

Abstract
A library of approximately 2000 small molecules biased toward inhibition of histone deacetylases was assayed for antimalarial activity in a high-throughput P. falciparum viability assay. Active compounds were cross-analyzed for induction of histone hyperacetylation in a human myeloma cell line to identify HDAC inhibitors with selectivity for P. falciparum over the human host. To verify on-target selectivity, pfHDAC-1 was expressed and purified and a biochemical assay for pfHDAC-1 activity was established.
The overwhelming majority of morbidity and mortality associated with human malaria is caused by the apicomplexan parasite Plasmodium falciparum. Widespread resistance to mainstay therapies such as chloroquine,(1) atovaquone,(2) pyrimethamine,(3) and sulfadoxine(4) has highlighted the need for new drugs. In addition to problems with resistance, the current suite of approved antimalarial drugs is limited to only a few targets within the parasite. Here, we address this issue through the identification and characterization of P. falciparum histone deactylase-1 (pfHDAC-1a) inhibitors that are potent against cultured parasite strains but do not perturb mammalian histone acetylation.
The histone epigenetic code is a key regulator of eukaryotic gene expression.(5) The reversible acetylation of lysine residues within histone tails is regulated by histone acetyltransferase (HAT) and histone deacetylase (HDAC) activity. HATs catalyze the acetylation of histone tails causing localized relaxation of chromatin and transcriptional activation of nearby genes, while HDACs catalyze the deacetylation of acetylated histones leading to transcriptional repression.(6) Equilibrium between the activities of HATs and HDACs must be maintained for proper transcriptional activity and cellular function. Mammalian HDACs are divided into four major classes based on size, cellular localization, catalytic domain, sequence homology, and mechanism of action. Classes I, II, and IV are zinc-dependent hydrolases, whereas class III enzymes, also called sirtuins, form an unrelated NAD-dependent subfamily. Class I HDACs are generally located in the nucleus and are relatively small in size; class II HDACs are present in the nucleus and cytoplasm and are generally larger.(7)
Disregulation of HDAC activity is an important therapeutic target. For example, HDAC inhibition has been shown to repress the transcription of tumor suppressor genes associated with the progression of various leukemias.8,9 The activity of class I and II HDACs can be inhibited by binding the zinc-containing tubular pocket of the enzyme.(10) These inhibitors can be classified into several groups: short-chain fatty acids such as butyrate and valproic acid; hydroxamates such as trichostatin A 3 (TSA), suberoylanilide hydroxamic acid 4 (SAHA), and LBH-589 5; benzamides such as MS-275 6; cyclic tetrapetides such as apicidin 7; and electrophilic ketones such as trifluoromethylketones.8,114, the most thoroughly characterized of these inhibitors, was recently approved by the Food and Drug Administration for the treatment of cutaneous T-cell lymphoma.(12) Although 4 is an effective HDAC inhibitor, it shows little species or isoform selectivity. Selective inhibition of specific HDACs can be achieved by structural modification of the recognition cap or metal-chelating functional group that is characteristic of most known HDAC inhibitors.(13)
Targeting of HDACs in apicomplexan protozoans, including the malaria parasite, has been previously investigated for drug discovery and development.14,15 The malaria parasite undergoes significant morphological changes during its asexual life cycle in humans and during transmission from the insect vector to the human host, and appropriate control of histone acetylation is certain to be vital for parasite survival. The HDAC inhibitor 7, which elicits an increase in P. falciparum histone acetylation concomitant with reduced parasite proliferation, provided the initial proof of concept for the essentiality of HDAC function in the parasite.(16) Unfortunately, unfavorable pharmacological properties limited the further development of 7 as an antimalarial agent.
