

Abstract
Axonal dysfunction is the major phenotypic change in many neurodegenerative diseases, but the processes underlying this impairment are not clear. Modifier of cell adhesion (MOCA) is a presenilin binding protein that functions as a guanine nucleotide exchange factor for Rac1. The loss of MOCA in mice leads to axonal degeneration and causes sensorimotor impairments by decreasing cofilin phosphorylation and altering its upstream signaling partners LIM kinase and p21-activated kinase, an enzyme directly downstream of Rac1. The dystrophic axons found in MOCA-deficient mice are associated with abnormal aggregates of neurofilament protein, the disorganization of the axonal cytoskeleton, and the accumulation of autophagic vacuoles and polyubiquitinated proteins. Furthermore, MOCA deficiency causes an alteration in the actin cytoskeleton and the formation of cofilin-containing rod-like structures. The dystrophic axons show functional abnormalities, including impaired axonal transport. These findings demonstrate that MOCA is required for maintaining the functional integrity of axons and define a model for the steps leading to axonal degeneration.
Keywords: MOCA, DOCK180, guanine nucleotide exchange factor, axonal degeneration, neurodegeneration, sensorimotor defects
Introduction
Axonal dystrophy, characterized by swellings and varicosities that interrupt the continuity of axons of dysfunctional or degenerating neurons, is a hallmark of many neurological disorders, including Alzheimer's disease (AD), Huntington's disease, amyotrophic lateral sclerosis, and Charcot-Marie-Tooth disease (Medana and Esiri, 2003; Coleman, 2005). Axonal dystrophy also occurs in response to inflammation, metabolic disturbances, myelin disorders, and ischemia (Medana and Esiri, 2003; Coleman, 2005). Axonal swellings often contain aberrant accumulations of axonal cargos such as organelles, vesicles, and cytoskeletal proteins such as neurofilaments (NFs) and microtubules (Chevalier-Larsen and Holzbaur, 2006; Duncan and Goldstein, 2006). Some axonal swellings are filled with autophagic structures resulting from the failure of lysosomal-mediated organelle transport and protein degradation (Nixon, 2006; Rubinsztein, 2006). However, the mechanisms underlying the formation of the axonal pathologies that are shared among many neurological diseases are not well understood.
Modifier of cell adhesion (MOCA, DOCK3) was discovered as a presenilin (PS) binding protein (PBP), and is only expressed in neurons and in the testis (Kashiwa et al., 2000). It is a member of DOCK180 family of proteins and functions as a guanine nucleotide exchange factor (GEF) (Côté and Vuori, 2002; Grimsley et al., 2004; Namekata et al., 2004; Meller et al., 2005). Mutations in the human MOCA gene have been linked to attention deficit hyperactivity disorder (ADHD) (de Silva et al., 2003). MOCA regulates metabolism of cell adhesion proteins, such as amyloid precursor protein (APP) and N-cadherin, and is also involved in modulating neurite growth (Chen et al., 2002, 2005). MOCA is found in AD tangles and regulates tau phosphorylation (Chen et al., 2001). In the present study, we generated a moca gene knock-out mouse line and demonstrate that the deletion of MOCA leads to a central axonal dystrophy, a concomitant loss of sensorimotor function, and impaired axoplasmic flow, all of which are common to many age-associated neurological diseases. In addition, we demonstrated an alteration of actin dynamics regulated by cofilin and its related kinase pathway that links MOCA to the cytoskeleton network required for axoplasmic function.
Materials and Methods
Reagents, antibodies, and animal care.
An affinity-purified polyclonal antibody that recognizes amino acid residues 2012–2027 of MOCA was generated in rabbits (Kashiwa et al., 2000). Other antibodies used in the present study include the following: glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (catalog #G8795), NF-68 (catalog #N5139) (Sigma-Aldrich); SMI-31 (catalog #SMI-31R), SMI-32 (catalog #SMI-32R) (Covance), APP (22C11, catalog #MAB348), choline acetyltransferase (catalog #AB143), NF-150 (catalog #AB1981), phosphorylated cofilin (catalog #AB3831), GFAP (catalog #AB5804), ubiquitin (catalog #MAB1510) (Millipore), cofilin (612144 from BD Biosciences), mAb 22 (from M. Maloney, Stanford University, Palo Alto, CA); phosphorylated cofilin (catalog #3313), LIM kinase 1 (LIMK1) (catalog #3842), phosphorylated LIMK (catalog #3841), p21-activated kinase (PAK)1/2/3 (catalog #2604, Cell Signaling), phosphorylated PAK1/2/3 pSer141 (catalog #44-940G, Invitrogen); and Iba1 (catalog #019-19741, Wako). Fluoro Jade C and Fluoro-Gold (fluorogold) were purchased from Millipore and FluoroChrome, respectively.
All procedures for animal studies adhered to the Guide for the Care and Use of Laboratory Animals and were approved by the Salk Institute Animal Care and Use Committee. Mice were housed in a temperature- and humidity-controlled environment on a 12 h light/dark cycle with free access to food and water.
Construction of the moca−/− mouse line.
A moca gene knock-out (moca−/−) targeting vector was made to include a 5′ arm containing a 6 kb HindIII-StuI fragment upstream of exon 2 that was fused in frame to the β-galactosidase (β-gal) gene and a 3′ arm containing a 3.1 kb EcoR I fragment in the downstream of exon 2. A pgk-neo cassette [used for positive selection of embryonic stem (ES) cells] was placed between the 5′ arm and the 3′ arm, and a tk cassette (used for negative selection of ES cells) was placed downstream of the 3′ arm. The resulting construct is shown in supplemental Figure S1A (available at www.jneurosci.org as supplemental material) and is predicted to disrupt most of exon 2 of the moca gene as well as its downstream region. Because the β-gal gene is fused to the first 24 aa (including those derived from the first exon of the moca gene), the expression of β-gal gene is driven by the moca gene promoter. The targeting construct was introduced via electroporation into ES cells, and homologous recombination was verified by Southern blot analysis (supplemental Fig. S1B,C, available at www.jneurosci.org as supplemental material). The chimeric mice were generated and mated with C57BL/6 mice to produce hemizygous MOCA knock-out (moca+/−) mice. The hemizygous mice were identified by PCR genotyping and further crossed to each other to produce homozygous moca−/− mice (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). Moca−/− mice were backcrossed with C57BL/6 mice for six generations to produce a mouse line with the same C57BL/6 genetic background. C57BL/6 mice were chosen because this mouse line is most commonly used, and has a relatively known genetic background and an average performance on various behavioral tests. Loss of MOCA expression in the moca−/− mice was verified by Western blotting and by the expression of β-Gal.
Rotarod test.
The rotating rod apparatus (Harvard Apparatus) was used to assess motor performance and measure the ability of mice to improve motor skill performance with training. Mice were placed on the elevated accelerating rod (rotarod, 3 cm in diameter) beginning at 5 rpm/min for four trials per day for three consecutive days. Each trial lasted a maximum of 10 min, during which time the rotating rod underwent a linear acceleration from 4 to 40 rpm over the first 5 min of the trial and then remained at maximum speed for the remaining 5 min. Animals were scored for their latency (in seconds) to fall (height = 20 cm) for each trial. Animals rested a minimum of 10 min between trials to avoid fatigue.
Open field test.
The open field test was performed by using MED Associates hardware and the Activity Monitor software according to the manufacturer's instructions (MED Associates). To perform the test, each 2-month-old mouse from either the control or mutant group was placed in the testing chamber, and the activity was tracked by the Activity Monitor software (MED Associates). The time of each testing session was 100 min and the sample interval (the time of each segment) was 5 min. The multiple behavioral parameters were derived from the test, but only the travel distance was analyzed in the present study.
Light microscopic evaluation of muscle, sciatic nerve, and dorsal and ventral roots.
The fresh gastrocnemius, gluteal, and triceps muscles from both moca−/− and wild-type mice were collected and frozen in isopentane precooled in liquid nitrogen or immersion-fixed in 10% formalin before use. Frozen muscle sections (8-μm-thick) were evaluated by hematoxylin and eosin staining, and ATPase reactions at pH 9.8 and after preincubation at pH 4.6 and 4.3. The grading system used for statistical purposes was 0 = no abnormalities and 1 = abnormalities observed, such as the presence of internal nuclei, vacuoles, lack of normal striation, glycogen or mitochondrial aggregates, myonecrosis, basophilic or eosinophilic sarcoplasm, fibrosis, and fat accumulation.
Sciatic nerves were either processed for paraffin sections or immersion-fixed in 2.5% glutaraldehyde in 0.1 m phosphate buffer before processing for plastic sections. L5 dorsal and ventral roots were obtained from animals perfused through the heart with 2.5% phosphate-buffered glutaraldehyde. The vertebral columns were removed and fixed overnight in 2.5% phosphate-buffered glutaraldehyde (4°C) and roots were dissected out the next day. Sciatic nerves as well as dorsal and ventral roots were postfixed in osmium tetroxide, and dehydrated in serial alcohol solutions and propylene oxide before embedding in Araldite resin. Sections (1 μm) were stained with toluidine blue acid fuschin or p-phenylenediamine for light microscopy.
