

Summary
The second messenger calcium plays an essential role in mediating the TCR signaling pathway leading to cytokine production and T cell clonal expansion. The immunosuppressive drugs cyclosproin A (CsA) and FK506 have served both as therapeutic agents and as molecular probes for unraveling the protein phosphatase calcineurin as a rate-limiting enzyme involved in the transmission of calcium signal from the cytosol into the nucleus to reprogram gene expression. The use of mouse knockout models has helped to verify and further elucidate the functions of different isoforms of calcineurin in both helper T cell activation and thymocyte development. In addition to calcineurin, three other classes of calmodulin-binding proteins have also been shown to play important roles in calcium signaling in T cells. Thus, Cabin1 and class II HDACs have been found to constitute a novel calcium-signaling module in conjunction with the transcription factor myocyte enhance factor family and the transcriptional coactivator p300 to suppress and activate cytokine gene transcription in a calcium-dependent manner. The calmodulin-dependent protein kinases (CaMK) II and IV were also shown to play negative and positive regulatory functions, respectively, in TCR-mediated cytokine production. The crosstalks among these and other signal transducers in T cells form an extensive non-linear signaling network that dictates the final outcome of the TCR signaling pathway.
Keywords: calcineurin, cabin1, histone deacetylases, carabin, MEF2, kinases
Introduction
Calcium ion is a universal second messenger that plays important roles in a multitude of signal transduction pathways and other cellular processes throughout biology, including the intracellular signaling pathway emanating from T-cell receptor (TCR) during the activation of peripheral CD4+ T lymphocytes. A common sensor for intracellular calcium ions is calmodulin (CaM). In resting T cells in which the concentration of free cytosolic calcium ions is low at about 10-7 M, the four calcium ion-binding sites of calmodulin are partially occupied. Upon TCR signaling and the ensuing membrane proximal signaling cascades involving protein tyrosine kinases ζ-associated protein of 70 kDa (ZAP70) and phospholipase Cγ1 (PLCγ1), the newly generated second messenger inositol-1,4,5-triphosphate (IP3) binds to its receptors on the endoplasmic reticulum (ER) membrane and releases calcium from the ER into the cytosol, which in turn triggers the movement of the ER calcium sensor STIM1 towards Orai/CRACM SOC channels on the plasma membrane, leading to the activation of CRAC channels. The influx of calcium from extracellular milieu via CRAC channels into the cytosol leads to the occupation of all four calcium-binding sites in calmodulin, which undergoes a significant conformational change, binding to and activating several target signaling proteins to further transmit the signal into the nucleus, ultimately culminating in the transcriptional activation of cytokine genes in the nucleus.
For years, the mechanism by which the calcium signal is transmitted from the cytosol into the nucleus remained a mystery. One of the first target proteins linked to the transmission of calcium, and calmodulin-dependent signaling in T cells was identified as the protein phosphatase calcineurin (CN) as part of an effort to elucidate the mechanism of action of the immunosuppressive drugs CsA and FK506 (1, 2). It is now clear that calcineurin is a rate-limiting signal transducer during T cell activation. Prior to the identification of calcineurin as the calcium, calmodulin-dependent protein phosphtase, several calmodulin-dependent kinases were known. Two of them, CaMKII and CaMKIV, have been shown to play negative or positive roles in T cells calcium signaling, respectively. More recently, another family of calmodulin-interacting transcriptional repressors, Cabin1 and class II histone deacetylases (HDACs), have been shown to form, together with the myocyte enhance factor 2 (MEF2) family of transcription factors, yet another calcium signaling module within the nuclear compartment to regulate cytokine gene expression.
A total of at least four classes of calmodulin-binding proteins have been shown to be involved in converting calcium signal from the cytosol into a nuclear transcriptional output. They include the CaM-dependent protein phosphatase calcineurin, the CaM-dependent protein serine/threonine kinases, the transcription corepressor Cabin1 and class II HDACs (Fig. 1). While most of these signaling proteins relay the calcium signal to a specific subset of transcription factors involved in cytokine gene transcription, there is also extensive crosstalk between them, which contributes to the ultimate transcriptional outcome. This review is intended to provide an overview of our current understanding of how each of these four classes of calmodulin-binding proteins are involved in intracellular calcium signaling downstream of TCR during the activation of peripheral CD4+ T-helper cells.
Fig. 1. Four classes of calmodulin-binding signal transducers in T cells.
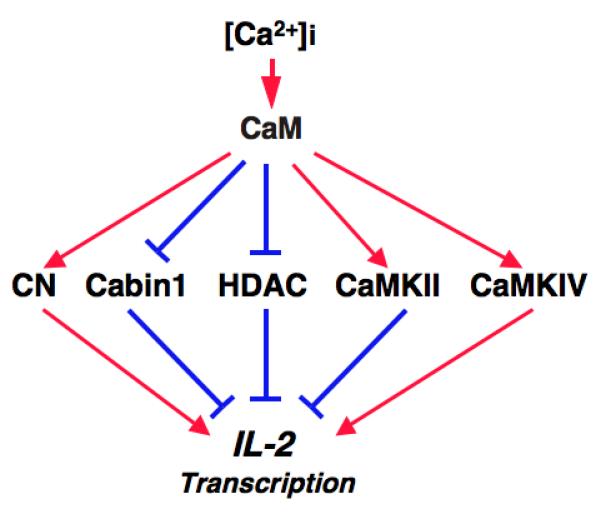
Color codes: Red, activation; blue, inhibition. Abbreviations: CaM, calmodulin; CN, calcineurin, HDAC, histone deacetylases; CaMK, CaM-dependent protein kinases.
Calcineurin
CN was initially identified as a protein with high affinity for calmodulin that inhibited the stimulation of Ca2+-dependent cyclic nucleotide phosphodiesterase by calmodulin (3), which was found to be an abundant calmodulin-binding protein in the brain (4). Its intrinsic calcium and calmodulin-dependent phosphatase activity was discovered years later (5). The first clue that calcineurin may play an important role in calcium signaling in T cells came from biochemical studies aimed at identifying calmodulin-binding proteins in T and B cells that led to the identification of calcineurin as a major calmodulin-interacting protein in those cell types (6). That calcineurin is a key transducer of calcium signaling in T cells was first demonstrated when it was identified as a common target of two important immunosuppressive drugs, cyclosporine A (CsA) and FK506 (7). As both CsA and FK506 had been shown to selectively block calcium-dependent activation of the interleukin-2 (IL-2) gene, it was logical and reasonable to speculate that their molecular target, CN, was likely to play an important role in mediating calcium signaling in T cells, especially given its known dependence on both calcium and calmodulin for activation.
Pharmacological and genetic evidence that CN is essential in mediating calcium signaling in peripheral T cells
The immunosuppressive drugs CsA and FK506 work by inhibiting the protein phosphatase activity of calcineurin in an unprecedented manner (7). Upon entering T cells, they bind to the abundant cytosolic receptor proteins cyclophilin (for CsA) and FKBP (for FK506), respectively (8-10). The resulting binary complexes, cyclophilin-CsA and FKBP-FK506, then bind to the composite surface of CNA and CNB to form a ternary complex (7). Although the cyclophilin-CsA and FKBP-FK506 complexes do not directly interact with the catalytic domain of calcineurin, their sheer sizes in front of the active site serve to sterically obstruct the access of protein substrates to the active site (11-14). The potent inhibition of TCR-mediated transcriptional activation of IL-2 and other cytokines by CsA and FK506 suggested that calcineurin plays a critical role in mediating the intracellular signaling pathway.
The two subunits of calcineurin exist in multiple isoforms (15). In the human and other mammalian genomes, three isoforms of the catalytic subunit of calcineurin have been identified, CNAα, β, and γ. Two isoforms of the regulatory subunit of calcineurin, CNB1 and CNB2, have also been reported. Although calcineurin is ubiquitously expressed in all cell and tissue types, CNAγ and CNB2 are found almost exclusively in the testis. Thus, only CNAα, β and CNB1 are relevant in T cells. One of the apparent questions is whether the two catalytic subunits of calcineurin play redundant or distinct functions in T-cell signaling.