Genome sequencing of Plasmodium falciparum uncovered one class I HDAC, two class II HDACs, and two class III sirtuins. Only one of the class III enzymes, P. falciparum silent information regulator 2 (pfSir2; PlasmoDB gene ID, PF13_0152), has been definitively shown to possess HDAC activity.17,18 The putative class I and II HDACs have not yet been examined in sufficient detail to confirm actual HDAC activity. Expression and purification of class I HDACs have generally afforded greater success than the class II enzymes, and thus, we focused our study on the sole class I HDAC, pfHDAC-1 (PlasmoDB gene ID, PFI1260c). The enzyme is a 51 kDa nuclear protein that is expressed in gametocytes and mature blood stages of the malaria parasite life cycle and shares significant homology to all of the class I human HDACs.(19)
I. For expression and purification of pfHDAC-1, pfHDAC-1 was recombinantly expressed and purified from S2 insect cells. The cDNA encoding the PfHDAC-1 was shuttled into the pAc5.1 expression vector using Gateway cloning (Invitrogen) with an engineered HPC4 epitope tag at the C-terminus for purification. S2 cells were co-transfected with this vector plus pCoBlast (Invitrogen), and a stable pool of transfectants was generated using blasticidin as the selective antibiotic.
II. For biochemical characterization of recombinant pfHDAC-1, the endogenous histone substrate from P. falciparum is not conveniently available to perform a detailed kinetic analysis of pfHDAC-1. Therefore, we investigated the possibility of measuring enzyme activity using a series of artificial substrates that resemble an N-acetylated lysine residue and that have been demonstrated to be processed by mammalian and bacterial class I or class II HDACs.20,21
Of the two substrates that were examined for recognition by pfHDAC-1 only 1 was efficiently catalyzed (Table 1). The Michaelis−Menten model was fitted to the data which afforded the kinetic constants kcat,1 = 0.19 ± 0.01 s−1 and Km,1app = 30 ± 2 μM. The binding affinity and catalytic efficiency of 1 to pfHDAC-1 are comparable to those observed with class I HDACs from other organisms. The known HDAC inhibitors 3−7 were tested for the inhibition of pfHDAC-1 activity and parasite proliferation (Table 2). 3−5 and 7 were found to be inhibitory in both assays, whereas 6 did not demonstrate significant inhibition of either pfHDAC-1 activity or parasite growth. Interestingly, o-aminoanilide based compounds such as 6 have been documented to exhibit selectivity for human class I over class II enzymes.(22) The class III specific HDAC inhibitor splitomicin was also determined to be inactive in enzyme and parasite inhibition assays.
Table 1. Substrate-Specificity of pfHDAC-1.
| substrate | Kmapp (μM) | kcat (s−1) |
|---|---|---|
| Ac-Leu-Gly-Lys(Ac)-AMC 1 | 30 ± 2 | 0.19 ± 0.01 |
| Boc-Lys(trifluoroacetyl)-AMC 2 | NAa | NA |
No activity observed with 2 in the presence of excess substrate and pfHDAC-1.
Table 2. Inhibition of pfHDAC-1 Activity and P. falciparum 3D7 Proliferation by Known HDAC Inhibitors.
| IC50 (nM) |
||
|---|---|---|
| compd | pfHDAC-1 | P. falciparum 3D7 |
| 3 | 0.6 ± 0.1 | 16 ± 7 |
| 4 | 59 ± 6 | 210 ± 30 |
| 5 | 1.8 ± 0.2 | 21 ± 3 |
| 6 | 940 ± 90 | >10000 |
| 7 | 1 ± 0.1 | 33 ± 5 |
| splitomicin | >10000 | >10000 |
III. For inhibition of pfHDAC-1 and parasite proliferation, the archetypal chemical structure for a HDAC inhibitor consists of a metal chelating group joined by a carbon chain linker to a hydrophobic capping element that confers potency and selectivity (Figure 1).(23)
Figure 1.

HDAC-biased chemical library. The archetypal HDAC inhibitor, and the basis for this focused library, consists of a capping element joined to a metal chelating moiety by a hydrophobic linker. Diversity was introduced at all three of these motifs with the final library consisting of ∼2000 chemical members.