Computer-assisted analyses of myelinated fiber size–frequency distributions were performed on the sciatic nerve and L5 dorsal and ventral roots. For the sciatic nerve of each animal, a single thick section, sampled from the tibial fascicle midway between the sciatic notch and popliteal fossa, was analyzed as described in detail previously (Mizisin et al., 1997). Size–frequency distributions for dorsal and ventral roots were based on single sections from each animal sampled 5 mm proximal to the dorsal root ganglion. Briefly, video images were obtained with an Olympus BH-2 light microscope and attached Cohu 5000 series television camera interfaced with a Hewlett Packard Compaq computer running NIH Image 1.55 software. Using the 40× objective, the final magnification of the digitized image was equivalent to 4.05 pixels/μm. Nonoverlapping fields were sampled by systematic serpentine progression across all fascicles. Myelinated fibers were individually identified and selected before being sorted with an automated process into bins based on axonal diameter.
Immunohistochemistry.
Brains were fixed with 4% paraformaldehyde in 100 mm sodium tetraborate, pH 9.5, for 3 h, cryoprotected with 20% sucrose-potassium-PBS (KPBS), and cryostat sectioned into coronal (30 μm) sections. Sections were submerged in 0.3% H2O2 for 10 min to eliminate endogenous peroxidase activity and treated with 1% borate to eliminate free paraformaldehyde. Sections were incubated with primary antibody in 0.3% Triton X-100 in KPBS plus 2% filtered serum or BSA overnight at 4°C, and with secondary antibodies (1:1000) in 0.3% Triton X-100 for 1 h at room temperature. After incubation with secondary antibody and ABC reagent (Vector Laboratories), sections were developed using metal-enhanced DAB solution. Sections were mounted to slides, dried, dehydrolyzed, treated with xylene, and covered using dibutyl phthalate xylene. For β-galactosidase histochemistry, sections were further fixed in 10% formalin plus 2 mm MgCl2 for 5 min, rinsed two times with KPBS plus 2 mm MgCl2, and permeabilized in permeabilization buffer (0.01% sodium desoxycholate, 0.02% NP-40, 2 mm MgCl2) for 10 min. Sections were reacted with reaction mix containing 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside in permeabilization solution overnight at 37°C. After rinsing in KPBS, sections were mounted to slides, counterstained with neutral red and dehydrated, and covered by coverslips as described above. Images were captured by a Hamamatsu digital camera connected to a Leica Leitz DM IRB microscope, and image analysis on sections was performed using Openlab software (Improvision) or NIH Image software.
Primary neuronal culture and immunostaining.
The cortex from the wild-type and moca−/− mice was dissected and dissociated. The cortical cells were placed in poly-d-lysine-coated coverslips and cultured in Neurobasal medium supplemented with B27. The neuronal cells were fixed in 4% paraformaldehyde in PBS plus 0.25 μm sucrose for 15 min, washed by PBS, and permeabilized by 0.5% Triton X-100. The fixed cells were blocked in 5% BSA in PBS for 1 h and incubated with primary antibodies followed by fluorescent-conjugated secondary antibodies (Molecular Probes). The cells were then mounted with antifading media containing 4% N-propyl gallate (Sigma-Aldrich). The cells were examined with a Leica TCS SP2 AOBS confocal laser scanning microscope. The images were taken and analyzed by the corresponding software with pixel intensity within a linear range.
Subcellular fractionation.
Subcellular fractionation of spinal cord extracts was performed as described with some modification (Kim et al., 2000; Lah and Levey, 2000; Leem et al., 2002; Gandy et al., 2007). Briefly, 0–30% continuous iodixanol gradients were formed in diluent buffer (10 mm HEPES, pH 7.3, 1 mm EDTA, 0.25 m sucrose containing 1 mm Na3VO4 and 10 mm NaF, and a protease inhibitor mixture) by overlayering 2.5, 5, 7.5, 10, 12.5, 15, 20, 25, and 30% iodixanol (Optiprep; Nycomed Pharma AS) in centrifuge tubes. The centrifuge tubes were equilibrated for 3–4 h at 4°C before use. The spinal cords from the control and mutant mice were collected and homogenized with a Dounce-type homogenizer in homogenization buffer [containing (in mm): 20 HEPES, pH 7.3, 40 KCl, 5 EGTA, 5 MgCl2 containing 1 Na3VO4 and 10 NaF, and a protease inhibitor mixture) at 4°C. Homogenates were spun at 1000 × g for 10 min and the supernatants were centrifuged at 65,000 × g for 60 min. The resulting pellets were suspended in diluent buffer, placed on the top of iodixanol gradients, and subjected to centrifugation at 38,000 rpm (SW41 Beckman rotor), 4°C for 90 min. Seventeen fractions were collected and analyzed by Western blotting.
Western blot analysis.
Cells or brain tissues were lysed in lysis buffer [1% Triton X-100, 50 mm HEPES, pH 7.5, 50 mm NaCl, 5 mm EDTA, 1 mm Na3VO4, 50 mm NaF, 10 mm Na4P2O7, plus a mixture of protease inhibitors (Roche Complete Mini)]. Protein concentrations were determined by Coomassie Plus (Pierce). The same amount of protein from each sample was separated on Novex precast polyacrylamide 10% gels (Invitrogen) and transferred to Immobilon membranes (Millipore). The membranes were blocked with 5% nonfat milk in Tris-buffered saline for 1 h at room temperature. After overnight incubation at 4°C with primary antibodies, the immunoreactive bands were detected with horseradish peroxidase-conjugated secondary antibodies (Bio-Rad, 1:20,000), using an ECL kit (GE Healthcare Bio-Sciences). For immunoprecipitations, equal amounts of protein from cell lysates were immunoprecipitated with selected antibodies at 4°C overnight. Either anti-mouse or anti-rabbit IgG agarose (25 μl) (Roche) was then added to each sample and incubated at 4°C for 2 h on a rocker platform. The immunoprecipitates were collected by centrifugation and washed four times with the washing buffer (0.1% Triton, 20 mm HEPES, 150 mm NaCl, 10% glycerol). The agarose beads were resuspended in 30 μl of SDS-PAGE sample buffer and boiled for 3 min to release the proteins. After 1 min of centrifugation, the supernatants were separated on 10% or 12% Tris-glycine gels.
Electron microscopy.
Moca−/− and wild-type mice were anesthetized and perfused transcardially with 0.5% glutaraldehyde and 2% paraformaldehyde in PBS, pH 7.4. Tissue blocks were postfixed for 1–3 h in the same fixative buffer at 4°C and rinsed in 0.05 m KPBS at pH 7.4. Serial 50-μm-thick vibratome sections of the spinal cord were cut and fixed in 1% osmium tetroxide with 1.5% potassium ferricyanide, dehydrated with ethanol and propylene oxide, and infiltrated with Spurr's resin. The vibratome sections were flat-embedded between two glass microscope slides, one coated with Liquid Release Agent (Electron Microscopy Sciences), and polymerized overnight at 70°C. After the coated slide was removed, a BEEM capsule was placed over each section, filled with Poly/Bed 812 (Polysciences), and polymerized overnight. Capsules were removed by concurrent warming and prying. Thin sections were collected onto 200-mesh copper grids, counterstained with uranyl acetate and lead citrate, and examined in a JEOL 100CXII transmission electron microscope. Images were collected as TIFF files with a MegaView III digital camera (SoftImaging System).
Fluorogold neurotracer experiments.
Twelve-month-old mutant and wild-type mice were used for the determination of retrograde transport using the neurotracer fluorogold. Mice were anesthetized and the left lower hind limb was positioned by strapping to a platform. A small stab incision 2–3 mm long was made in the skin to expose the underlying muscle at the widest point of the lower limb (∼7–10 mm distal to the femorotibial joint). A 10 μl Hamilton syringe was used to deliver 1 μl of a 2% solution of fluorogold (w/v) in 0.9% saline at a medial, central, and lateral site ∼2 mm deep in the gastrocnemius muscle of the mouse. Mice were killed at 46 h, the lumbar intumescence of the spinal cord was harvested and fixed in 4% paraformaldehyde made in PBS, and floated in 20% sucrose overnight until the cords sank. Longitudinal and coronal sections were prepared by standard methods and subjected to histochemistry analyses described as above. The total number of motor neurons accumulating fluorogold in their cell bodies was counted for each mouse. The mean number of cell bodies accumulating fluorogold was calculated for wild-type versus mutant mice (12 months old) (n = 6 for each group) for the 46 h time point.
