1-min intervals. Cell movement with changing shape was analyzed using NIH image analysis. Images in the lowerpanel were created with false colors according to the movement and change in shape of cells. An increase in membrane ruffling was seen by an increase in red color, and a decrease is indicated by presence of blue color as observed in the bar on the side of Figure 4. Increased membrane ruffling was seen when cells were incubated with HGF or EGF and an additive effect was observed by a combination of both
c-Met kinase inhibitor SU11274 and EGFR kinase inhibitors Tyrphostin AG1478 or gefitinib synergize to inhibit proliferation of NCI-H2170 NSCLC cell lines
Due to the common signaling pathways that are regulated by HGF and EGF, we were interested in studying if the inhibitors of these RTKs, Tyrphostin AG1478, and SU11274 could potentially be synergistic in their ability to inhibit proliferation of NSCLC cells. The IC50 for each drug was first determined by MTT assays (data not shown) and was determined to be 4 µM for SU11274 and 1 µM for Tyrphostin AG1478. Both drugs were used independently and simultaneously in the same dosing. Interestingly, a synergistic effect on inhibition of cell proliferation was seen in the presence of SU11274 and Tyrphostin. 0.5 µM Tyrphostin and 2 µM SU11274 inhibited growth by 21.5% and 25.5%, respectively; a combination of both tyrosine kinase inhibitors inhibited growth by 65.2%. There was no indication of antagonism with any of the doses utilized.
SU11274 and gefitinib induce apoptosis in NSCLC cell line H358 in a synergistic fashion
Growth factor signal pathways are one of the main regulators of cell proliferation and apoptosis, and hence, we studied the effect of a combination of an EGFR inhibitor (gefitinib) and c-Met inhibitor (SU11274) on apoptosis in H358 cells. The cells were treated as described earlier with Iressa and SU11274 alone and in combination after which cells were stained with propidium iodide and the sub G0 DNA content was determined by FACS analysis. As shown in Figure 5A and 5B, Iressa and SU11274 have a synergistic effect on apoptosis.
Figure 5.
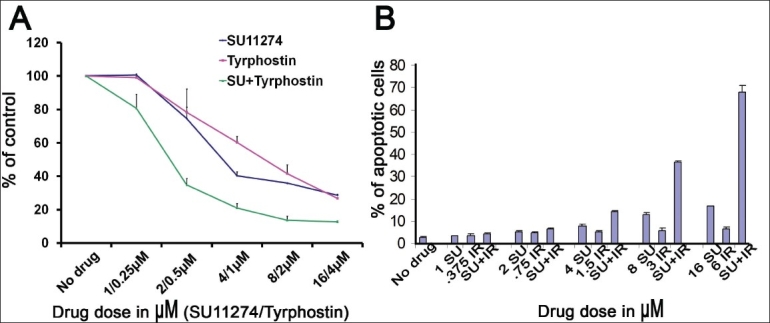
SU11274 and Tyrphostin AG1478 inhibit growth in a synergistic manner in H2170. SU11274 and gefitinib induce apoptosis in a synergistic manner in H358 cells. (A) Intially, studies were performed on a combination of drugs and the IC50for SU11274 and Tyrphostin AG1478 for the cell line H2170 was determined; further, the drugs were diluted in medium with 10% FBS. H2170 cells (5000 cells/well) were plated in a 96-well plate, and after 24 h, wells were treated with varying concentrations of tyrphostin, SU11274 or both in medium with 10% FBS. At 72 h, cell viability was performed by an MTT assay. Percentage of viable cells were determined relative to the control which had no additional growth factors. As shown in (A), synergistic effect on inhibition of cell proliferation was observed with 0.5 µM Tyrphostin and 2 µM SU11274. (B) H358 cells were plated in 6-well plates and treated after 24 h with various concentrations of Iressa and SU11274 as indicated. After 72 h of treatment, cells were collected by trypsinization, stained with propidium iodide, and the sub-G0 DNA content was determined by FACS analysis and analyzed by Cell-Quest software. As shown in Figure 5B, gefitinib and SU11274 have a synergistic effect on apoptosis
Discussion
Receptor tyrosine kinase (RTK) inhibition is becoming very important in therapy for cancer. We identify here that both EGFR and c-Met RTKs were expressed in lung cancer, and they can cooperate to enhance cell biological functions such as proliferation, cell motility, and downstream signal transduction (such as the Akt/S6 kinase, ERK1/2, and focal adhesion protein pathways). Importantly, inhibition of both the receptors leads to enhanced growth inhibition and apoptosis.