Given the effectiveness of hydroxamate-based HDAC inhibitors, it was hypothesized that selectivity could be engineered for pfHDAC-1 and the malaria parasite with a core scaffold containing a hydroxamate metal chelating unit. Although not species selective, the spectrum of activity already observed for 3−5 supported this model (Table 2). For this reason, a ∼2000 member HDAC-biased chemical library was sequentially screened for antimalarial efficacy and inhibition of pfHDAC-1 activity. Chemical diversity within the library was introduced at the recognition cap or metal chelating portion of the molecule, and the linker length between these two groups was varied from four to six carbons (Figure 1).(24)
We have previously analyzed this library for the alteration of in vivo bulk histone acetylation in mammalian cells (Supporting Information Figure 1). Many compounds were found to potently inhibit parasite proliferation and pfHDAC-1 activity; however, only 17 displayed antiparasitic activity coupled with minimal perturbation of mammalian histone acetylation (Table 3). Inhibition of P. falciparum proliferation was biased toward compounds with ortho-substitution (bromine, hydroxyl) in the cap region of the core scaffold.
Table 3. Inhibition of P. falciparum Growth and pfHDAC-1 Activity by Cherry-Picked Hits from the HDAC-Biased Chemical Library.

| compd | R | n | pfHDAC-1, IC50 (nM) | fold change in MM.1S histone acetylationa | P. falciparum3D7, IC50 (nM) |
|---|---|---|---|---|---|
| 8 | 3-hydroxyphenyl | 5 | 59 ± 6 | 1.2 | 99 ± 12 |
| 9 | 2,5-dihydroxyphenyl | 6 | 110 ± 13 | 1.1 | 142 ± 28 |
| 10 | 6-bromobenzo[d][1,3]dioxol-5-yl | 5 | 20 ± 2 | 1.0 | 59 ± 15 |
| 11 | 2,4,6-trishydroxyphenyl | 6 | 43 ± 5 | 1.1 | 50 ± 6 |
| 12 | 2-bromo-5-methoxyphenyl | 5 | 22 ± 2 | 1.1 | >500 |
| 13 | 4-(dimethylamino)-2-hydroxyphenyl | 5 | 37 ± 4 | 1.2 | 24 ± 2 |
| 14 | 2-hydroxynaphthalen-1-yl | 5 | 41 ± 4 | 1.0 | 20 ± 2 |
| 15 | 2-bromo-5-hydroxyphenyl | 5 | 49 ± 5 | 1.0 | 35 ± 4 |
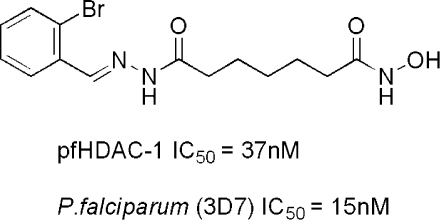
| 16 | 2-bromophenyl | 5 | 37 ± 4 | 1.1 | 15 ± 2 |
| 17 | 2-bromo-4-hydroxy-5-methoxyphenyl | 5 | 59 ± 7 | 1.0 | 47 ± 7 |
| 18 | 2-chlorophenyl | 5 | 90 ± 10 | 0.9 | 288 ± 20 |
| 19 | 4-(1H-imidazol-1-yl)phenyl | 5 | 15 ± 2 | 1.0 | 57 ± 11 |
| 20 | 2-bromopyridin-3-yl | 5 | 36 ± 6 | 0.9 | 22 ± 5 |
| 21 | 2-bromo-4-hydroxyphenyl | 4 | 106 ± 11 | 1.1 | 53 ± 13 |
| 22 | 2-bromo-5-hydroxyphenyl | 4 | 89 ± 9 | 1.1 | 30 ± 7 |
| 23 | 2-bromo-4-hydroxy-5-methoxyphenyl | 4 | 59 ± 5 | 1.2 | 498 ± 168 |
| 24 | 4-boronophenyl | 4 | 45 ± 5 | 1.0 | 68 ± 8 |
The fold change in MM.1S histone acetylation was measured at a compound concentration of 0.2 μM.
The presence of a hydroxamic acid as a metal chelating ligand was necessary for activity. The ideal linker length for selective compounds was found to be five methylene units in contrast to the six methylene units preferred for compounds inducing histone hyperacetylation in mammalian cells.