Sciatic nerve ligation.
Control and mutant mice were anesthetized and the sciatic nerve was exposed at the level of the hip flexure 3 cm distal to the L4-L5 DRGs. Two ligations, 2.5 mm apart, were placed on the sciatic nerve with surgical sutures. The wound was closed and the animals were allowed to recover for 6 or 24 h. Subsequently, ligated sciatic nerves were used for immunoblotting. In addition, the sciatic nerve was transected in one group of animals.
Statistical analysis.
The behavioral performance scores of each mouse group in the tests as well as the sciatic nerve and root morphometry data were presented as mean ± SEM. Any differences between groups were examined by performing either a Student's t test, a Wilcoxon signed ranks test, or a one-way ANOVA followed by a post hoc test, as indicated in each related figure legend. A p value <0.05 was considered significant.
Results
MOCA knock-out mice have sensorimotor impairments
A moca−/− mouse line was established in which the loss of MOCA expression was verified by Western blotting and by the expression of β-gal (Fig. 1A,B). β-gal expression is detected in all regions of the CNS, consistent with the MOCA gene expression pattern shown previously (Kashiwa et al., 2000; Namekata et al., 2004).
Figure 1.

Expression of MOCA in the CNS and behavioral phenotypes of the moca−/− mouse line. A, The expression of MOCA in the wild-type, hemizygous, and homozygous knock-out mice. B, The expression of β-galactosidase in several representative brain structures of the wild-type (top) and homozygous knock-out (bottom) mice, including the motor cortex (CTX), caudate putamen (CP), cerebellum (Cb), and spinal cord (Sc). The last panel shows the β-gal expression pattern in the ventral horn of the spinal cord (blue) counterstained with neutral red. Scale bar, 500 μm (left three panels) or 50 μm (the right color panel). C, D, A clasping phenotype is shown in a mutant mouse (moca−/−) at the age of 20 months when its tail is lifted (D), a behavior that is not present in control (C). E, Motor performance was assessed by the rotarod test. The average time on the rod for each mouse group (2 months of age) is shown as the mean ± SD. Statistical analysis was performed by one-way ANOVA (F = 18, **p < 0.01). F, The motor learning ability was measured by continuing the training of the mice on rotarod performance. Four trials were performed daily for three consecutive days using three groups of animals at the age of 16 months. Data are presented as the mean ± SD. The data were subjected to a mixed ANOVA in which days were considered the repeated measure. Only the control group at the age of 16 months shows a significant difference (F = 4.42; **p = 0.02). G, The difference of the locomotor activities in the mutant and control mice was assessed by an open field test. The travel distance in every 5 min for each mouse group (2 months, n = 12) is shown as the mean ± SD. The statistics were performed by a Wilcoxon signed ranks test for related samples. *p < 0.05.
Moca−/− mice have a normal lifespan and successfully produce offspring. No significant difference in body weight is found between mutant and wild-type groups at any age. The common clinical features of the mutant mice are gait abnormalities, including limb weakness, ataxia, and an impaired ability to swim. The mutant mice also show spinning and clasping phenotypes when their tails are lifted (Fig. 1C,D). Therefore, the mutant mice may have defects in sensory and/or motor function.
The motor performance of moca−/− mice was further examined using the rotarod test. The abnormal behaviors are detectable using this procedure at 2 months of age (Fig. 1E). The average time on the rod for the moca−/− mouse group at 2 months of age was 31 s, compared with 65 s for the control group and 46 s for the moca+/− group. To evaluate motor learning ability, we measured the number of trials needed for animals to reach the maximal latency to fall off the accelerating rotarod. Four trials were performed daily for three consecutive days using three groups of animals at two different ages. Although rotarod performance tended to improve after 3 d of training trials in all groups tested, only the control group at the age of 16 months showed a significant difference (Fig. 1F). It follows that MOCA deficiency causes impaired motor performance with partial retention of motor learning ability.
We next performed an open field test to evaluate activities related to spontaneous locomotion. Over a 100 min monitoring period, there was no significant difference between control and homozygous mutant groups in total travel distance (data not shown). However, when the data were analyzed by splitting travel distance into successive 5 min time bins, the distance traveled every 5 min by each group differs significantly between 45 and 95 min (Fig. 1G). Therefore, moca−/− mice have decreased overall activity compared with control mice, which may reflect weakness caused by the loss of MOCA.
The muscles, dorsal and ventral roots, and sciatic nerve of moca−/− mice are not consistently altered
To determine whether locomotor impairments in moca−/− mice reflect neuromuscular impairment, skeletal muscle and sciatic nerve were examined. The muscles were stained with hematoxylin and eosin to evaluate the basic tissue organization and cellular structure, and myofibrillar ATPase reactivity was performed to identify fast-twitch and slow-twitch muscle fibers. Compared with wild-type mice, an excessive variability in myofiber size was found in some, but not all, 2-year-old moca−/− mice, with atrophic fibers of both fiber types having a round to polygonal shape (supplemental Fig. S2A–D, available at www.jneurosci.org as supplemental material).
Morphometric analysis of the L5 dorsal and ventral roots revealed a shift in the size–frequency distribution toward larger fibers in the moca−/− mice compared with moca+/+ mice that was reflected by increases in mean axonal diameter and the relative frequency of large fibers (supplemental Table S1 and Fig. S3, available at www.jneurosci.org as supplemental material). The apparent shift toward larger fibers in moca−/− mice was not associated with a change in myelinated fiber number or density in either the dorsal or ventral roots (supplemental Table S2, available at www.jneurosci.org as supplemental material). In contrast, there was a nonsignificant trend for a shift in the size–frequency distribution of the more distal mixed sciatic nerve of moca−/− mice toward smaller fibers compared with moca+/+ mice that was associated with trends for a decrease in mean axonal diameter and an increase in the relative frequency of small fibers (supplemental Fig. S2 and Table S1, available at www.jneurosci.org as supplemental material).
Moca−/− mice accumulate aberrant NFs in the spinal cord
To determine whether any changes in the CNS were associated with the MOCA deficiency, the brain and spinal cord structure and morphology were examined first by Nissl staining. The mutant cortex and cerebellum are slightly smaller compared with the age-matched controls. However, the overall brain and spinal structure is intact in the moca−/− mice without apparent loss or disorganization of any major brain or spinal cord region. We next assessed nerve fibers in the CNS of MOCA-knock-out mice. Because abnormal NF accumulation is a marker for axon pathology (Coleman, 2005; Petzold, 2005), we examined the patterns of NF expression in the brain and spinal cord. No significant change was observed in the cortex, but pathological changes were seen consistently in other areas, including the cerebellum, brainstem, habenula, and spinal cord. Abnormal axonal spheroids are stained by NF antibodies in all these structures (Fig. 2, and below), which are consistent with the behavioral abnormalities seen in MOCA-knock-out mice. Because the spinal cords of the MOCA mutant mice show the most pronounced pathology, subsequent analyses are focused on this tissue.
Figure 2.
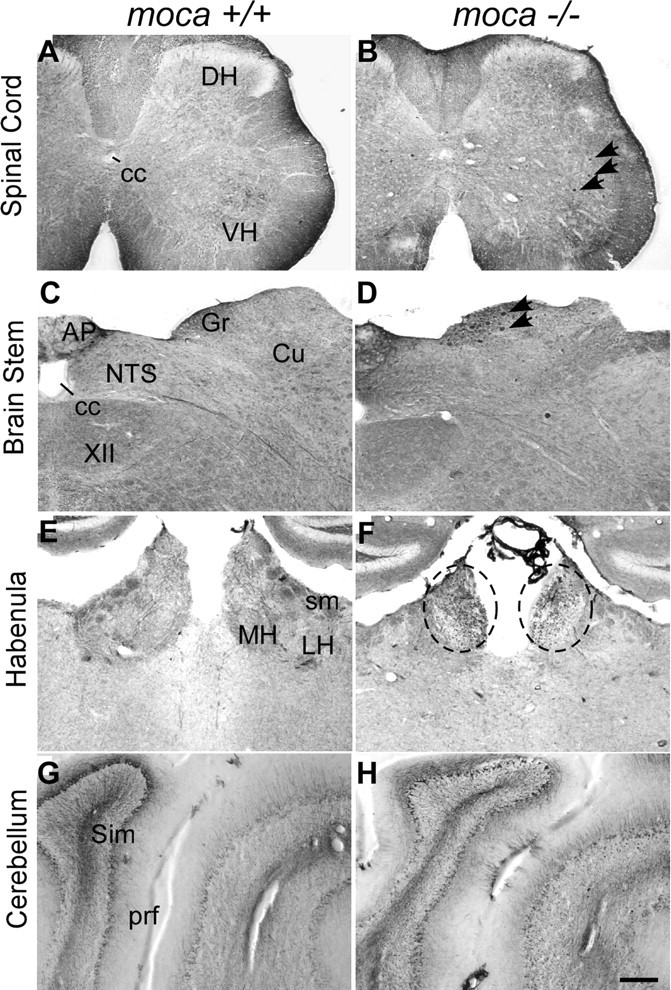
A–G, Abnormal axonal swellings/spheroids (indicated by arrows or circles) stained by an antibody against NF-68 are present in the spinal cord (B), the dorsal column of the brainstem (D), the habenula of moca−/− mice (F), and the cerebellum (H), but not in the corresponding areas of control mice (A, C, E, G). DH, Dorsal column; VH, ventral column; cc, central canal; AP, area postrema; Gr, gracile nucleus; Cu, cuneate nucleus; NTS, nucleus of the solitary tract; XII, hypoglossal nucleus; MH, medial habenular nucleus; LH, lateral habenular nucleus; sm, stria medullaris; prf, primary fissure; Sim, simple lobule. Scale bar, 200 μm.