Using isoform-specific antibodies, the relative abundance of the two isoforms of CNA was assessed in different tissues. It was found that the β isoform was far more abundant than the α isoform in T cells, suggesting that CNAβ is likely to play a more critical role in calcium signaling in T cells (16). Indeed, subsequent knockout studies revealed significant differences in functions of the two isoforms in T-cell signaling and development with CNAβ playing a much more critical role. Both CNAα and CNAβ knockout animals survived to adulthood, with no gross developmental abnormalities, indicating that neither isoform of calcineurin alone is essential for development (17, 18). T cells from CNAα-deficient mice exhibited no defects in proliferative response to stimulation by mitogens and TCR agonists in vitro. Furthermore, these responses to TCR stimulation remained sensitive to CsA and FK506, suggesting that the remaining calcineurin activity is important and crucial for TCR signaling. However, the T-cell response to antigens in vivo was found to be defective (17), suggesting that CNAα is required for TCR signaling in vivo. It is worth pointing out that the T-cell response to antigen in vivo was assessed by response of the T cells isolated from antigen-exposed animals to restimulation with antigens. A defect observed in such an assay may be alternatively interpreted as a consequence of anergy induction when the T cells were activated by antigen in the absence of CNAα in vivo. In contrast to the subtler T-cell phenotype in CNAα knockout mice, the phenotype of CNAβ knockout animal was pronounced and unmistakable (18). The peripheral T cells from CNAβ-/- animals showed clear defects in proliferation and IL-2 production in response to TCR agonists. Moreover, CNAβ knockout animals were permissive to xenografts of tumor cell lines, similar to, albeit to a slightly lesser extent, those treated with CsA. These results, together with the mild phenotype of CNAα knockout mice, strongly suggested that of the two isoforms of calcineurin A, CNAβ plays a significantly more important role than CNAα in mediating intracellular calcium signaling, consistent with their relative abundance in T cells (16).
In addition to defects in peripheral T-cell activation, significant decreases in the total CD3, CD4+, and CD8+ T-cell populations were also seen in CNAβ null animals, suggesting that CNAβ is required for T-cell development (18). These results were corroborated and further extended in another study using thymocyte-specific conditional CNB knockout (19). As CNB is required for activity by both isoforms of CNA, the CNB null thymocytes were completely devoid of calcineurin activity. The CNB-deficient thymocytes were found to be defective in positive but not negative, selection, which explains the decrease in single positive T-cell population in CNAβ knockout animals (18). The definitive lack of defect in negative selection CNAβ knockout thymocytes also put to rest one of the long-standing questions on the role of calcineurin in that process. Earlier studies using immature T-cell lines had suggested that calcineurin might be involved in negative selection through induction of the orphan hormone receptor Nur77 (20, 21), as induction of Nur77 had been shown to be sensitive to inhibition by CsA, thus implicating a role of calcineurin in thymocyte apoptosis. However, based on the phenotype of CNB knockout mice, the in vitro model does not recapitulate the thymocyte negative selection in vivo and the physiological relevance of members of the Nur77 family in double positive thymocyte apoptosis remains unclear.
Mechanisms of regulation and substrate recognition
Calcineurin is composed of two subunits, the catalytic A subunit and the regulatory B subunit. The primary sequence of CNA can be divided into four distinct domains, a catalytic domain at the very N-terminus, the B subunit-binding domain, a calmodulin-binding domain and a C-terminal autoinhibitory domain. In crystal structures, the calmodulin-binding domain is invisible due to conformational flexibility, while the remaining three domains exist as dividable structural motifs (12). The catalytic domain of calcineurin is highly similar in structure to that of PP1 (22, 23). Unlike PP1, however, the B-binding domain and the bound regulatory B subunit lie immediately C-terminal to the catalytic domain, which has been speculated to confer a much narrower substrate specificity to calcineurin in comparison to other serine/threonine phosphatases. The calmodulin-binding domain and the C-terminal autoinhibitory domain together form an intramolecular calcium-sensing ‘on and off’ molecular switch. Thus, in the absence of calcium signaling, the C-terminal autoinhibitory domain is bound to the active site as a pseudo substrate, keeping calcineurin inactive. Upon calcium signaling, activated calmodulin binds to the calmodulin-binding domain adjacent to the autoinhibitory domain, causing dissociation of the autoinhibitory domain from the active site of calcineurin and activation of calcineurin towards cellular substrates. This type of calcium sensing switch using a combination of a calmodulin-binding domain and an autoinhibitory domain is also seen in the calmodulin-dependent kinases.
The major substrate for calcineurin in T cells is the nuclear factor of activated T cells (NFAT) family of transcription factors. In resting T cells, NFAT is extensively phosphorylated and resides in the cytosol. At least 13 sites of phosphorylation have been identified (24). A number of kinases have been shown to phosphorylate NFAT, including casein kinase 1 (CK1) (25, 26), MEKK (25), protein kinase A (27), glycogen synthase kinase 3 (GSK3) (28), as well as the recently identified dual specificity tyrosine-phosphorylation regulated kinases (DYRK) 1A and 2 (29). Some of these kinases, such as DYRK1A and 2 and PKA, appear to serve a priming role, while CK1 and GSK-3 act to phosphorylate the majority of sites. The constitutively active nature of most of the NFAT kinases ensure a default cytosolic localization of NFAT and apparent recycling of NFAT from the nucleus back into the cytosol in a reversible fashion upon termination of calcium signaling (30, 31). This recycling of NFAT from the nucleus may be of physiological significance. While most naive cells eventually undergo apoptosis upon activation by antigen, a subset differentiates into memory T cells. It is conceivable that NFAT needs to be recycled back into the cytosol to resume its place in dormant memory T cells awaiting encounter with the same antigen.
Upon calcium signaling and calcineurin activation, NFAT undergoes dephosphorylation, leading to the exposure of its nuclear localization signal sequence and its nuclear translocation. The sites that undergo dephosphorylation by calcineurin spans a linear sequence of over 300 amino acid residues at the N-terminal regulatory domain of NFAT, making it unique among known substrates of calcinerin (24), which usually possess only one or a few sites for dephosphorylation (15). It is thus not surprising that there are multiple domains in NFAT that serve as ‘anchors’ for the substrate to interact with different sites of the phosphatase. One such anchoring sequence in NFAT contains the consensus sequence ‘PxIxIT’ (in which x can be any amino acid) lying N-terminal to the multiple dephosphorylation sites on the regulatory domain of NFAT (32, 33). Interestingly, this anchoring motif is bound to the catalytic domain at a docking site that is adjacent to the active site of calcineurin (34). The binding of NFAT to the docking site in calcineurin may help to enhance the efficiency of dephosphorylation at the multiple sites by calcineurin through an increase in the processivity of the process.
The PxIxIT anchoring peptide has been exploited in the development of novel inhibitors of calcineurin that are more specific for NFAT without affecting dephosphorylation of other substrates, which would likely have lower toxicity than CsA and FK506. Based on the observation that a peptide derived from the PxIxIT motif of NFAT was capable of blocking dephosphorylation of NFAT by calcineurin in vitro (33), the identification of peptides with higher affinity was achieved from screening a randomized peptide library based on the PxIxIT sequence (35). An optimized VIVIT peptide was shown, upon ectopic expression as part of a fusion protein with green fluorescence protein (GFP), to be capable of inhibiting NFAT-driven luciferase activity and the activation of NFAT-dependent cytokine genes without affecting other cytokines that requires calcineurin activity but not NFAT. Moreover, small molecules were found that interfered with the docking of NFAT via the PxIxIT motif to calcineurin and inhibited calcineurin-NFAT signaling (36). It remains to be seen whether such small molecules can be further improved to possess sufficient potency for evaluation as novel immunosuppressive agents with lower toxicity than CsA and FK506.
In addition to the PxIxIT motif, a second calcineurin-binding motif, calcineirn-binding region 2 (CNBR2), was also identified in NFAT (37). The existence of the second site was later confirmed in another isoform of NFAT (38). Significantly, the second site containing an ‘LxVP’ consensus sequence has higher intrinsic affinity for calcineurin than the corresponding PxIxIT motif. Unlike PxIxIT, the LxVP containing peptides not only blocked calcineurin-NFAT interaction but also inhibited the phosphatase activity of calcineurin in vitro (38). Judging from the sequences, it is unlikely that the second LxVP containing calcineurin-binding motif will interact with the same docking site as the PxIxIT motif. It remains to be determined which site in calcineurin is bound by the second LxVP motif. The presence of two distinct calcineurin-binding motifs in NFAT that bind to different sites on calcineurin further highlights the unusual interaction between calcineurin and NFAT and may hint at a dynamic movement of the substrate relative to calcineurin as NFAT undergoes dephosphorylation by calcineurin at different phosphorylation sites within a fairly large regulatory domain.
Mechanism of inhibition of calcineurin by immunophilin-drug complexes
Several initial biochemical observations suggested an unprecedented mode of inhibition of calcineurin by the immunophilin-drug complexes. First, it is not the drugs alone but rather the cyclophilin-CsA and FKBP-FK506 complexes that bind to and inhibit the protein phosphatase activity of calcineurin, raising the question of whether cyclophilin and FKBP participate in direct interaction with calcineurin or simply served in an allosteric fashion to fix the drugs in certain conformations that are conducive to interactions with calcineurin. Second, the immunophilin-drug complexes have higher affinity for the calcium and calmodulin-activated form of calcineurin as judged by the relative amounts of calcineurin retained on the immunophilin-drug affinity resin in the absence and presence of calcium and calmodulin. Third, the binding of cyclophilin-CsA or FKBP-FK506 to calcineurin stimulated, rather than inhibited, the intrinsic enzymatic activity of calcineurin when a small substrate, p-nitrophenyl phosphate, was used as a substrate. On the other hand, they inhibited the phosphatase activity of calcineurin when a phospho-RII polypeptide was used as a substrate. Last but not least, the two structurally distinct immunophilin-drug complexes, cyclophilin-CsA and FKBP-FK506 competed for an overlapping binding site on calcineurin as their association with calcineurin is mutually exclusive. The attainment of crystal structures of calcineurin by itself and its complexes with the immunophilin-drug complexes offered answers to all those questions.