Transcriptional regulation orchestrates the multiple morphological transformations that occur throughout the complex developmental stages of the P. falciparum life cycle. HDACs participate in the reversible acetylation of lysine residues within histone tails to control chromatin unwinding and DNA transcription. Inhibition of HDAC activity has already proven useful for the treatment of various cancers and may also be valuable for the development of antimalarial chemotherapies. Here, we have expressed recombinant pfHDAC-1, the sole class I isoform identified by homology in P. falciparum, and confirmed that the protein has in vitro HDAC activity. The known HDAC inhibitor 4 elicits hyperacetylation of P. falciparum histones and retards parasite proliferation. Unfortunately 4 is neither species nor HDAC-isoform selective, and yet specificity for pfHDAC-1 and P. falciparum was obtained herein by modifying the core scaffold. Seventeen compounds were found to significantly inhibit parasite proliferation and pfHDAC-1 activity, and yet these compounds did not perturb histone acetylation or inhibit whole-cell growth of mammalian MM.1S. These compounds generally contained bromine and hydroxyl substituted phenol derivatives as recognition caps and a hydroxamic acid as the metal chelating ligand. It is noted that the mechanism of action of these compounds is linked to pfHDAC-1 but is not necessarily limited to this target. The compounds may, for example, also target class II, other unidentified class I HDACs, or possibly even other metalloenzymes in the parasite. Nevertheless, chemical optimization of these compounds including replacement of the acyl hydrazone motive is underway for improved pharmacological properties such as solubility and serum stability. In vivo efficacy trials using a murine malaria model are planned for the near future.
Acknowledgments
This work was supported by the U.S. Public Health Service, NIH Grants R21NS053660 (J.C.) and NIH R21NS059404 (J.C.), and by the NSF Graduate Research Fellowship Program (V.P. and B.C.), Medicines for Malaria Venture, Harvard Malaria Initiative (D.F.W.), SPARC Grant 2737380 from the Broad Institute of MIT and Harvard, the Burroughs Welcome Fund (M.T.D.), and the Humanitarian Assistance for Neglected Diseases (HAND) Initiative of Genzyme Corporation. We thank Mary Lynn Baniecki for providing screening data.
Supporting Information Available
Details of the expression and purification of pfHDAC-1, pfHDAC-1 enzyme characterization, inhibition assays, and the antimalarial activity of candidate compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: pfHDAC-1, P. falciparum histone deactylase-1; AMC, 4-methyl-7-aminocoumarin; HDAC, histone deactylase; HAT, histone acetyltransferase; TSA, trichostatin A; SAHA, suberoylanilide hydroxamic acid.
Supplementary Material
References
- Peters W. Drug-resistant malaria. Lancet 1969, 2, 54. [DOI] [PubMed] [Google Scholar]
- Srivastava I. K.; Morrisey J. M.; Darrouzet E.; Daldal F.; Vaidya A. B. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol. Microbiol. 1999, 33, 704–711. [DOI] [PubMed] [Google Scholar]
- Young M. D.; Contacos P. G.; Stitcher J. E.; Millar J. W. Drug resistance in Plasmodium falciparum from Thailand. Am. J. Trop. Med. Hyg. 1963, 12, 305–314. [DOI] [PubMed] [Google Scholar]
- Hess U.; Timmermans P. M.; Jones M. Combined chloroquine/Fansidar-resistant falciparum malaria appears in East Africa. Am. J. Trop. Med. Hyg. 1983, 32, 217–220. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Bernstein B. E.; Tong J. K.; Schreiber S. L. Genomewide studies of histone deacetylase function in yeast. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 13708–13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoretti I. V.; Lee Y. M.; Goodson H. V. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [DOI] [PubMed] [Google Scholar]
- Minucci S.; Pelicci P. G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. 2006, 6, 38–51. [DOI] [PubMed] [Google Scholar]