Series of sections through spinal cord were stained with different NF antibodies including NF-68 (Fig. 3A,B), which recognizes the NF light chain; SMI-31 (Fig. 3C,D), which recognizes phosphorylated NF heavy chain (NF-200), the active form of the protein; and SMI-32 (Fig. 3E,F), which recognizes nonphosphorylated NF-200. Abnormal axonal spheroids stained by all three NF antibodies are present in the spinal cord of 20-month-old mutant mice but not in age-matched controls. These abnormal axonal spheroids are spread throughout the gray matter and occasionally are seen in the white matter of the mutant spinal cord; the NF inclusions are rarely seen within neuronal cell bodies. The NF-containing spheroids have different shapes and sizes ranging from 5 to 20 μm in diameter (Fig. 3G). Larger spheroids often have a fibrillary appearance. Similar spheroids are not seen in the mutant spinal cord on postnatal day 1 (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material), but are present as early as 2 months of age (supplemental Fig. S4D, available at www.jneurosci.org as supplemental material). These abnormal NF aggregates suggest widespread axonal dystrophy in moca−/− mice and that the defects become apparent only after birth.
Figure 3.

A–J, Abnormal axonal swellings/spheroids representing degenerative axons are present in the spinal cord of moca−/− mice at the age of 20 months (B, D, F) but not in the age-matched controls (A, C, E). These abnormal axonal spheroids contain aggregates of neurofilaments. The coronal sections of the spinal cord are stained with different neurofilament antibodies including NF-68 (A, B), which recognizes the NF light chain; SMI-31 (C, D), which recognizes phosphorylated NF heavy chain; and SMI-32 (E, F), which recognizes nonphosphorylated NF heavy chain. G, Closer views of axonal spheroids of different sizes. H, Western blotting patterns of NFs from different brain regions of control (+/+) and moca−/− (−/−) mice. CTX, cerebral cortex; STR, striatum; CB, cerebellum; SC, spinal cord. Arrows indicate the degraded products of nonphosphorylated NF-200. GAPDH is the loading control. I, J, Activation of microglia in moca−/− mice. The activation of microglia is present in the spinal cord of moca−/− (J) but not in control (I) mice at the age of 20 months. Scale bar, 50 μm.
In addition to immunostaining, we examined the expression of the three NF subunits in the cortex and the spinal cord by Western blotting. The levels of NF-68 and NF-150 in the mutant mice are similar to those in age-matched controls (Fig. 3H). There are also no differences in the levels of the full-length phosphorylated (detected by antibody SMI-31) and nonphosphorylated (antibody SMI-32) NF-200 in moca−/− mice relative to controls. However, two protein fragments with molecular weights of 25 and 50 kDa, respectively, react with the antibody to nonphosphorylated NF-200 only in the mutant spinal cord (Fig. 3H, arrows). Therefore, NF-200 metabolism may be affected in the spinal cords of mutant mice.
Because inflammatory responses are commonly associated with axonal damage, we examined microglial activation by staining with an antibody against ionized calcium-binding adaptor molecule 1 (Iba1). Iba1 is a Ca2+-binding protein produced by monocytes and microglia, and alterations in its expression are associated with inflammation (Ito et al., 1998). Iba1-immunoreactive cells in the spinal cord of moca−/− mice at both 20 (Fig. 3I,J) and 2 (supplemental Fig. S4F, available at www.jneurosci.org as supplemental material) months of age display thickening and retraction of their processes, indicative of microglial activation. These data demonstrate that there is both neurofilament accumulation and microglial activation in the spinal cord of moca−/− mice. Both may contribute to the progressive damage and dysfunction resulting from MOCA deficiency.
Moca−/− mice have disorganized cytoskeletons and an abnormal accumulation of autophagic vacuoles in the spinal cord
To clarify the nature of the defect in moca−/− mice, we examined axons by electron microscopy. The spinal cord of wild-type mice shows the normal appearance of axonal structures with key axoplasmic features including microtubules and NFs (Fig. 4A,B). We did not see a disorganization of cytoskeletal components, or abnormal vesicle and organelle accumulations in the wild-type mice. In contrast, the spinal cord of moca−/− mice shows several different types of abnormal axonal morphologies involving both vesicles and structural proteins. Large numbers of organelles and vesicles consisting of haphazardly arranged vesicles, mitochondria, sporadic multilamellar bodies, and vacuoles accumulate in spinal cord axonal swellings of moca−/− mice (Fig. 4C,D). Degenerating mitochondria and lysosomal electron-dense bodies are common signs of axonal dystrophy (Yu et al., 2005). Some of the structures also fit the criteria used to define autophagic vacuoles (AVs) including a size >0.25 μm in diameter, a double limiting membrane, and the presence of multiple membranous organelle-derived structures within a single vacuole (Glaumann et al., 1981; Herrera et al., 2006). AVs found in dystrophic axonal swellings of moca−/− mice represent both early and late stages of autophagy (Fig. 4C,D) (Yu et al., 2005). The early-stage vacuoles (Fig. 4D, labeled orange 1) include double membrane-bound vacuoles containing engulfed organelles. The late-stage vacuoles (Fig. 4D, labeled blue 2) include residual bodies containing the engulfed degraded organelles as well as translucent and dense multivesicular and multilamellar bodies with single and double outer membranes. These abnormal accumulations of AVs are not present in wild-type mice.
Figure 4.
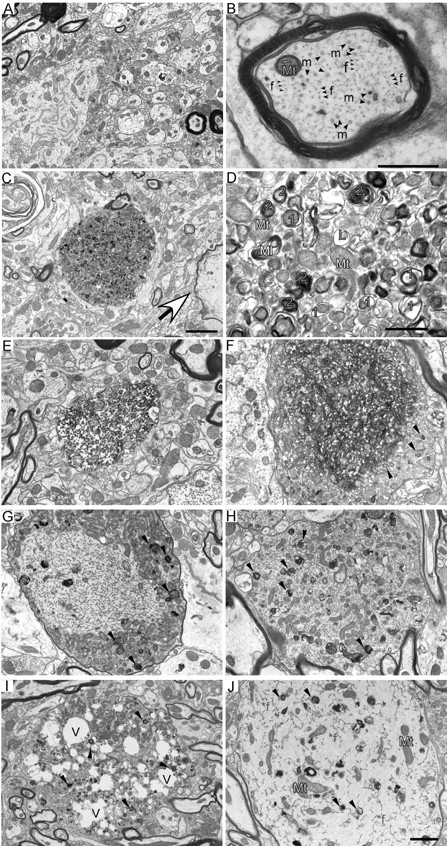
A–D, Autophagic vacuoles are present in spinal cord dystrophic axons of 20-month-old moca−/− mice. A, A general view of the wild-type spinal cord showing the normal appearance of myelinated and nonmyelinated axons. B, A closer look at the axonal cytoskeleton and organization showing microtubules (m, arrow) are often in bundles and more irregularly spaced compared with evenly distributed NFs (f, arrow). Mt (blue), Mitochondria. C, A typical axonal swelling showing accumulation of AVs (dashed circle) and a degenerated axon (arrowhead) in the spinal cord of moca−/− mice. D, Accumulation of AVs. Mt (blue), Mitochondria; 1 (orange), double-membrane autophagosomes representing early stages of autophagy; 2 (blue), residual bodies containing degraded organelles, representing late stages of autophagy; L (pink), lysosomes, before fusion with autophagosomes; Ml (yellow), multilaminar body. Scale bars: 2 μm (A, C) or 500 nm (B, D). E–J, Various pathological morphologies of axonal swellings in the spinal cord of 20-month-old moca−/− mice. E, Disorganized axoplasm. F, Disorganized axonal cytoskeletal components are tangled together, and mitochondria are buried in these abnormal structures. The organelles and AVs accumulate at the edge of the tangled axonal cytoskeleton (arrows). G, Hypertrophy of NFs is shown in the central part of axonal swellings with AVs and organelles accumulated at the edge (arrows). H, Various axoplasmic abnormalities are present in the axonal swelling including a hypertrophic cytoskeleton, organelles, and AVs (arrows). I, An axonal swelling has many vacuoles (V) along with accumulated organelles and AVs (arrows). J, An axonal swelling showing hypertrophic NFs (*f), and diffused organelles and AVs (arrows).