From the structure of calcineurin in its inactive form, three of the four domains were visible, the catalytic domain, the B subunit binding domain along with the bound CNB and the autoinhibitory domain bound to the active site of the enzyme (12). Apparently, the calmodulin-binding domain does not assume a single stable conformation in the absence of bound calmodulin in solution. This structure also confirmed the previous model of the role of the autoinhibitory domain as a pseudo substrate to block the active site of CN. The structure of the FKBP-FK506-calcineurin and cyclophilin-CsA-calcineurin ternary complexes revealed the unprecedented mode of inhibition of calcineurin by the immunophilin-drug complexes (12-14, 22). Rather than directly binding to the active site of calcineurin, the FKBP-FK506 and cyclophilin-CsA complexes are bound to the composite surface formed by the B subunit-binding domain in the form of an amphipathic peptide and the bound CNB. In addition to the interactions between the drugs and calcineurin, both cyclophilin and FKBP form direct interactions with calcineurin as well. Thus, CsA and FK506 can be viewed as pro-drugs. The cyclophilin-CsA and FKBP-FK506 complexes are the true drugs that bring about inhibition of the target calcineurin. Neither the drugs alone nor the immunophilins alone are sufficient to cause significant inhibition of calcineurin. Although the interactions between immunophilin-drug complexes are confined to the composite surface of CNA-CNB outside of the catalytic and autoinhibitory domains, it is conceivable that when the inactive form of calcineurin is constrained in such a way that restricts the conformation of the CNA-CNB composite surface flanked by the two domains to lower its affinity for calcineurin. This unusual mode of binding explains why FKBP-FK506 and cyclophilin-CsA have opposite effects on the phosphatase activity of calcineurin towards the small p-nitrophenyl phosphate and protein based phospho-RII peptide (7). As FKBP-FK506 and cyclophilin-CsA do not directly interact with the active site of calcineurin, they do not affect the access of p-nitrophenyl phosphate. However, the bulk is sufficient to block the access of larger protein substrates such as NFAT to the active site of calcineurin. In this sense, the immunophilin-drug complexes are allosteric inhibitors of calcineurin. A somewhat unexpected view came from the comparison of the two immunophilin-drug-calcineurin ternary complexes (Fig. 2). Surprisingly, the two distinct immunophilin-drug complexes are bound to the same CNA-CNB composite surface. Despite the lack of similarity between cyclophilin-CsA and FKBP-FK506 complexes, calcineurin employs a number of common residues, particularly hydrophobic ones, to interact with both complexes. In addition, a distinct set of residues from the two subunits of calcineurin are used to specifically interact with each of the two immunophilin-drug complexes, solving the problem of the convergent recognition of the same site of CN by the two different immunophilin-drug complexes.
Fig. 2. Overlay of the CyP-CsA-CN and FKBP-FK506-CN ternary complexes.
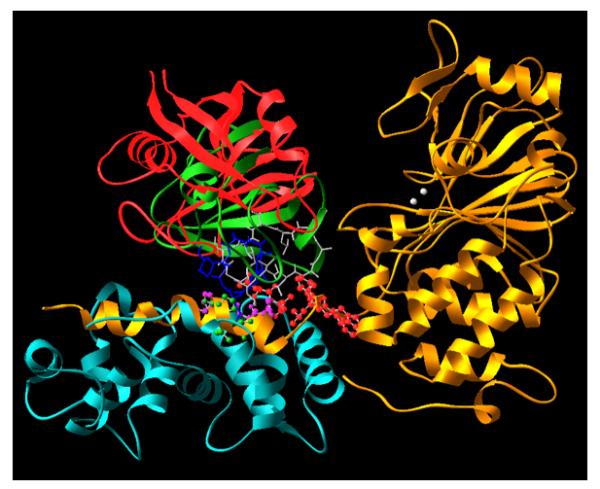
The two complexes were overlaid with maximal overlap in the backbone of calcineurin A and B subunits. Color code: Gold, CNA subunit; white, Zn2+ and Fe2+ at the active site of CNA; cyan, CNB; Red, CyP; green, FKBP12; blue, FK506; gray, CsA. The red, purple and green balls at the interface between CN and immunophilin-drug complexes are resides from CN that interact with either FKBP-FK506 or CyP-CsA.
Mechanism of immunosuppression by CsA and FK506 in vivo
Given the strong genetic evidence that calcineurin is indispensable for TCR-mediated cytokine production, it is straightforward to explain how these drugs manifest their immunosuppressive effects. Since CN is ubiquitously expressed in all tissues and mediates calcium signaling pathways in a wide range of physiological processes, a broader question arises as to how these drugs elicit selective inhibition of T cells in the immune system. The fact that both CsA and FK506 have side effects, particularly nephrotoxicity and neurotoxicity, suggests that these drugs are not specific for the immune system and they affect other physiological systems. It has been shown that the nephrotoxicity of CsA correlates with its immunosuppressive activity based on the correlation of these two activities of a number of CsA analogs, suggesting that inhibition of calcineurin in the kidney by these drugs is responsible for this side effect (39). As one of the most abundant proteins in the brain, it is not surprising that inhibition of calcineurin by these drugs, particularly by FK506, which crosses the brain blood barrier with higher efficiency than CsA, would account for the neurotoxicity. Another important factor influencing the efficacy of immunosuppression and side effects of these drugs is the relative abundance of calcineurin in different tissues and cell types. The relatively moderate to low levels of calcineurin in T cells render them more susceptible to CsA and FK506 in comparison to neuronal cells that express much higher levels of CN. Thus, at the existing drug regimen, the plasma concentrations of the drugs are sufficiently high to inhibit calcineurin activity in T cells and the relevant kidney cells to near completion while they are not high enough to completely block calcineurin activity in neuronal and many other tissues where calcineurin also play important functions.
While CN is the key target for CsA and FK506 for their inhibition of intracellular calcium signaling in T cells, these drugs also bind and inhibit the functions of their respective immunophilin receptors. Both cyclophilin and FKBP exist in multiple isoforms. A general biochemical function of all immunophilins is the intrinsic peptidyl-prolyl cis-trans isomerase activity, which has been implicated in protein folding (9, 10, 40). In addition, some of the isoforms have each a unique cellular function. For example, cyclophilin D has been shown to be involved in regulating mitochondria permeability transition (41, 42). FKBP12.6 has been shown to be an integral part of the ryanodine receptor that regulates the release of calcium from sarcoplasmic reticulum in cardiac muscle (43, 44). It is inevitable that CsA and FK506 will affect the functions of many, if not all, of these individual immunophilins. Additional experiments will be required to determine whether inhibition of the different isoforms of immunophilins has a synergistic, additive or antagonistic effect on the inhibition of calcineurin in T cells and other cell types.
Endogenous protein regulators of calcineurin
As mentioned above, calcineurin A contains a built-in on-and-off switch in the form of its autoinhibitory domain and the adjacent calmodulin-binding domain, which allows for the activation of calcineurin by calmodulin upon calcium signaling. In addition, a large number of proteins have been reported to interact with calcineurin (45). Some of the calcineurin-interacting proteins, such as AKAP79, FKBP12, calsarcin and Bcl-2, appear to serve as scaffold proteins for calcineurin and their relevance to calcineurin’s function in T cells remains to be verified (reviewed in 45). For other calcineuin-interacting proteins, including Cabin1/cain, Carabin, RCANs, FKBP38, CHP, and A238L, inhibition of the intrinsic protein phosphatase activity of calcineurin has been demonstrated in vitro. Among this latter group, three proteins, Cabin1, Carabin and RCAN1 (or calcipressin 1), have been further investigated for their roles in TCR signaling at both cellular and animal levels (Fig. 3).
Fig. 3. The endogenous protein inhibitors of calcineurin.
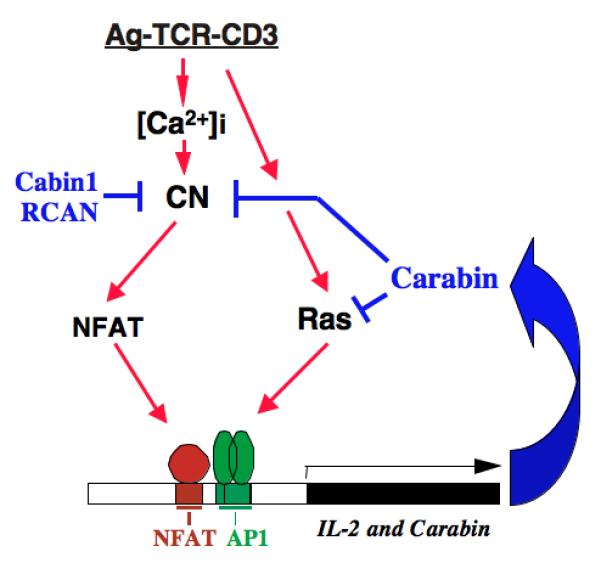
Highlighted are three well-characterized endogenous protein inhibitors of calcineurin, Cabin1, RCAN and Carabin. Color code: red, activation; blue, inhibition.