c-Met RTK has been shown to play an important role in the pathogenesis and progression of a number of solid tumors.[30] It also has potent angiogenic properties.[31] Previously, we have shown that c-Met can be overexpressed, activated (as measured by phosphorylation of the catalytic domain as well as juxtamembrane domain), mutated (in the semaphorin or juxtamembrane domains), and/or amplified in lung cancers.[32,33] Also, inhibition of c-Met with siRNA or small molecule inhibitors leads to decreased cell growth and apoptosis of lung cancer cells.[33] We have previously shown that there also can be synergism of c-Met inhibition with mTOR inhibition (with rapamycin) in activated c-Met cells, including lung cancer.[34] Importantly, we show here that inhibition of c-Met is synergistic with EGFR inhibition. This was reflected by the effects on cell growth as well as apoptosis. EGFR plays a crucial role in the progression of lung cancer, and therapeutic inhibition has come to clinical fruition with agents such as erlotinib, gefitinib, and cetuximab.[35,36]
EGFR can be targeted in a number of tumors, especially in lung cancer.[35,36] There have been somatic mutations of EGFR in the tyrosine kinase domain reported and amplification of the gene; these have been linked to therapeutic response to small molecule inhibitors.[9,10] An emerging theme has also been resistance to EGFR inhibition and the necessity of other combinational strategies. With the data presented here, the combination of EGFR and c-Met inhibition would be a useful strategy to pursue in clinical trials. Also, it would be interesting to determine how this would relate to combination with cytotoxic chemotherapies such as cisplatin, taxanes, and pemetrexed (all effective therapies in lung cancer).
Since EGFR can be mutated in the tyrosine kinase domain[9,10] and c-Met in the non-tyrosine kinase domains in NSCLC,[33] it would be important to study the biological/biochemical effects and therapeutic inhibition of these mutations in combination. It would also be important to study if mutations of c-Met and EGFR in NSCLC are concordant or mutually exclusive in NSCLC. For example, in a majority of NSCLC, K-ras mutations and EGFR appear to be mutually exclusive.[37] We have certainly studied the EGFR/c-Met pathways in NSCLC, and it would be important to study the biological and signal transduction pathways in normal human bronchial epithelial cells with and without smoking to determine if smoking also plays an important role in the cross-talk between these two receptors.
Lung cancers have remarkable ability for metastasis. For example, over 1/3 of patients with NSCLC present with metastatic disease beyond the chest cavity.[38] RTKs play an important role in metastases-especially c-Met[39] and EGFR.[40] Cell motility and migration of lung cancer cells are the first initial mechanisms of metastasis. We show here that cell motility and membrane ruffling of NSCLC cells was enhanced in combination with c-Met and EGFR activation. It would now also be important to determine the signal transduction pathways involving the focal adhesion proteins in the context of EGFR/c-Met signaling and determine the effects in vivo as related to metastasis. Ultimately, one could envision that inhibition of both of these RTKs could lead to decreased metastasis in lung cancers.
Conclusion
c-Met and EGFR receptors are widely expressed in several NSCLC cell lines. In NSCLC, HGF and EGF exhibited synergistic effects on proliferation, phosphorylation of c-Met and also important signaling intermediates. Interestingly, c-Met and EGFR inhibitors synergistically inhibited cell proliferation and promoted apoptosis in NSCLC. Additive effects of HGF and EGF on cell motility were also observed. These studies demonstrate that HGF and EGF tyrosine kinase inhibitors could be synergistic targets in the treatment of NSCLC
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
The study is in part supported by grants from NIH (2P01HL058064-13, Garcia), NCI (CA125541-02, CA129501-01, CA100750-05), American Lung Association, Kate McMullen Foundation, MARF (Jefferey P. Hayes Memorial), Cancer Research Foundation (Goldblatt Award), and V-Foundation (Guy Geelerd Memorial Foundation) to RS.
Contributor Information
Neelu Puri, Email: neelupur@uic.edu.
Ravi Salgia, Email: rsalgia@medicine.bsd.uchicago.edu.