Polymerized and disorganized axonal cytoskeletal components are also observed in axonal swellings of the mutant spinal cord (Fig. 4E,F). These abnormal structures are often surrounded by AVs (arrows) or mixed with neurofilamentous hypertrophy, a form of terminal degeneration described in experimental autoimmune encephalomyelitis (Brown and Sawchenko, 2007) (Fig. 4G,H). Finally, we observed highly deteriorated axonal morphologies, including swellings containing many vacuoles distributed among the cytoskeletal components, organelles, and AVs (Fig. 4I), or neurofilamentous hypertrophy within the axoplasm in which little axon cytoskeletal structure remains (Fig. 4J). These latter morphologies clearly represent axonal atrophy and degeneration in the spinal cord of moca−/− mice.
Accumulation of polyubiquitinated proteins in moca−/− mice
The proteasome–ubiquitin pathway can play a role in axon degeneration, because its disruption leads to abnormal protein accumulation in axons (Ehlers, 2004). To determine whether this pathway is affected in moca−/− mice, we examined ubiquitinated proteins by Western blotting. There is a dense smear of high molecular weight ubiquitinated proteins in the spinal cord protein extracts of mutant mice, but much less in age-matched controls (Fig. 5A). There is less aggregated material in extracts of cortex from moca−/− mice, although there is an unknown 25 kDa protein in the cortex that is always highly ubiquitinated in MOCA-deficient mice. The high molecular weight protein bands in moca−/− mice represent aggregated ubiquitinated proteins similar to those observed in many cases of neurodegeneration (Rubinsztein, 2006). The protein extracts were further examined after separation into Triton X-100-soluble and -insoluble fractions. The expression of the polyubiquitin-conjugated proteins is significantly increased in both soluble and insoluble fractions of the spinal cord of mutant mice (Fig. 5B,C), with a smaller difference in the cortex (except for the 25 kDa protein noted above). In addition, some dystrophic neurite structures were labeled by the ubiquitin antibody in older moca−/− mice (20 months), but none were detected in younger mice (2 months) (Fig. 5D–G), showing age-dependent accumulation of ubiquitinated proteins in the mutant mice. These data suggest that protein accumulation in axons may contribute to age-dependent changes in axon morphology and function.
Figure 5.
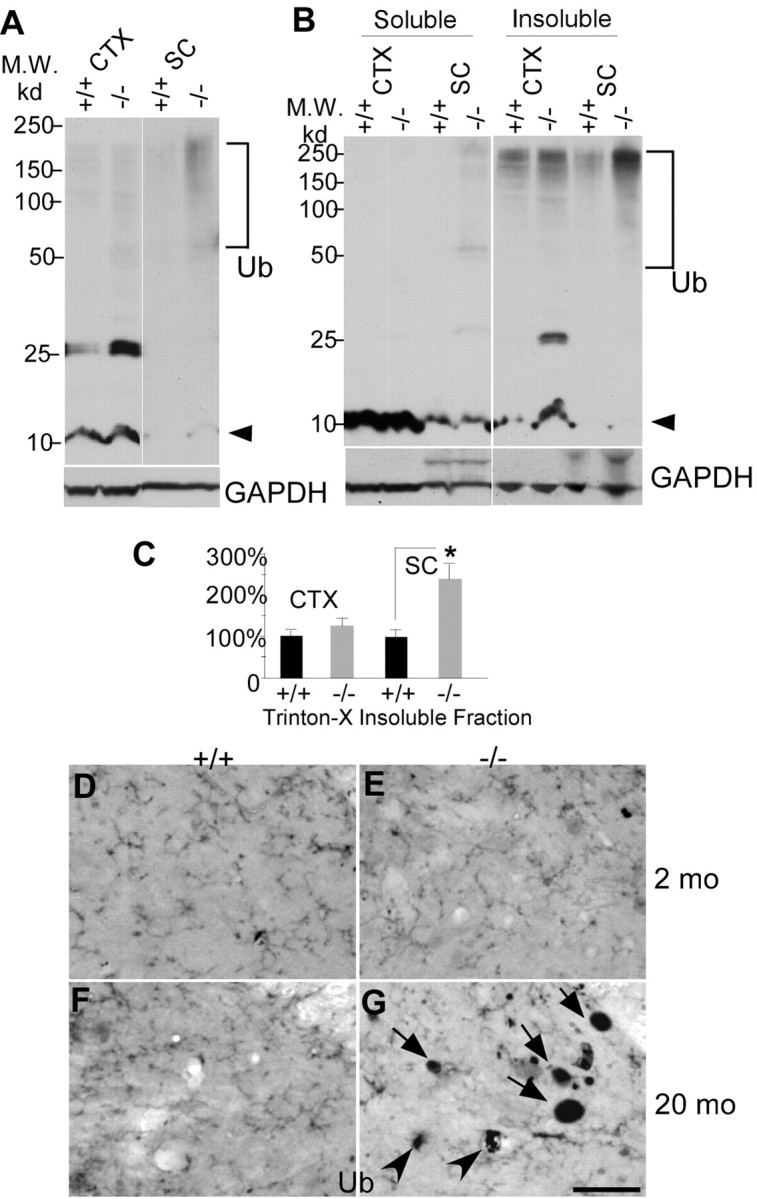
Accumulation of polyubiquitinated proteins in the spinal cord of 12-month-old moca−/− mice. A, Western blotting patterns of ubiquitinated proteins from different brain regions of control (+/+) and moca−/− (−/−) mice. CTX, cerebral cortex; SC, spinal cord. Arrowhead indicates monomer ubiquitins. Polyubiquitin-conjugated high molecular weight proteins are indicated by the bracket. B, A representation of Triton X-100-soluble and -insoluble fractions of protein extracts followed by immunoblotting with ubiquitin antisera. C, The data are quantified from six independent experiments. GAPDH is the loading control. The percentage change in the expression of polyubiquitin-conjugated high molecular weight proteins (bracket) in the Triton X-100-insuluble fraction was normalized to GAPDH and shown as the mean ± SD. Statistical analysis was done by a Student's t test (n = 6, *p < 0.05). D–G, Ubiquitin staining of the posterior funicular gray area of the spinal cord at the age of 2 months (D, E) and at the age of 20 months (F, G). Abnormal axonal spheroids stained by ubiquitin are present in moca−/− mice at the age of 20 months (G) but not at the age of 2 months (E). D, F, the age-matched controls. Scale bar, 50 μm. Ub, ubiquitin-containing aggregates.
Impaired axonal transport in moca−/− mice
We next asked whether the abnormal features in moca−/− mice correlate with axon functional alterations. Retrograde tracer studies were performed to assess axonal transport. Moca−/− mice and age-matched wild-type controls were injected with the retrograde neurotracer fluorogold in the gastrocnemius muscle and killed 46 h later to compare the number of motor neuron cell bodies that had accumulated tracer (LaMonte et al., 2002). Longitudinal sections of spinal cord were made and the accumulation of the fluorogold tracer was examined in the motor neuron pools that innervate the injected muscle (Fig. 6A,B). Sections from wild-type animals revealed heavy labeling of motor neuron columns, whereas those from moca−/− mice of the same age contained fewer and less intensely labeled cells (Fig. 6B). To obtain quantitative estimates, the numbers of fluorogold-labeled cells were counted in cross-sections of the spinal cord (n = 6). Loss of MOCA causes an average 55% reduction in the number of labeled cells in the spinal cord of mutant mice relative to controls (Fig. 6E). To ask whether this decrement is caused by the loss of motor neurons, a series of coronal sections of lumbar spinal cords derived from age-matched moca+/+ and moca−/− mice were stained with hematoxylin and eosin, and using an antibody against choline acetyltransferase (ChAT), a marker for spinal motor neurons. We did not find any significant difference in motor neuron numbers in the spinal cord between groups (Fig. 6C,D). In addition, we probed for degeneration of perikaryal elements in the spinal cord by staining with Fluoro-Jade C. Fluoro-Jade C is a polyanionic fluorescein derivative that specifically binds to degenerating neurons (Schmued et al., 2005). We failed to observe any remarkable or consistent staining with Fluoro Jade C in the spinal cord of either wild-type or mutant mice, confirming the absence of motor neuron degeneration. These data suggest that impaired retrograde transport resulting from the MOCA deficiency does not cause loss of motor neurons in the spinal cord. Therefore, it is most likely that the MOCA deficiency causes an impairment of retrograde axonal transport, which is consistent with the observed axonal pathology.
Figure 6.