Cabin1 was initially identified as one of two hits from a mouse T-cell cDNA library in a yeast two-hybrid screen for calcineurin-binding proteins and was named as such for its Calcineurin binding activity (46). A rat homolog was independently identified in search for new target of the FKBP-FK506 complex other than calcineurin and was named cain, for its calcineurin inhibitory activity (47). Similar to calcineurin, Cabin1 is ubiquitously expressed in all tissues and cell types examine, including spleen, leukocytes, and T cells (46). In sharp contrast to its relatively large size with 2,220 amino acids, the domain that is both necessary and sufficient to bind calcineurin only occupies a small domain close to the C-terminus (46, 47). Interestingly, this domain contains the so-called PxIxIT motif (PEITVT in Cabin1) that is a calcineurin-binding motif in the NFAT family of calcineurin substrates (32). It is thus not surprising that overexpression of the C-terminal calcineurin-binding domain of Cabin1 is sufficient to block TCR-mediated IL-2 promoter-driven luciferase reporter genes (46). These observations suggest that one of the functions of Cabin1 may be to set a higher threshold for activation of calcineuin and to help maintain calcineurin in an inactive state in resting T cells. This hypothesis, however, remains to be proven, as the Cabin1ΔC knockout animals, which express a C-terminally truncated mutant of Cabin1 missing the calcineurin-binding domain, exhibited no gross defects in overall calcineurin activity in comparison to wild type control (48). Nor can this hypothesis be refuted since the calcineurin enzymatic assay using cell lysates in vitro is not sensitive and could have hiden a difference in cellular calcineurin activity in vivo. In a series of subsequent experiments, Cabin1 was found to possess yet another important function, as a calcium-dependent transcriptional corepressor of the transcription factor myocyte enhencer factor 2, which led to the identification of an entirely distinct calcium signaling module that also plays an important role in calcium signaling in T cells (vide infra).
Carabin was identified from the same yeast two-hybrid screen that led to the identification of Cabin1 (46). Although Carabin binds to and inhibits the phosphatase activity of calcineurin, similar to Cabin1, it differs from Cabin1 in several aspects (49). First, unlike Cabin1, the minimal calcineurin-binding domain in Carabin does not contain the PIxIxT motif that is shared with the calcineurin substrate NFAT. Carabin appears to directly bind to the active site of calcineurin and is expected to inhibit a broader range of substrates in addition to NFAT. Second, Carabin is a dual functional protein with a second functional domain being a Ras/Rab GAP. It possesses Ras GAP activity both in vitro and in vivo and selectively inhibited, upon ectopic overexpression in T cells, the phosphorylation and activation of ERKs without affecting the activity of JNK or p38. Carabin is thus a dual functional inhibitor of the intracellular signaling pathway through inhibition of both calcineurin and Ras. Third, Carabin expression is itself regulated by the calcineurin-NFAT pathway. In resting T cells, Carabin is expressed at relatively low levels. Upon TCR signaling, Carabin is induced over a period of 8-12 h before its level decreases. This induction by TCR agonists is sensitive to inhibition by CsA, consistent with the presence of multiple NFAT consensus binding sites present in the Carabin promoter. The calcium-calcineurin-NFAT dependent induction of Carabin makes it a unique endogenous inhibitor of calcineurin, placing it in a negative regulatory loop in the downstream calcium signaling cascade (Fig. 3). The negative regulatory role of Carabin in calcium-calcineurin signaling in T cells was supported by the dramatic upregulation of IL-2 transcription in response to antigen or TCR agonists in human and mouse T cells in which Carabin was knocked down using lentivirus-mediated shRNA.
Unlike Cabin1 and Carabin that are expressed only in higher eukaryotes, the RCAN family of proteins is conserved from humans to yeast (50-52). RCANs are a family of at least three proteins in mammals, including RCAN1 (also known as calcipressin 1, DSCR1, MCIP1, ADPT78, or CALP1), RCAN2 (also known as DSCR1L1, MCIP2, ZAKI-4 or CALP2), and RCAN3 (also known as DSCR1L2, MCIP3 or CALP3). A unique feature of RCANs among all known calcineurin-interacting proteins is that they appear to play both positive and negative regulatory roles in calcineurin activity in certain cell types depending in part on their cellular concentrations. Thus, knockout of RCAN from either yeast or mice led to a significant decrease in calcineurin activity in yeast and muscle cells and so did overexpression of RCANs (51, 53). Another interesting feature of RCANs is that their expression appeared to be regulated by calcium and calcineurin in both yeast and mouse muscle cells, suggesting the RCANs can serve in a negative feedback loop in a manner similar to that of Carabin. Somewhat surprisingly, neither of those two features was observed for RCAN1 in T cells (54). The total calcineurin activity was increased, rather than decreased, in RCAN1-deficient mice, possibly due to compensation by RCAN2 or RCAN3. Moreover, there was no evidence that RCAN1 level increased upon TCR signaling.
While the RCAN1 knockout animals developed normally and there was no gross defects in thymocyte development as judged by the total number of double and single positive T cells in the periphery, they displayed a unique Th1 deficiency, revealing an important function of RCAN1 in setting up the threshold of TCR signaling and the differential sensitivity of different target genes to the strength of calcineurin signaling (54). It was found that the Th1 cells underwent premature apoptosis upon TCR signaling due to abnormal upregulation of Fas ligand. In addition, the Th1-specific cytokine IFN-γ was also significantly downregulated in RCAN1 null T cells. The underlying cause of this unique phenotype was elegantly shown to be the change in threshold of calcineurin activation in RCAN1 knockout cells in comparison to that of wild type T cells. Thus, one of the key functions of RCAN1 is to maintain a relatively higher threshold for calcineurin activation to ensure that an appropriate set of calcineurin target genes will be expressed under given strength of TCR stimulation. It also highlighted the role of the degree of activation of calcineurin in preferentially affecting the expression of FasL over other target genes. In contrast to RCAN1, the physiological functions of other members of RCAN family, RCAN2 and RCAN3, in TCR signaling remain to be determined.
The missing link between calcium and calcineurin during TCR signaling
It is well established that calmodulin and calcineurin interpret the increase in intracellular calcium signaling during TCR signaling to cause reprogramming in gene expression through dephosphorylation of NFAT and other substrates of calcineurin. The influx of calcium through the plasma membrane of T cells has two components, one being the net amount of calcium ions entering the cytosol of T cells and the other being the calcium oscillation frequency. Importantly, it has been shown that the activation of NFAT is dependent on the oscillation frequency in T cells (55). Thus, artificially decreasing the oscillation frequency can lead to a dramatic decrease in NFAT reporter gene activation. More recently, it has been shown that perturbation of the voltage-gated potassium channel, Kv1.3, by small ligand can impair activation of calcineurin by altering the calcium oscillation frequency in Jurkat T cells (56). How calmodulin and calcineurin sense the appropriate calcium oscillation frequency, however, remains an outstanding question. It is worth pointing out that in addition to calmodulin, the regulatory B subunit of calcineurin is also a calcium binding protein with four EF hands. To date, the precise role of CNB in calcineurin activity other than its stabilizing effect has remained unknown. It is tempting to speculate that the calcium-binding CNB subunit may play a part in sensing calcium oscillation frequency during calcium signaling.
Cabin1, class II HDACs and MEF2
MEF2 is a family of four transcription factors, MEF2A-D, that were initially implicated in gene transcription leading to muscle cell differentiation (57). Although they were found to be ubiquitously expressed in all tissues and cell types including T cells, their role in calcium signaling and particularly in T cells remained unknown. It was after MEF2 was identified as an interacting protein of Cabin1 (and HDACs), and through the ensuing characterization of the interaction between Cabin1 and MEF2 that a role of MEF2 in calcium signaling in general, and in mediating intracellular calcium signaling in T cells in particular, was unraveled using TCR-mediated T-cell hybridoma apoptosis as a model system (58-60).