References
- 1.Tsao MS, Liu N, Chen JR, Pappas J, Ho J, To C, et al. Differential expression of Met/hepatocyte growth factor receptor in subtypes of non-small cell lung cancers. Lung Cancer. 1998;1:1–16. doi: 10.1016/s0169-5002(98)00007-5. [DOI] [PubMed] [Google Scholar]
- 2.Spaulding DC, Spaulding BO. Epidermal growth factor receptor expression and measurement in solid tumors. Semin Oncol. 2002;5:45–54. doi: 10.1053/sonc.2002.35647. [DOI] [PubMed] [Google Scholar]
- 3.Sridhar SS, Seymour L, Shepherd FA. Inhibitors of epidermal-growth-factor receptors: A review of clinical research with a focus on non-small-cell lung cancer. Lancet Oncol. 2003;7:397–406. doi: 10.1016/s1470-2045(03)01137-9. [DOI] [PubMed] [Google Scholar]
- 4.Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R. Role of the hepatocyte growth factor receptor, c-Met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev. 2002;1:41–59. doi: 10.1016/s1359-6101(01)00029-6. [DOI] [PubMed] [Google Scholar]
- 5.Maulik G, Kijima T, Ma PC, Ghosh SK, Lin J, Shapiro GI, et al. Modulation of the c-Met/hepatocyte growth factor pathway in small cell lung cancer. Clin Cancer Res. 2002;2:620–7. [PubMed] [Google Scholar]
- 6.Danilkovitch-Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Invest. 2002;7:863–7. doi: 10.1172/JCI15418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carpenter G. Receptors for epidermal growth factor and other polypeptide mitogens. Annu Rev Biochem. 1987;56:881–914. doi: 10.1146/annurev.bi.56.070187.004313. [DOI] [PubMed] [Google Scholar]
- 8.Yarden Y. The EGFR family and its ligands in human cancer: Signaling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37:S3–8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 9.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;21:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 10.Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;5676:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 11.To CT, Tsao MS. The roles of hepatocyte growth factor/scatter factor and met receptor in human cancers. Oncol Rep. 1998;5:1013–24. doi: 10.3892/or.5.5.1013. [DOI] [PubMed] [Google Scholar]
- 12.Faletto DL, Tsarfaty I, Kmiecik TE, Gonzatti M, Suzuki T, Vande Woude GF. Evidence for non-covalent clusters of the c-met proto-oncogene product. Oncogene. 1992;6:1149–57. [PubMed] [Google Scholar]
- 13.Iyer A, Kmiecik TE, Park M, Daar I, Blair D, Dunn KJ, et al. Structure, tissue-specific expression, and transforming activity of the mouse met protooncogene. Cell Growth Differ. 1990;2:87–95. [PubMed] [Google Scholar]
- 14.Rong S, Bodescot M, Blair D, Dunn J, Nakamura T, Mizuno K, et al. Tumorigenicity of the met proto-oncogene and the gene for hepatocyte growth factor. Mol Cell Biol. 1992;11:5152–8. doi: 10.1128/mcb.12.11.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stella MC, Comoglio PM. HGF: A multifunctional growth factor controlling cell scattering. Int J Biochem Cell Biol. 1999;12:1357–62. doi: 10.1016/s1357-2725(99)00089-8. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt L, Junker K, Weirich G, Glenn G, Choye P, Lubensky I, et al. Two North American families with hereditary papillary renal carcinoma and identical novel mutations in the MET proto-oncogene. Cancer Res. 1998;8:1719–22. [PubMed] [Google Scholar]
- 17.Christensen JG, Schreck R, Burrows J, Kuruganti P, Chan E, Le P, et al. A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003;21:7345–55. [PubMed] [Google Scholar]
- 18.Sattler M, Pride YB, Ma P, Gramlich JL, Chu SC, Quinnan LA, et al. A novel small molecule met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase. Cancer Res. 2003;17:5462–9. [PubMed] [Google Scholar]
- 19.Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. JBC. 2000;12:8806–11. doi: 10.1074/jbc.275.12.8806. [DOI] [PubMed] [Google Scholar]