A–H, Impaired axonal transport in moca−/− mice. Longitudinal views of fluorogold labeling in the lumbar 5 region of spinal cord of control (A) and moca−/− (B) mice at the age of 12 months. C, D, ChAT staining of the cross-sections of spinal cord of control (C) and moca−/− (D) mice. The percentage change of fluorogold labeling was quantified and represented in E. The fluorogold-labeled cells for each mouse group are shown as the mean ± SD. Statistical analysis was done by a Student's t test (n = 6, **p < 0.01). F, G, Schematic presentation of sciatic nerve ligation (F) and the APP expression levels of different parts in the axon (G). The ligation sites are indicated by arrows. The proximal (P part) and distal (D part) segments (on the cell body end) relative to the ligation sites, and the remaining distal site (toward the cell body end; E part) were collected for Western blot analysis after the surgery. The levels of APP in each axonal part were analyzed by using 22C11 antibody. H, The percentage of APP expression in the proximal (P part) and distal (D part) segments relative to the controls of similar segments of wild-type animals was quantified by NIH Image tools and the statistics were performed by a Student's t test. The data are presented as mean ± SD (n = 6, **p < 0.01). Scale bar, 50 μm.
Because APP is transported by an anterograde mechanism (Koo et al., 1990), we ligated sciatic nerve and examined levels of APP in different segments of the nerve to determine whether the loss of MOCA also affects anterograde transport in moca−/− mice. Sciatic nerves were ligated in the middle, and segments proximal and distal to the point of ligature were assayed for APP after 6 and 24 h by Western blotting. Six pairs of control and mutant mice were used for the analysis. Because the pattern of APP accumulation was very similar at 6 and 24 h after ligation, 6 h after ligation was used for collecting tissues. In moca+/+ mice, we observed a substantial increase of APP on the site proximal to the ligation point, accompanied by a striking depletion in the distal half (Fig. 6F,H). In contrast, the level of APP in the proximal site is significantly less in the moca−/− mice compared with that in the control, whereas the APP level in the distal site of mutant mice is consistently higher than that seen in controls. The level of a slow transport marker, β-tubulin, remained unchanged. The loss of MOCA results in an approximately 40% decrease in APP accumulation proximal to the ligation site, whereas APP levels in the cell body segment of the sciatic nerve of moca−/− mice are ∼8 times greater than in the control mice. These results show that anterograde axonal transport of APP is impaired in moca−/− mice.
Alteration in cofilin activity caused by the loss of MOCA
To identify a possible mechanism for the cytoskeletal disorganization and impaired axonal transport in moca−/− mice, we fractionated the spinal cord using 0–30% continuous iodixanol density gradients and asked whether there is an aberrant distribution of cargo and structural proteins in moca−/− mice (Fig. 7A). In control mice, actin is distributed throughout the gradient, with the higher densities in the first few “light” fractions. In contrast, much less actin is found in both the intermediate and heaviest fractions from moca−/− mice. The distribution pattern of β-tubulin is similar to that of actin, but with a much smaller decrease in levels in the intermediate fractions from moca−/− mice. There are no reproducible changes in the distribution patterns of a marker for neurofilaments, NF-68, and the synaptic protein synaptophysin, in moca−/− mice relative to controls. These data suggest that the MOCA deficiency alters protein-protein interactions or the polymerization of proteins within the spinal cord and that actin is the most robustly affected protein.
Figure 7.
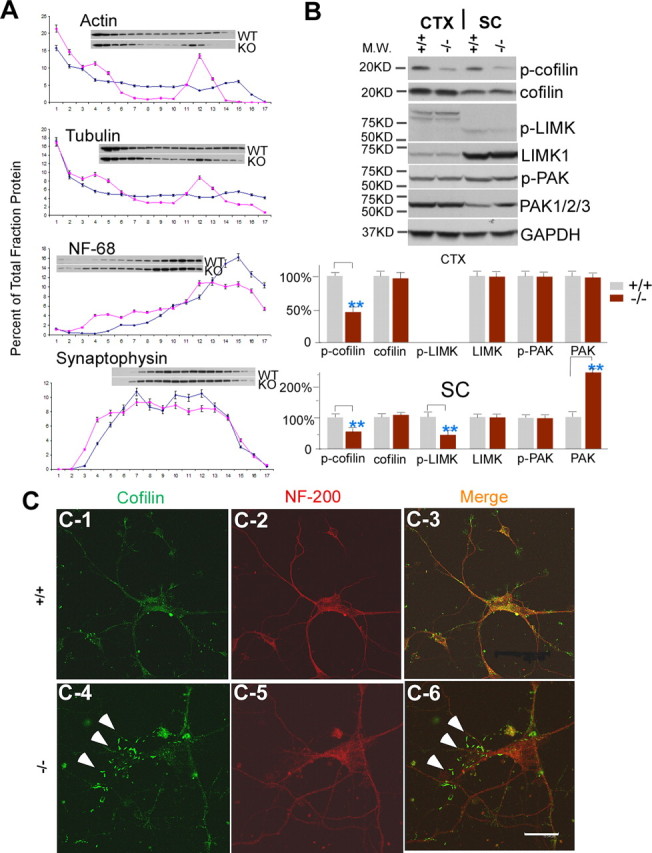
Alterations in actin dynamics are mediated by cofilin in moca−/− mice. A, A representative spinal cord subcellular fractionation using a 0–30% continuous iodixanol gradient (n = 3). Each fraction is presented as the percentage of total protein (sum of OD in scanned blot) for each protein. B, Western blotting patterns of cofilin, phosphorylated cofilin, LIMK1, phosphorylated LIMK, PAK1/2/3, and phosphorylated PAK from the cerebral cortex and spinal cord of control (+/+) and moca−/− (−/−) mice. GAPDH was used as a loading control. The percentage change in the expression level for each protein is normalized to GAPDH and shown as the mean ± SD. Statistical analysis was done by a Student's t test (n = 6, **p < 0.01). CTX, cerebral cortex; SC, spinal cord. C1–C6, Immunostaining of cultured cortical neurons with the antibodies recognizing cofilin (green) and NF-200 (red), and merged (overlap yellow). The formation of rod-like structures (arrows) stained by the cofilin antibody is present in cultured moca−/− (−/−) neurons (C4, C6). The rod-like structures are largely confined to the thinner neurites, as previously shown (Minamide et al., 2000). Scale bar, 15 μm.
Cofilin is a major actin-depolymerizing factor in the mammalian CNS and is a target of many signaling pathways that perturb the actin cytoskeleton (Ono, 2003; Sarmiere and Bamburg, 2004). Importantly, the regulation of cofilin is downstream of Rac1, and MOCA is a GEF that mediates Rac1 activity (Grimsley et al., 2004). Cofilin inclusions are found in AD brains, and the formation of rod-like structures containing cofilin is associated with neurodegeneration (Minamide et al., 2000). Therefore, we asked whether the MOCA deficiency leads to an alteration of cofilin activity, which is modulated by its phosphorylation. Western blots were performed using specific antibodies recognizing cofilin and phosphorylated-cofilin (Ser3). Levels of cofilin expression in the spinal cord and the cortex of moca−/− mice are similar to those of control mice (Fig. 7B). However, the levels of phosphorylated cofilin are reduced by ∼50% in the cortex and spinal cord of moca−/− mice relative to controls (Fig. 7B). These data suggest that the loss of MOCA causes an increase in cofilin activity, which may have a direct impact on actin dynamics.
A pathological alteration in cofilin activity results in the appearance of rod-like structures in cultured neurons (Minamide et al., 2000). Therefore, this assay was used to confirm the phosphorylation data. Cortical neurons derived from the control and moca−/− mice were cultured for 3 d and stained with cofilin and neurofilament antibodies. In control neurons, cofilin is evenly distributed (Fig. 7C1–3). However, in moca−/− neurons, there were rod-like structures positively stained in thin neurites by the cofilin antibody (Fig. 7C4,6). Approximately 50% of the moca−/− neurons contained the rod-like structures in the mutant cultures, whereas essentially none were present in cultured control neurons. These data suggest that the MOCA deficiency causes the dysfunction and abnormal distribution of cofilin similar to that reported previously in cultured neurons after the induction of neurodegeneration (Minamide et al., 2000).