MEF2 is composed of a highly conserved N-terminal MADS/MEF2S domain responsible for homo-or heterodimerization and DNA binding and a divergent C-terminal domain involved in transactivation that is unique for each isoform. It is constitutively bound to DNA in the nucleus regardless of the activation status of T cells (60). In the absence of calcium signaling, it is associated with Cabin1 along with its associated class I HDACs and a histone methyltransferase or class II HDACs (59, 61). Together, the HDAC and methyltransferase-containing ternary complex on MEF2 silence the promoter containing MEF2 binding sites. Upon an increase in intracellular calcium concentration during TCR signaling, the nuclear subset of calmodulin binds to Cabin1 and class II HDACs, releasing them from MEF2 (58, 62). This leads to the association of the transcriptional coactivator p300 (59). The calcium-dependent dynamic switch in partners from HDAC/methyltransferase to HAT is made possible by another built-in molecular switch in the form of a merged overlapping MEF2-binding domain and calmodulin-binding domain in Cabin1 and class II HDACs, which converts MEF2 from a transcriptional repressor to a transcriptional activator (Fig. 4). These findings also revealed an important role of MEF2 in sensing and transducing calcium signal in various cellular processes. They pointed to the existence of another calcium signaling module consisting of MEF2, Cabin1/class II HDACs and p300 that is independent of the well-established calcineurin-NFAT signaling module (Fig. 4). In addition to extensive biochemical evidence for the MEF2-based calcium signaling module, the crystal structure of the Cabin1-MEF2-DNA complex provided further support for this model (63).
Fig. 4. The calcineurin-NFAT vs. MEF2-Cabin1/class II HDAC signaling modules in the regulation of IL-2 transcription.
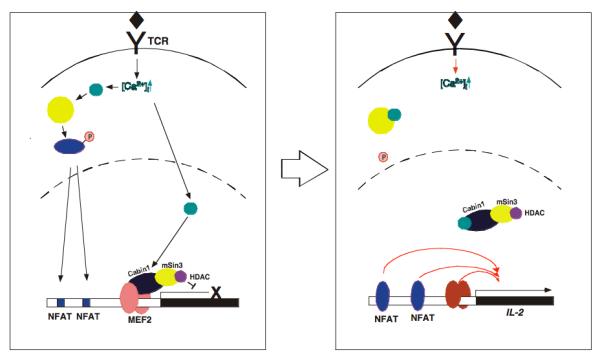
The calcineurin-NFAT signaling module works through dephosphorylation of NFAT and its nuclear translocation into the nucleus to bind the IL-2 promoter. In contrast, the MEF2-Cabin1/class II HDAC signaling module operates via calmodulin-dependent release of Cabin1 or class II HDACs from MEF2, making room for association by p300 (See Fig. 5 for more detail).
The role of the MEF2-Cabin1-p300 in calcium sensing and signaling was initially identified and characterized in the context of TCR-mediated thymocyte apoptosis and the induction of the orphan nuclear receptor Nur77 (58, 60). In an effort to assess the importance of Cabin1 in thymocyte apoptosis, a knockout mouse strain was produced that expressed a truncation mutant of Cabin1 lacking the C-terminal calcineurin-binding domain and the MEF2-binding domain (48). Given the critical role of both calcineurin and MEF2 in mediating Nur77 expression and apoptosis of T-cell hybridoma, it was expected that deletion of the two C-terminal domains would cause a defect in thymocyte negative selection. Surprisingly, the Cabin1ΔC mutant displayed no deficiency in thymocyte development with normal populations of double positive thymocytes and single positive T cells (48). These observations ruled out a role of Cabin1 and MEF2 as well as Nur77 in TCR-mediated thymocyte apoptosis and negative selection. Unexpectedly, when the response of peripheral CD4+ T cells were stimulated with either PMA/ionomycin or anti-CD3 antibodies, a dramatic increase in the production of IL2, IFN-γ and other cytokines was observed relative to the same T-cell population isolated form wildtype animals, suggesting a previously unknown role of Cabin1 and MEF2 in signal transduction in helper T cells (48). As both the calcineurin-binding domain and the MEF2-binding domain were missing in the Cabin1ΔC mutant animal, the abnormal upregulation of cytokine production could have been attributed to either an upregulation of calcineurin or MEF2, the latter due to the absence of the associated Cabin1-HDAC repressive complex. An examination of NFAT, however, revealed no difference in its dephosphorylation and nuclear translocation between the Cabin1ΔC mutant and wild type animals, leaving the dysregulation of MEF2 as the remaining cause of the observed phenotype.
Although MEF2 has been found to be expressed in T cells, it was not known to be involved in TCR signaling leading to cytokine gene expression. In particular, MEF2 sites had not been found in the well-characterized IL-2 promoter or promoters of other cytokines that were upregulated in the Cabin1ΔC T cells. Thus, the hint from the Cabin1ΔC mutant animal raised another question as to how MEF2 participates in the regulation of cytokine gene expression. A reexamination of the IL-2 proximal promoter revealed a site that bears some, but not all, characteristics of a consensus MEF2 binding site (64). This site happens to be in a region of long-standing interest and confusion as it contains a TATA element upstream of the established TATA box. It had been thought that this region might represent a unique second TATA box in the IL-2 gene. The sequence, ‘CATAATATTT’ in the human gene and ‘CATATTATTT’ in the mouse gene, is reminiscent of the consensus MEF2 binding sequence ‘G(A/T)8C’ in that they do contain a purine base pair followed by at least eight contiguous pyrimidine base pairs. But, they lack a purine cap at the 3′ end. Using gel mobility shift assay with oligonucleotide probes corresponding to the putative human MEF2 binding sequence, it was shown that it indeed bound to MEF2 specifically (64). Using a promoter sequence with a mutation in the putative MEF2-binding site, it was shown by CHIP assay that this site is required for MEF2 binding. Most importantly, when MEF2D, the most abundant isoform of MEF2 in peripheral T cells, was knocked down using lentivirus-mediated shRNA, it was found that levels of IL-2 transcripts as well as secreted IL-2 in response to stimulation by anti-CD3 and anti-CD28 antibodies were dramatically reduced in primary human T lymphocytes. These results established a key role of MEF2 and its partner proteins Cabin1/mSin3/HDAC1/2 in the regulation of calcium-dependent transcription of IL-2 and other cytokines. The association of the repressive MEF2-Cabin1-mSin3-HDAC/methyltransferase complex with the IL-2 promoter in the resting naïve T cells also offers an explanation on how this and the promoters of other cytokines are kept in a silenced state in the absence of TCR activation and the ensuing calcium signaling. The demonstration of MEF2 as a critical factor independent of NFAT for TCR-mediated cytokine production also suggests that MEF2 may serve as an alternative target for discovering and developing novel immunosuppressive agents.
In addition to Cabin1, class II HDACs including HDAC4, 5, 7, and 9, have been shown to directly bind to MEF2 via the same N-terminal MADS/MEF2S domain (65-68). More recent structural studies revealed that class II HDACs binds to the same pocket in MEF2 as Cabin1 (63, 69). Interestingly, HDAC4, and by prediction based on sequence similarity, all class II HDACs, binds to calmodulin via similar bifunctional domains that also mediate their interaction with MEF2 (62). Like Cabin1, the binding of class II HDACs is mutually exclusive to their binding to MEF2. Thus, upon calcium signaling, class II HDACs are dissociated from MEF2 in a similar manner as the Cabin1-mSin3-HDAC1/2 complex. In addition to regulation directly by calmodulin, class II HDACs as well as Cabin1, are subject to regulation by CaMKIV and other kinases, which constitute another mechanism of regulation of their interactions with MEF2. The regulation of the interaction between class II HDACs in the context of muscle cell differentiation by CaMKIV has been extensively investigated (70-72). However, the role of class II HDACs in calcium signaling in T cells remains to be investigated, especially in light of the existence of multiple members within the class II HDAC family and their potential redundancy with the Cabin1-mSin3-HDAC1/2 complex. The generation of conditional knockout animals for different members of class II HDAC family and different isoforms of MEF2 offers new opportunities to address those questions.
Calmodulin-dependent kinase II and IV
Like calcineurin, Ca2+/calmodulin-dependent kinases (CaMKs) are also activated upon calcium influx during TCR signaling. Similar to calcineurin, a C-terminal autoinhibitory domain and an adjacent calmodulin-binding domain form an intramolecular calcium-sensing on-and-off switch for this family of kinases. Two CaMKs have been shown to play important roles in mediating intracellular TCR signaling, CaMKII and CaMKIV. While CaMKIV plays a positive role in mediating calcium signaling in T cells, CaMKII has been shown to play a negative function in the same process.
The effect of CaMKIV on calcium-dependent activation of the IL-2 and other cytokine promoters is mediated in large part through the transcription factor CREB, which upon phosphorylation and activation by CaMKIV, turns on the expression of a number of immediate early genes including Jun and Fos, which bind to the IL-2 promoter in a cooperative manner with NFAT to drive transcription (73).
Given the essential role of Jun and Fos in mediating IL-2 transcription, one would predict that inhibition of CaMKIV should lead to inhibition of TCR-mediated IL-2 expression. That expectation was fulfilled in a transgenic model which expressed a catalytically inactive and dominant negative mutant of CaMKIV specifically in thymic T cells (74). T cells from the transgenic animal suffered dramatic reduction in IL-2 production upon stimulation with either anti-CD3 or PMA/ionomycin. In a separate study, CaMKIV deficient mice were generated and TCR signaling was examined (75). Surprisingly, naive CD4+ T cells from the CaMKIV-/- mice displayed normal production of cytokines, accompanied by normal level of induction of immediate early genes and CREB phosphorylation. Furthermore, defects in TCR-mediated IL-2 production were seen only in a subset of what appeared to be memory T cells. The results from the CaMKIV knockout mice suggested that CaMKIV may not be essential for TCR-mediated expression of immediate early genes including Fos and Jun, which could be attributable to the existence of a redundant pathway or kinase.