- Richon V. M.; Emiliani S.; Verdin E.; Webb Y.; Breslow R.; Rifkind R. A.; Marks P. A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 3003–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnin M. S.; Donigian J. R.; Cohen A.; Richon V. M.; Rifkind R. A.; Marks P. A.; Breslow R.; Pavletich N. P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature (London) 1999, 401, 188–193. [DOI] [PubMed] [Google Scholar]
- Ciossek T.; Julius H.; Wieland H.; Maier T.; Beckers T. A homogeneous cellular histone deacetylase assay suitable for compound profiling and robotic screening. Anal. Biochem. 2008, 372, 72–81. [DOI] [PubMed] [Google Scholar]
- Marks P. A.; Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [DOI] [PubMed] [Google Scholar]
- Koeller K. M.; Haggarty S. J.; Perkins B. D.; Leykin I.; Wong J. C.; Kao M. C.; Schreiber S. L. Chemical genetic modifier screens: small molecule trichostatin suppressors as probes of intracellular histone and tubulin acetylation. Chem. Biol. 2003, 10, 397–410. [DOI] [PubMed] [Google Scholar]
- Andrews K. T.; Tran T. N.; Lucke A. J.; Kahnberg P.; Le G. T.; Boyle G. M.; Gardiner D. L.; Skinner-Adams T. S.; Fairlie D. P. Potent antimalarial activity of histone deacetylase inhibitor analogues. Antimicrob. Agents Chemother. 2008, 52, 1454–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinke P. T.; Colletti S. L.; Doss G.; Myers R. W.; Gurnett A. M.; Dulski P. M.; Darkin-Rattray S. J.; Allocco J. J.; Galuska S.; Schmatz D. M.; Wyvratt M. J.; Fisher M. H. Synthesis of apicidin-derived quinolone derivatives: parasite-selective histone deacetylase inhibitors and antiproliferative agents. J. Med. Chem. 2000, 43, 4919–4922. [DOI] [PubMed] [Google Scholar]
- Darkin-Rattray S. J.; Gurnett A. M.; Myers R. W.; Dulski P. M.; Crumley T. M.; Allocco J. J.; Cannova C.; Meinke P. T.; Colletti S. L.; Bednarek M. A.; Singh S. B.; Goetz M. A.; Dombrowski A. W.; Polishook J. D.; Schmatz D. M. Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 13143–13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty S. P.; Saikumari Y. K.; Bopanna M. P.; Balaram H. Biochemical characterization of Plasmodium falciparum Sir2, a NAD(+)-dependent deacetylase. Mol. Biochem. Parasitol. 2008, 158, 139–151. [DOI] [PubMed] [Google Scholar]
- Merrick C. J.; Duraisingh M. T. Plasmodium falciparum Sir2: an unusual sirtuin with dual histone deacetylase and ADP-ribosyltransferase activity. Eukaryotic Cell 2007, 6, 2081–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi M. B.; Lin D. T.; Chiang P. H.; Goldman N. D.; Fujioka H.; Aikawa M.; Syin C. Molecular cloning and nuclear localization of a histone deacetylase homologue in Plasmodium falciparum. Mol. Biochem. Parasitol. 1999, 99, 11–19. [DOI] [PubMed] [Google Scholar]
- Lahm A.; Paolini C.; Pallaoro M.; Nardi M. C.; Jones P.; Neddermann P.; Sambucini S.; Bottomley M. J.; Lo Surdo P.; Carfi A.; Koch U.; De Francesco R.; Steinkuhler C.; Gallinari P. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 17335–17340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener D.; Wirsching F.; Riester D.; Schwienhorst A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem. Biol. 2003, 10, 61–68. [DOI] [PubMed] [Google Scholar]
- Khan N.; Jeffers M.; Kumar S.; Hackett C.; Boldog F.; Khramtsov N.; Qian X.; Mills E.; Berghs S. C.; Carey N.; Finn P. W.; Collins L. S.; Tumber A.; Ritchie J. W.; Jensen P. B.; Lichenstein H. S.; Sehested M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [DOI] [PubMed] [Google Scholar]
- Butler K. V.; Kozikowski A. P. Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr. Pharm. Des. 2008, 14, 505–528. [DOI] [PubMed] [Google Scholar]
- Vegas A. J.; Bradner J. E.; Tang W.; McPherson O. M.; Greenberg E. F.; Koehler A. N.; Schreiber S. L. Fluorous-based small-molecule microarrays for the discovery of histone deacetylase inhibitors. Angew. Chem., Int. Ed. 2007, 46, 7960–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.