Cofilin phosphorylation is mediated by upstream signals that include the activation of Rho/Rac GTPases, PAK, and LIMK (Ridley, 2006). PAKs are primary targets for Rho/Rac GTPases; most of the closest relatives of Rac1 and Cdc42 activate at least one of these kinases (Ridley, 2006). The phosphorylation and activation of LIMKs is regulated by PAKs (Edwards et al., 1999; Gohla and Bokoch, 2002). Because MOCA activates Rac1 and modifies the actin cytoskeleton in cultured neurons (Grimsley et al., 2004; Chen et al., 2005), we asked whether the overactivation of cofilin caused by the MOCA deficiency is the result of a change in the PAK–LIMK pathway. Western blots were performed using specific antibodies recognizing LIMK1 and 2, phosphorylated LIMK (Thr508 and Thr505 for LIMK1 and 2, respectively), PAK1/2/3, and phosphorylated PAK (Ser144, Ser141, or Ser139 for PAK1, 2, or 3). Consistent with the decrease in cofilin phosphorylation, the levels of phosphorylated LIMK are also reduced by ∼50% in the spinal cord of moca−/− mice relative to control animals, but not in the cortex, where minimal unphosphorylated protein could be detected (Fig. 7B). In contrast, the overall level of phosphorylated PAK remains similar between the control and moca−/− groups in the cortex and spinal cord, but the expression level of PAK1/2/3 is increased by two- to threefold in the spinal cord. These data show that the PAK–LIMK pathway is affected by the loss of MOCA, and that the changes in this pathway are more pronounced in the spinal cord than in the cortex. A different pathway from MOCA to cofilin must be operational in the cortex because cofilin phosphorylation in cortex and spinal cord are similarly decreased.
Discussion
To clarify the function of MOCA, we have generated a moca−/− mouse line and analyzed the resultant phenotype. The mice have sensorimotor impairments and structural changes that include axonal swellings containing NFs and AVs in several brain regions including the spinal cord and cerebellum. These changes are associated with impaired axonal transport. Because axonal pathology is the major abnormality found in moca−/− mice, it is concluded that MOCA is essential for maintaining the functional integrity of axons and that its loss causes pronounced central axonal pathology and behavioral abnormalities.
The only mutations of the MOCA gene linked to a human disease to date are associated with ADHD (de Silva et al., 2003). A chromosome disruption was found in an Australian family, and all individuals carrying this mutation display the ADHD phenotype. The disruption was caused by the insertion of a gene called SLC9A9 (a sodium/hydrogen exchanger protein) into the MOCA genomic locus. However, it is not clear whether this disruption causes a loss or gain of either gene's function, or which gene is responsible for the phenotype. We performed several behavioral tests including the startle response (data not shown), open field, and rotarod tests to clarify the phenotypic changes caused by the loss of MOCA. All of these procedures demonstrate that moca−/− mice are physically less active compared with age-matched controls and have various sensory (ataxia and proprioceptive deficits) and motor (weakness) impairments. Therefore, it appears that the loss of MOCA leads to impaired sensorimotor behaviors, but the ADHD-like phenotype cannot be formally excluded.
The most striking pathology in moca−/− mice is in the spinal cord, which has axonal swellings containing abnormal accumulations of NFs indicative of dystrophic axons. NFs are the primary constituents of the axon cytoskeleton and are formed by heteropolymerization of the NF triplet proteins, NF-L, NF-M, and NF-H (Hoffman and Lasek, 1975; Hirokawa et al., 1984). NFs work together with actin filaments and microtubules to maintain neuronal morphology and function (Lee and Cleveland, 1996). Because NFs have a long half-life relative to other proteins, damaged NFs are likely to accumulate, leading to axonal pathology (Lee and Cleveland, 1996). We did not observe motor neuron death in moca−/− mice or loss of myelinated fibers in the dorsal and ventral roots, but NF spheroids accumulate in axonal fibers and only rarely appear in cell bodies, suggesting that the axon is the major target affected by the loss of MOCA.
Electron micrographs show that cellular organelles and AVs accumulate in some abnormal axonal swellings in moca−/− mice. These morphological changes are almost identical to those found in the spinal cord of the APP/PS1ki mouse model of AD (Wirths et al., 2007). This mouse model carries the mutant AD genes including human APP751 with Swedish and London mutations, and murine PS1 with two familial AD-linked mutations (PS1M233T and PS1L235P). Because immature AVs move along axons by regulated retrograde transport (Hollenbeck, 1993) and the defective AV transport may impede lysosomal degradation, the accumulation of AVs in moca−/− mice is most likely linked to impaired axonal transport.
The autophagic pathway is involved in the turnover and recycling of cytoplasm and organelles engulfed within double-membrane vesicles, known as autophagosomes (Shintani and Klionsky, 2004). Similar to the AV morphologies observed in dystrophic neurites of AD transgenic mice, axonal swellings in moca−/− mice also contain autophagosomes and other immature AVs, suggesting impaired maturation of nascent AVs (Yu et al., 2005). Autophagy plays a major role in the clearance of most long-lived and unfavorable proteins that aggregate in cells as a consequence of aging and disease (Rubinsztein, 2006; Simonsen et al., 2007). A deficiency of autophagy in neural cells causes neurodegeneration and leads to the accumulation of ubiquitin-containing inclusion bodies (Hara et al., 2006; Komatsu et al., 2006; Simonsen et al., 2007). We also find that polyubiquitin-conjugated proteins are elevated in the spinal cord of moca−/− mice and that this effect increases with age, suggesting that their accumulation is a consequence of inefficient or overwhelmed autophagic activities. In addition to the disorganization of the axonal cytoskeleton and the accumulation of AVs, we observed axonal morphologies indicative of advanced axonal degeneration in the spinal cord and brainstem of moca−/− mice. Impaired axonal transport is a common feature of many neurological diseases. Our data from fluorogold-labeling and sciatic nerve ligation studies demonstrate impaired axonal transport in moca−/− mice. The accentuation of proximo-distal tapering of peripheral myelinated fibers in the roots and sciatic nerve may be a structural correlate of the axonal transport abnormalities in moca−/− mice, because impaired axonal transport in experimental diabetic neuropathy is associated with an increase in axonal caliber proximally and a decrease distally (Medori et al., 1988). APP is transported in a fast anterograde manner within axons (Koo et al., 1993; Kamal et al., 2000; Lazarov et al., 2005; Stokin et al., 2005). PS is involved in APP trafficking (Naruse et al., 1998; Cai et al., 2003) and PS mutations impair APP axonal transport (Pigino et al., 2003; Lazarov et al., 2007). We show here that axonal transport of APP is defective in moca−/− mice, consistent with our previous observation showing that MOCA interacts with PSs and regulates APP metabolism (Kashiwa et al., 2000; Chen et al., 2002).
Because the axonal transport system comprises various cargos, motor proteins, cytoskeleton components, and accessory regulatory molecules, disruption of any of these components could result in defective transport. Our electron micrographs show that axonal cytoskeletal components are disorganized in the spinal cord and brainstem of moca−/− mice. To explore the cause for the initiation of cytoskeletal disorganization, we have focused on the actin cytoskeleton because of the aberrant distribution of actin in iodixanol gradients of spinal cord. The actin cytoskeleton has been directly linked to the abnormal accumulation of NFs and is required for NF transport (Yang et al., 1999; Jung et al., 2004). The disruption of BPAG1 (bullous pemphigoid antigen 1), a protein involved in anchoring neuronal intermediate filaments to the actin cytoskeleton, leads to axonal swellings containing NF aggregates similar to those in moca−/− mice (Yang et al., 1999). We have previously shown that MOCA colocalizes with actin fibers and that MOCA is involved in regulating neurite outgrowth (Chen et al., 2005). In addition, MOCA is a GEF for the Rho/Rac GTPases, which play major roles in regulating the actin cytoskeleton (Ridley, 2006). Therefore, MOCA may be required for the maintenance of the actin cytoskeleton and its loss may lead to defects in the actin dynamics, NF accumulation, and defective axonal transport.
Actin dynamics are regulated by a plethora of actin-binding proteins under the control of specific signaling pathways. Among this group, cofilin has been implicated in neurite outgrowth and axonal transport (Bray et al., 1992; Meberg and Bamburg, 2000), and cofilin and its related upstream signals have been implicated in the generation of dystrophic neurites (Minamide et al., 2000; Zhao et al., 2005; Maloney and Bamburg, 2007). Cofilin is the major actin-depolymerizing factor in the CNS and is required for the proper assembly and function of actin fibers (Sarmiere and Bamburg, 2004). The activity of cofilin is regulated by LIM kinase. LIM kinase phosphorylates cofilin and inactivates it, leading to an increase in polymerized actin (Arber et al., 1998). LIM kinase is, in turn, a substrate for PAK, which is activated by Rho/Rac GTPases (Edwards et al., 1999; Gohla and Bokoch, 2002). We show here that the MOCA deficiency causes the formation of cofilin-containing rod-like structures in cultured cells, with a corresponding decrease in cofilin phosphorylation in both the spinal cord and cortex, suggesting an increase in cofilin activity in moca−/− mice. In addition, LIM kinase phosphorylation is decreased in the spinal cord, but not in the cortex of these animals. These data suggest that the actin cytoskeleton in the spinal cord is affected by the loss of MOCA via the disruption of the LIM kinase–cofilin pathway. Although there is a similar decrease in cofilin phosphorylation in spinal cord and cortex, and cultured cortical neurons display cofilin abnormalities, there are no significant pathological alterations in the cortex. This difference may be attributable to the relative lengths of axons and transport distances, or unknown differences in signaling pathways affected by MOCA.