In contrast to the observations made in CaMKIV knockout mice, a significant defect in TCR-mediated IL-2 production in response to stimulation by anti-CD3 and anti-CD28 was observed in primary human T cells when the human counterpart was knocked down using lentivirus-mediated shRNA specific for CaMKIV (76). The discrepancy between human and mice T cells in their dependency on CaMKIV could be due to differences in the intrinsic wiring of the intracellular signaling network between the species. Alternatively, it may be do to a compensatory kinase that was induced during the relatively long-term development of the CaMKIV null mice as opposed to the more acute downregulation of human CaMKIV by shRNA. Parallel knockdown experiments using murine CD4+ T cells will be required to distinguish between the different scenarios.
Unlike CaMKIV, the multifunctional CaMKII appears to play a negative, rather than positive, role in the TCR-mediated IL-2 production (77, 78). Thus, ectopical expression of a constitutively active and calcium-independent form of CaMKII suppressed IL-2 promoter activation in response to stimulation by TCR agonists. This antagonism by CaMKII was seen with luciferase reporter genes for several transcription factors involved in IL-2 promoter activation, including NFAT and AP-1, without affecting reporters for c-fos or β-actin, although the extent of the inhibition for the individual transcription factor reporters was less than the IL-2 reporter (77). Importantly, the constitutively active form of CaMKII was found to be capable of blocking IL-2 promoter activation by the constitutively active form of calcineurin (77, 78), suggesting that the site of interference by CaMKII occurs either on calcineurin itself or downstream of it. It has been speculated that CaMKII may work by competing with calcineurin to phosphorylate NFAT (77). That CaMKII is not among the multitude of NFAT kinases identified to date does not support this hypothesis. Another possibility is that CaMKII may directly modulate the phosphatase activity of calcineurin through phosphorylation. Although CaMKII has been shown to phosphorylate calcineurin in vitro, this has not been demonstrated in vivo. Thus, it remains an open question as to how CaMKII inhibits calcineurin signaling with respect to IL-2 transcription.
Crosstalk between different calcium signaling transducers and signal integration
As we learn more about signal transduction pathways, it has become clear that almost none of the signaling pathways is linear as we knew them and they are formed by a network of signal transducers that also crosstalk to each other in a ‘horizontal’ fashion. Extensive crosstalk has also been seen even among the calcium signal transducers discussed above.
Calcineurin/NFAT and MEF2 via NFAT-MEF2-p300 interaction
MEF2 has relatively low intrinsic transactivation capacity, necessitating recruitment of other transcription factors and coactivators to induce robust gene activation, as has been seen in muscule cells (57). In T cells, at least in the context of the expression of the immediate early gene Nur77, it has been shown that MEF2 synergizes with NFAT to activate Nur77 transcription (60). Thus, upon the release of Cabin1 along with its associated mSin3 and HDAC1/2, MEF2 is bound to p300. The resultant MEF2-p300 complex only mildly activated Nur77 transcription. The concurrent activation of the calcineurin pathway led to the arrival of NFAT in the nucleus, which forms a ternary complex with both MEF2 and p300, stabilizing the MEF2-p300 complex. The newly formed MEF2-NFAT-p300 ternary complex on the Nur77 promoter causes a significant upregulation of transcription. It is worth noting that in this ternary complex, NFAT works in a new capacity as a transcriptional coactivator independent of DNA binding. Thus, the two seemingly independent calcium-signaling modules are capable of crosstalk to cause synergistic activation of target genes.
MEF2 and NFAT, via AP-1 transcription
In addition to their direct protein-protein interactions described above, MEF2 also indirectly crosstalks with NFAT through its regulation of AP-1. MEF2 is known to be involved in the transcriptional activation of a number of immediate early genes including Nur77 and c-jun (79, 80). As a key component of AP-1, the level of jun dictates the extent of NFAT-dependent activation of IL-2 promoter and those of other cytokines due to the configuration of the binding sites of NFAT and AP-1, which are in close proximity such that they bind to DNA in a cooperative manner (81, 82). Although it has not been extensively probed in peripheral T cells, it is anticipated that MEF2 has a profound effect on the expression levels of AP-1 and consequently impinge on the activity of NFAT in addition to its direct binding to the IL-2 enhancer to remodel the chromatin in a calcium-dependent manner.
CaMKIV and MEF2 via phosphorylation of Cabin1 and class II HDACs
There is extensive evidence that CaMKIV as well as other kinases positively regulates MEF2 through phosphorylation of class II HDACs in muscle cells, and regulates their subsequent nuclear export from the nucleus into the cytosol by 14-3-3 (70-72). This was also found to be true for Cabin1 (76). Thus, Cabin1 is phosphorylated by CaMKIV upon TCR signaling, leading to the creation of a docking site for 14-3-3τ. The association of 14-3-3τ with phosphorylated Cabin1 causes its nuclear export, effectively excluding Cabin1 from MEF2. Although it was shown that the CaMKIV-mediated and 14-3-3 dependent nuclear export of Cabin1 worked synergistically with calmodulin to directly compete with MEF2 for Cabin1 to dissociate Cabin1 from MEF2, whether Cabin1 and class II HDACs play additional role in the cytosol to further regulate other cellular events remains unknown.
Integration of signals from different individual calcium signal transducers
One of the most important outputs of calcium signaling in helper T cells is the production of IL-2 and other cytokines to amplify the immune response through the ensuing clonal expansion. As such, a key site of signal integration is at the promoter of IL-2 and other cytokines in the nucleus. A survey of what has been learnt about the calcium signal transducers alone and their mode of signal transmission is already giving rise to an extensive two or even three dimensional signaling network with many cross connections among the different signal transducers. Some of the connections are perhaps to enable functional redundancy, thus adding robustness to the signaling system while others are likely there to form an intricate circuit that can sense and interpret subtle differences in the extracellular antigen signal and mount a transcriptional response that also takes into consideration the intrinsic states of the responding cell. Only when most, if not all, the players involved in such a signaling network are fully understood as individual components of the entire network, can one begin to make sense of the integration of the signal to cause gene transcription essential for an appropriate T-cell response.
Fig. 5. Signal integration between the MEF2-Cabin1-p300 signaling module and the CN-NFAT signaling module.
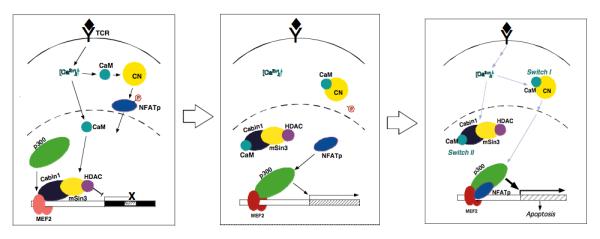
Calcium signal bifurcates with one activating calcineurin in the cytosol and the other activating MEF2 in the nucleus. Upon release of the Cabin1-mSin3-HDAC1/2 complex from MEF2, p300 is bound to the same pocket of MEF2. The newly translocated NFAT then forms a ternary complex with both MEF2 and p300 to stabilize the transcriptionally active complex, causing robust transcription of target genes.
Fig. 6. Crosstalk among the known calcium signal transducers in T cells and their signaling network.

Single-headed arrow signifies activation; blunt ended lines represents inhibition; double-headed arrows highlight mutual interaction.
Acknowledgments
I am grateful to all coworkers who have contributed to the work described in this review and to NIGMS for financial support. The careful proofreading of the manuscript by Woon-Kai Low, Benjamin Nacev, and Richard Ren is also gratefully acknowledged.