- 20.Reznik TE, Sang Y, Ma Y, Abounader R, Rosen EM, Xia S, et al. Transcription-dependent epidermal growth factor receptor activation by hepatocyte growth factor. Mol Cancer Res. 2008;1:139–50. doi: 10.1158/1541-7786.MCR-07-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonine-Summers AR, Aakre ME, Brown KA, Arteaga CL, Pietenpol JA, Moses HL, et al. Epidermal growth factor receptor plays a significant role in hepatocyte growth factor mediated biological responses in mammary epithelial cells. Cancer Biol Ther. 2007;4:561–70. doi: 10.4161/cbt.6.4.3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spix JK, Chay EY, Block ER, Klarlund JK. Hepatocyte growth factor induces epithelial cell motility through transactivation of the epidermal growth factor receptor. Exp Cell Res. 2007;15:3319–25. doi: 10.1016/j.yexcr.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu KP, Yu FS. Cross talk between c-Met and epidermal growth factor receptor during retinal pigment epithelial wound healing. IOVS. 2007;5:2242–8. doi: 10.1167/iovs.06-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;5827:1039–43. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 25.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. PNAS. 2007;52:20932–7. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;6:1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 27.Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, et al. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide: Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;1:217–21. [PubMed] [Google Scholar]
- 28.Ciardiello F, De Vita F, Orditura M, Tortora G. The role of EGFR inhibitors in nonsmall cell lung cancer. Curr Opin Oncol. 2004;2:130–5. doi: 10.1097/00001622-200403000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;27:3787–800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 30.Christensen JG, Burrows J, Salgia R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2004;1:1–26. doi: 10.1016/j.canlet.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 31.Puri N, Khramtsov A, Ahmed S, Nallasura V, Hetzel JT, Jagadeeswaran R, et al. A selective small molecule inhibitor of c-Met, PHA665752, inhibits tumorigenicity and angiogenesis in mouse lung cancer xenografts. Cancer Res. 2007;7:3529–34. doi: 10.1158/0008-5472.CAN-06-4416. [DOI] [PubMed] [Google Scholar]
- 32.Ma PC, Kijima T, Maulik G, Fox EA, Sattler M, Griffin JD, et al. c-MET mutational analysis in small cell lung cancer: Novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;19:6272–81. [PubMed] [Google Scholar]
- 33.Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;4:1479–88. doi: 10.1158/0008-5472.CAN-04-2650. [DOI] [PubMed] [Google Scholar]
- 34.Ma PC, Schaefer E, Christensen JG, Salgia R. A selective small molecule c-MET Inhibitor, PHA665752, cooperates with rapamycin. Clin Cancer Res. 2005;6:2312–9. doi: 10.1158/1078-0432.CCR-04-1708. [DOI] [PubMed] [Google Scholar]
- 35.Hahn O, Salgia R. Novel therapies in lung cancer. Hematol Oncol Clin North Am. 2005;2:343–67. doi: 10.1016/j.hoc.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 36.Ahmed SM, Salgia R. Epidermal growth factor receptor mutations and susceptibility to targeted therapy in lung cancer. Respirology. 2006;6:687–92. doi: 10.1111/j.1440-1843.2006.00887.x. [DOI] [PubMed] [Google Scholar]
- 37.Sugio K, Uramoto H, Ono K, Oyama T, Hanagiri T, Sugaya M, et al. Mutations within the tyrosine kinase domain of EGFR gene specifically occur in lung adenocarcinoma patients with a low exposure of tobacco smoking. Br J Cancer. 2006;6:896–903. doi: 10.1038/sj.bjc.6603040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caddy G, Conron M, Wright G, Desmond P, Hart D, Chen RY. The accuracy of EUS-FNA in assessing mediastinal lymphadenopathy and staging patients with NSCLC. Eur Respir J. 2005;3:410–5. doi: 10.1183/09031936.05.00092104. [DOI] [PubMed] [Google Scholar]
- 39.Lengyel E, Sawada K, Salgia R. Tyrosine kinase mutations in human cancer. Curr Mol Med. 2007;1:77–84. doi: 10.2174/156652407779940486. [DOI] [PubMed] [Google Scholar]
- 40.Maulik G, Kijima T, Salgia R. Role of receptor tyrosine kinases in lung cancer. Met Mol Med. 2003;74:113–25. doi: 10.1385/1-59259-323-2:113. [DOI] [PubMed] [Google Scholar]