References
- 1.Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- 2.Liu J. FK506 and cyclosporin, molecular probes for studying intracellular signal transduction. Immunol Today. 1993;14:290–5. doi: 10.1016/0167-5699(93)90048-P. [DOI] [PubMed] [Google Scholar]
- 3.Wang JH, Desai R. Modulator binding protein. Bovine brain protein exhibiting the Ca2+-dependent association with the protein modulator of cyclic nucleotide phosphodiesterase. J Biol Chem. 1977;252:4175–84. [PubMed] [Google Scholar]
- 4.Klee CB, Krinks MH. Purification of cyclic 3′,5′-nucleotide phosphodiesterase inhibitory protein by affinity chromatography on activator protein coupled to Sepharose. Biochemistry. 1978;17:120–6. doi: 10.1021/bi00594a017. [DOI] [PubMed] [Google Scholar]
- 5.Stewart AA, Ingebritsen TS, Manalan A, Klee CB, Cohen P. Discovery of a Ca2+- and calmodulin-dependent protein phosphatase: probable identity with calcineurin (CaM-BP80) FEBS letters. 1982;137:80–4. doi: 10.1016/0014-5793(82)80319-0. [DOI] [PubMed] [Google Scholar]
- 6.Kincaid RL, Takayama H, Billingsley ML, Sitkovsky MV. Differential expression of calmodulin-binding proteins in B, T lymphocytes and thymocytes. Nature. 1987;330:176–8. doi: 10.1038/330176a0. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Farmer JD, Jr., Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–15. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 8.Handschumacher RE, Harding MW, Rice J, Drugge RJ. Cyclophilin: A Specific Cytosolic Binding Protein for Cyclosporin A. Science (New York, NY. 1984;226:544–7. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- 9.Harding MW, Galat A, Uehling DE, Schreiber S. A receptor for the immuno-supressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature. 1989;341:758–60. doi: 10.1038/341758a0. [DOI] [PubMed] [Google Scholar]
- 10.Siekierka JJ, Hung SHY, Poe M, Lin CS, Sigal NH. A cytosolic binding protein for the immunosuppressant FK506 has peptidyl-prolyl isomerase activity but is distinct from cyclophilin. Nature. 1989;341:755–7. doi: 10.1038/341755a0. [DOI] [PubMed] [Google Scholar]
- 11.Griffith JP, et al. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell. 1995;82:507–22. doi: 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]
- 12.Kissinger CR, et al. Crystal structures of human calcineurin and the human FKBP12-FK506-calcineurin complex. Nature. 1995;378:641–4. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- 13.Huai Q, et al. Crystal structure of calcineurin-cyclophilin-cyclosporin shows common but distinct recognition of immunophilin-drug complexes. Proc Natl Acad Sci USA. 2002;99:12037–42. doi: 10.1073/pnas.192206699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin L, Harrison SC. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc Natl Acad Sci USA. 2002;99:13522–6. doi: 10.1073/pnas.212504399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klee CB, Draetta GF, Hubbard MJ. Calcineurin. Adv Enzymol Relat Areas Mol Biol. 1988;61:149–200. doi: 10.1002/9780470123072.ch4. [DOI] [PubMed] [Google Scholar]
- 16.Jiang H, Xiong F, Kong S, Ogawa T, Kobayashi M, Liu JO. Distinct tissue and cellular distribution of two major isoforms of calcineurin. Mol Immunol. 1997;34:663–9. doi: 10.1016/s0161-5890(97)00054-0. [DOI] [PubMed] [Google Scholar]
- 17.Zhang BW, et al. T cell responses in calcineurin A alpha-deficient mice. J Exp Med. 1996;183:413–20. doi: 10.1084/jem.183.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bueno OF, Brandt EB, Rothenberg ME, Molkentin JD. Defective T cell development and function in calcineurin A beta -deficient mice. Proc Natl Acad Sci USA. 2002;99:9398–403. doi: 10.1073/pnas.152665399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neilson JR, Winslow MM, Hur EM, Crabtree GR. Calcineurin B1 is essential for positive but not negative selection during thymocyte development. Immunity. 2004;20:255–66. doi: 10.1016/s1074-7613(04)00052-4. [DOI] [PubMed] [Google Scholar]
- 20.Woronicz JD, Calnan B, Ngo V, Winoto A. Requirement for the orphan steroid receptor Nur77 in apoptosis of T-cell hybridomas. Nature. 1994;367:277–81. doi: 10.1038/367277a0. [DOI] [PubMed] [Google Scholar]
- 21.Liu ZG, Smith SW, McLaughlin KA, Schwartz LM, Osborne BA. Apoptotic signals delivered through the T-cell receptor of a T-cell hybrid require the immediate-early gene nur77. Nature. 1994;367:281–4. doi: 10.1038/367281a0. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg J, Huang HB, Kwon YG, Greengard P, Nairn AC, Kuriyan J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase 1. Nature. 1995;376:745–53. doi: 10.1038/376745a0. [DOI] [PubMed] [Google Scholar]
- 23.Egloff MP, Cohen PT, Reinemer P, Barford D. Crystal structure of the catalytic subunit of human protein phosphatase 1 and its complex with tungstate. J Mol Biol. 1995;254:942–59. doi: 10.1006/jmbi.1995.0667. [DOI] [PubMed] [Google Scholar]
- 24.Okamura H, et al. Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Molecular cell. 2000;6:539–50. doi: 10.1016/s1097-2765(00)00053-8. [DOI] [PubMed] [Google Scholar]
- 25.Zhu J, et al. Intramolecular masking of nuclear import signal on NF-AT4 by casein kinase I and MEKK1. Cell. 1998;93:851–61. doi: 10.1016/s0092-8674(00)81445-2. [DOI] [PubMed] [Google Scholar]
- 26.Okamura H, Garcia-Rodriguez C, Martinson H, Qin J, Virshup DM, Rao A. A conserved docking motif for CK1 binding controls the nuclear localization of NFAT1. Molecular and cellular biology. 2004;24:4184–95. doi: 10.1128/MCB.24.10.4184-4195.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chow CW, Davis RJ. Integration of calcium and cyclic AMP signaling pathways by 14-33. Molecular and cellular biology. 2000;20:702–12. doi: 10.1128/mcb.20.2.702-712.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase 3. Science (New York, NY. 1997;275:1930–4. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 29.Gwack Y, et al. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature. 2006;441:646–50. doi: 10.1038/nature04631. [DOI] [PubMed] [Google Scholar]
- 30.Timmerman LA, Clipstone NA, Ho SN, Northrop JP, Crabtree GR. Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature. 1996;383:837–40. doi: 10.1038/383837a0. [DOI] [PubMed] [Google Scholar]
- 31.Shibasaki F, Price ER, Milan D, McKeon F. Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF AT4. Nature. 1996;382:370–3. doi: 10.1038/382370a0. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Cozar FJ, et al. Two-site interaction of nuclear factor of activated T cells with activated calcineurin. J Biol Chem. 1998;273:23877–83. doi: 10.1074/jbc.273.37.23877. [DOI] [PubMed] [Google Scholar]
- 33.Aramburu J, Garcia-Cozar F, Raghavan A, Okamura H, Rao A, Hogan PG. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Molecular cell. 1998;1:627–37. doi: 10.1016/s1097-2765(00)80063-5. [DOI] [PubMed] [Google Scholar]
- 34.Li H, Zhang L, Rao A, Harrison SC, Hogan PG. Structure of calcineurin in complex with PVIVIT peptide: portrait of a low-affinity signalling interaction. Journal of molecular biology. 2007;369:1296–306. doi: 10.1016/j.jmb.2007.04.032. [DOI] [PubMed] [Google Scholar]
- 35.Aramburu J, Yaffe MB, Lopez-Rodriguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science (New York, NY. 1999;285:2129–33. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- 36.Roehrl MH, Kang S, Aramburu J, Wagner G, Rao A, Hogan PG. Selective inhibition of calcineurin-NFAT signaling by blocking protein-protein interaction with small organic molecules. Proc Natl Acad Sci USA. 2004;101:7554–9. doi: 10.1073/pnas.0401835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J, Masuda ES, Tsuruta L, Arai N, Arai K. Two independent calcineurin-binding regions in the N-terminal domain of murine NF-ATx1 recruit calcineurin to murine NF ATx1. J Immunol. 1999;162:4755–61. [PubMed] [Google Scholar]
- 38.Martinez-Martinez S, Rodriguez A, Lopez-Maderuelo MD, Ortega-Perez I, Vazquez J, Redondo JM. Blockade of NFAT activation by the second calcineurin binding site. The Journal of biological chemistry. 2006;281:6227–35. doi: 10.1074/jbc.M513885200. [DOI] [PubMed] [Google Scholar]
- 39.Sigal NH, et al. Is cyclophilin involved in the immunosuppressive and nephrotoxic mechanism of action of cyclosporin A? The Journal of experimental medicine. 1991;173:619–28. doi: 10.1084/jem.173.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature. 1989;337:476–8. doi: 10.1038/337476a0. [DOI] [PubMed] [Google Scholar]
- 41.Tanveer A, et al. Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. European journal of biochemistry / FEBS. 1996;238:166–72. doi: 10.1111/j.1432-1033.1996.0166q.x. [DOI] [PubMed] [Google Scholar]
- 42.Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ. Cyclophilin D as a drug target. Current medicinal chemistry. 2003;10:1485–506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]
- 43.Jayaraman T, et al. FK506 binding protein associated with the calcium release channel (ryanodine receptor) The Journal of biological chemistry. 1992;267:9474–7. [PubMed] [Google Scholar]
- 44.Wehrens XH, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–40. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 45.Liu JO. Endogenous protein inhibitors of calcineurin. Biochem Biophys Res Commun. 2003;311:1103–9. doi: 10.1016/j.bbrc.2003.10.020. [DOI] [PubMed] [Google Scholar]
- 46.Sun L, Youn HD, Loh C, Stolow M, He W, Liu JO. Cabin 1, a negative regulator for calcineurin signaling in T lymphocytes. Immunity. 1998;8:703–11. doi: 10.1016/s1074-7613(00)80575-0. [DOI] [PubMed] [Google Scholar]
- 47.Lai MM, Burnett PE, Wolosker H, Blackshaw S, Snyder SH. Cain, a novel physiologic protein inhibitor of calcineurin. J Biol Chem. 1998;273:18325–31. doi: 10.1074/jbc.273.29.18325. [DOI] [PubMed] [Google Scholar]
- 48.Esau C, Boes M, Youn HD, Tatterson L, Liu JO, Chen J. Deletion of calcineurin and myocyte enhancer factor 2 (MEF2) binding domain of Cabin1 results in enhanced cytokine gene expression in T cells. J Exp Med. 2001;194:1449–59. doi: 10.1084/jem.194.10.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan F, Sun L, Kardian DB, Whartenby KA, Pardoll DM, Liu JO. Feedback inhibition of calcineurin and Ras by a dual inhibitory protein Carabin. Nature. 2007;445:433–6. doi: 10.1038/nature05476. [DOI] [PubMed] [Google Scholar]
- 50.Gorlach J, Fox DS, Cutler NS, Cox GM, Perfect JR, Heitman J. Identification and characterization of a highly conserved calcineurin binding protein, CBP1/calcipressin, in Cryptococcus neoformans. EMBO J. 2000;19:3618–29. doi: 10.1093/emboj/19.14.3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kingsbury TJ, Cunningham KW. A conserved family of calcineurin regulators. Genes Dev. 2000;14:1595–604. [PMC free article] [PubMed] [Google Scholar]
- 52.Rothermel B, Vega RB, Yang J, Wu H, Bassel-Duby R, Williams RS. A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J Biol Chem. 2000;275:8719–25. doi: 10.1074/jbc.275.12.8719. [DOI] [PubMed] [Google Scholar]
- 53.Vega RB, et al. Dual roles of modulatory calcineurin-interacting protein 1 in cardiac hypertrophy. Proc Natl Acad Sci USA. 2003;100:669–74. doi: 10.1073/pnas.0237225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryeom S, Greenwald RJ, Sharpe AH, McKeon F. The threshold pattern of calcineurin-dependent gene expression is altered by loss of the endogenous inhibitor calcipressin. Nat Immunol. 2003;4:874–81. doi: 10.1038/ni966. [DOI] [PubMed] [Google Scholar]
- 55.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–6. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- 56.Ren YR, et al. Clofazimine is a novel inhibitor of human Kv1.3 potassium channel and calcium signaling in T lymphocytes. PLoS One. 2008 doi: 10.1371/journal.pone.0004009. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Molkentin JD, Olson EN. Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc Natl Acad Sci USA. 1996;93:9366–73. doi: 10.1073/pnas.93.18.9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Youn HD, Sun L, Prywes R, Liu JO. Apoptosis of T cells mediated by Ca2+-induced release of the transcription factor MEF2. Science (New York, NY. 1999;286:790–3. doi: 10.1126/science.286.5440.790. [DOI] [PubMed] [Google Scholar]
- 59.Youn HD, Liu JO. Cabin1 represses MEF2-dependent Nur77 expression and T cell apoptosis by controlling association of histone deacetylases and acetylases with MEF2. Immunity. 2000;13:85–94. doi: 10.1016/s1074-7613(00)00010-8. [DOI] [PubMed] [Google Scholar]
- 60.Youn HD, Chatila TA, Liu JO. Integration of calcineurin and MEF2 signals by the coactivator p300 during T-cell apoptosis. EMBO J. 2000;19:4323–31. doi: 10.1093/emboj/19.16.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jang H, Choi DE, Kim H, Cho EJ, Youn HD. Cabin1 represses MEF2 transcriptional activity by association with a methyltransferase, SUV39H1. J Biol Chem. 2007;282:11172–9. doi: 10.1074/jbc.M611199200. [DOI] [PubMed] [Google Scholar]
- 62.Youn HD, Grozinger CM, Liu JO. Calcium regulates transcriptional repression of myocyte enhancer factor 2 by histone deacetylase 4. J Biol Chem. 2000;275:22563–7. doi: 10.1074/jbc.C000304200. [DOI] [PubMed] [Google Scholar]
- 63.Han A, Pan F, Stroud JC, Youn HD, Liu JO, Chen L. Sequence-specific recruitment of transcriptional co-repressor Cabin1 by myocyte enhancer factor 2. Nature. 2003;422:730–4. doi: 10.1038/nature01555. [DOI] [PubMed] [Google Scholar]
- 64.Pan F, Ye Z, Cheng L, Liu JO. Myocyte enhancer factor 2 mediates calcium-dependent transcription of the interleukin-2 gene in T lymphocytes: a calcium signaling module that is distinct from but collaborates with the nuclear factor of activated T cells (NFAT) J Biol Chem. 2004;279:14477–80. doi: 10.1074/jbc.C300487200. [DOI] [PubMed] [Google Scholar]
- 65.Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999;18:5099–107. doi: 10.1093/emboj/18.18.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lemercier C, Verdel A, Galloo B, Curtet S, Brocard MP, Khochbin S. mHDA1/HDAC5 histone deacetylase interacts with and represses MEF2A transcriptional activity. J Biol Chem. 2000;275:15594–9. doi: 10.1074/jbc.M908437199. [DOI] [PubMed] [Google Scholar]
- 67.Dressel U, Bailey PJ, Wang SC, Downes M, Evans RM, Muscat GE. A dynamic role for HDAC7 in MEF2-mediated muscle differentiation. J Biol Chem. 2001;276:17007–13. doi: 10.1074/jbc.M101508200. [DOI] [PubMed] [Google Scholar]
- 68.Zhang CL, McKinsey TA, Lu JR, Olson EN. Association of COOH-terminal-binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J Biol Chem. 2001;276:35–9. doi: 10.1074/jbc.M007364200. [DOI] [PubMed] [Google Scholar]
- 69.Han A, He J, Wu Y, Liu JO, Chen L. Mechanism of recruitment of class II histone deacetylases by myocyte enhancer factor 2. J Mol Biol. 2005;345:91–102. doi: 10.1016/j.jmb.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 70.McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–11. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc Natl Acad Sci USA. 2000;97:14400–5. doi: 10.1073/pnas.260501497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McKinsey TA, Zhang CL, Olson EN. Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol. 2001;21:6312–21. doi: 10.1128/MCB.21.18.6312-6321.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ho N, Gullberg M, Chatila T. Activation protein 1-dependent transcriptional activation of interleukin 2 gene by Ca2+/calmodulin kinase type IV/Gr. J Exp Med. 1996;184:101–12. doi: 10.1084/jem.184.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anderson KA, Ribar TJ, Illario M, Means AR. Defective survival and activation of thymocytes in transgenic mice expressing a catalytically inactive form of Ca2+/calmodulin-dependent protein kinase IV. Mol Endocrinol. 1997;11:725–37. doi: 10.1210/mend.11.6.0011. [DOI] [PubMed] [Google Scholar]
- 75.Anderson KA, Means AR. Defective signaling in a subpopulation of CD4(+) T cells in the absence of Ca(2+)/calmodulin-dependent protein kinase IV. Mol Cell Biol. 2002;22:23–9. doi: 10.1128/MCB.22.1.23-29.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pan F, Means AR, Liu JO. Calmodulin-dependent protein kinase IV regulates nuclear export of Cabin1 during T-cell activation. EMBO J. 2005;24:2104–13. doi: 10.1038/sj.emboj.7600685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nghiem P, Ollick T, Gardner P, Schulman H. Interleukin-2 transcriptional block by multifunctional Ca2+/calmodulin kinase. Nature. 1994;371:347–50. doi: 10.1038/371347a0. [DOI] [PubMed] [Google Scholar]
- 78.Hama N, Paliogianni F, Fessler BJ, Boumpas DT. Calcium/calmodulin-dependent protein kinase II downregulates both calcineurin and protein kinase C-mediated pathways for cytokine gene transcription in human T cells. J Exp Med. 1995;181:1217–22. doi: 10.1084/jem.181.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Han TH, Prywes R. Regulatory role of MEF2D in serum induction of the c-jun promoter. Mol Cell Biol. 1995;15:2907–15. doi: 10.1128/mcb.15.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coso OA, et al. Signaling from G protein-coupled receptors to the c-jun promoter involves the MEF2 transcription factor. Evidence for a novel c-jun amino-terminal kinase-independent pathway. J Biol Chem. 1997;272:20691–7. doi: 10.1074/jbc.272.33.20691. [DOI] [PubMed] [Google Scholar]
- 81.Chen L, Glover JN, Hogan PG, Rao A, Harrison SC. Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature. 1998;392:42–8. doi: 10.1038/32100. [DOI] [PubMed] [Google Scholar]
- 82.Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476–89. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]