The other upstream component of the LIM kinase pathway, PAK, is also altered in response to the MOCA deficiency. Total PAK (PAK1, 2, and 3) expression is increased in the spinal cord, but, again, not in the cortex. MOCA is a member of the DOCK180 family (Côté and Vuori, 2002) and the other members, such as DOCK4 and DOCK180, may partially replace the MOCA-mediated function after the cells lose MOCA. Therefore, the increase in the total PAK may be a compensatory reaction in response to the MOCA deficiency, and the cells need to make more PAKs to react with other members of the DOCK180 family to overcome the loss of MOCA. However, the ratio of phosphorylated PAK versus total PAK is reduced in the spinal cord, consistent with the reduced phosphorylation of cofilin and LIM kinase. Therefore, the loss of MOCA GEF activity alters the PAK–LIM kinase pathway in the spinal cord that regulates cofilin and the actin cytoskeleton. The intermediate pathway between MOCA and cofilin is likely to be different in the cortex. Our data are in line with a growing body of evidence supporting a link between alterations in the actin cytoskeleton and axonal transport defects in the early stages of many neurological diseases (Minamide et al., 2000; Zhao et al., 2006; Maloney and Bamburg, 2007).
Axonal dysfunction is not always associated with nerve cell loss, and in the majority of cases, only select subsets of neurons are affected. For example, in many mutations that are linked to motor neuropathies, axons are the only major disease target (Pasinelli and Brown, 2006). In human AD brains, loss of synapses is widely distributed, whereas neuronal loss is selective and localized (Masliah et al., 2006). Identifying a common pathway linking the initiation of axonal dysfunction with axonal degeneration should provide a significant advance in understanding the mechanisms involved in the neurodegenerative process and aid in the development of an early treatment for many neurological diseases. Based on our morphological and mechanistic evidence, we present a working model to delineate the likely progression of axonal degeneration occurring in moca−/− mice (Fig. 8). Initially, the loss of MOCA causes the disruption of the axonal cytoskeleton because of a dysregulation in actin polymerization through the PAK–LIM kinase–cofilin pathway. Altered actin polymerization leads to the disruption of axonal transport promoting further disorganization of structural proteins and the accumulation of organelles and AVs. Protein turnover is impaired because of the inability of protein clearance mechanisms, such as autophagy, to remove debris, leading to more abnormal protein aggregation and accumulation, and impaired axonal trafficking. The final steps are the vacuolization and hypertrophism of the axonal cytoskeleton, which leads to axonal degeneration. Therefore, moca−/− mice provide a novel model to study the connections between the distinct molecular and pathological features that define many neurological diseases.
Figure 8.
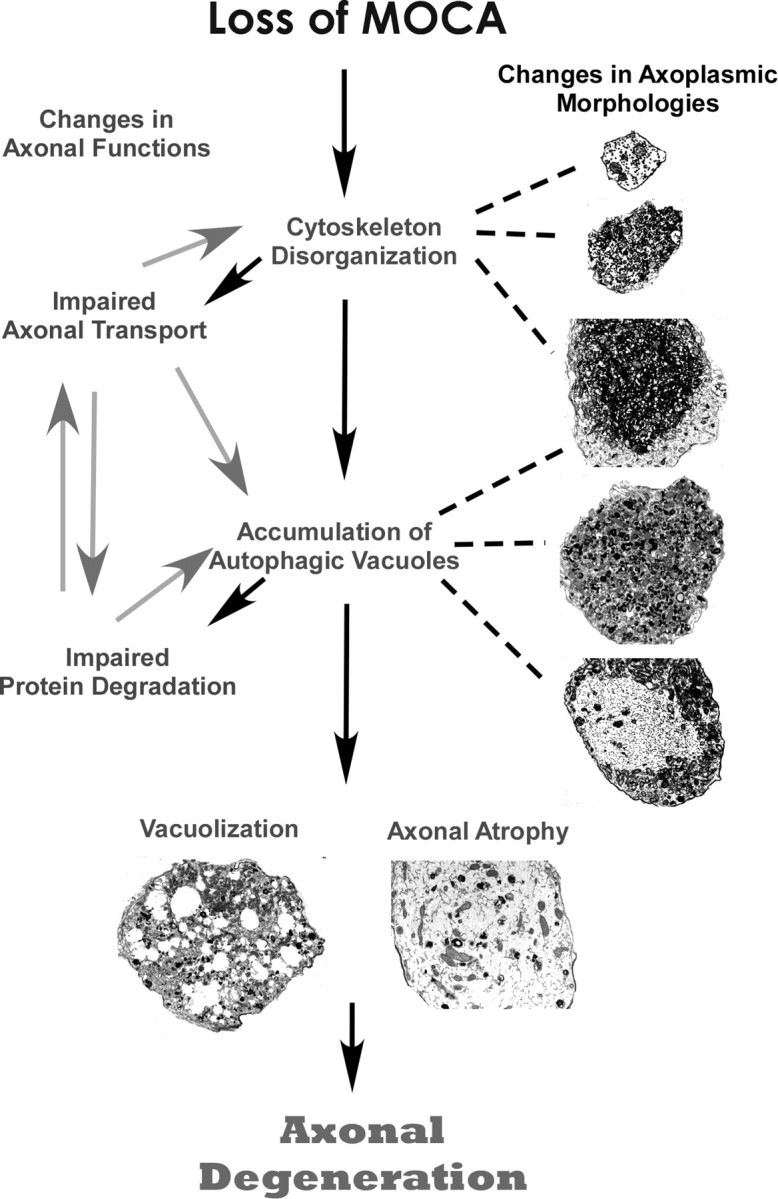
A model for the progression of axonal degeneration in moca−/− mice. See Discussion for details.
Footnotes
This work was supported by grants from the Alzheimer's Association and the Shirley Foundation for Alzheimer's Research to D.S., and by grants from the National Institutes of Health (NIH) to D.S. and P.E.S. Q.C. is supported by an NIH National Research Service Award fellowship and the Bundy Foundation. We are grateful to Drs. Pam Maher, Mark Geyer, Kuo-Fen Lee, Jan Lewerenz, Federico Herrera, Robert Cummings, Jordi Serrats, Robert Rissman, David Brown, Qingxian Lu, Ying Zhang, Zhijiang Chen, Michael Maloney, Victoria Risbrough, and Chunli Zhang for comments, discussions, and experimental assistance; and Carlos Arias and Kris Trulock for technical assistance.
References
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- Bray JJ, Fernyhough P, Bamburg JR, Bray D. Actin depolymerizing factor is a component of slow axonal transport. J Neurochem. 1992;58:2081–2087. doi: 10.1111/j.1471-4159.1992.tb10949.x. [DOI] [PubMed] [Google Scholar]
- Brown DA, Sawchenko PE. Time course and distribution of inflammatory and neurodegenerative events suggest structural bases for the pathogenesis of experimental autoimmune encephalomyelitis. J Comp Neurol. 2007;502:236–260. doi: 10.1002/cne.21307. [DOI] [PubMed] [Google Scholar]
- Cai D, Leem JY, Greenfield JP, Wang P, Kim BS, Wang R, Lopes KO, Kim SH, Zheng H, Greengard P, Sisodia SS, Thinakaran G, Xu H. Presenilin-1 regulates intracellular trafficking and cell surface delivery of β-amyloid precursor protein. J Biol Chem. 2003;278:3446–3454. doi: 10.1074/jbc.M209065200. [DOI] [PubMed] [Google Scholar]
- Chen Q, Yoshida H, Schubert D, Maher P, Mallory M, Masliah E. Presenilin binding protein is associated with neurofibrillary alterations in Alzheimer's disease and stimulates tau phosphorylation. Am J Pathol. 2001;159:1597–1602. doi: 10.1016/S0002-9440(10)63005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Kimura H, Schubert D. A novel mechanism for the regulation of amyloid precursor protein metabolism. J Cell Biol. 2002;158:79–89. doi: 10.1083/jcb.200110151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Chen TJ, Letourneau PC, Costa LdaF, Schubert D. Modifier of cell adhesion regulates N-cadherin-mediated cell-cell adhesion and neurite outgrowth. J Neurosci. 2005;25:281–290. doi: 10.1523/JNEUROSCI.3692-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier-Larsen E, Holzbaur EL. Axonal transport and neurodegenerative disease. Biochim Biophys Acta. 2006;1762:1094–1108. doi: 10.1016/j.bbadis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Côté JF, Vuori K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci. 2002;115:4901–4913. doi: 10.1242/jcs.00219. [DOI] [PubMed] [Google Scholar]
- de Silva MG, Elliott K, Dahl HH, Fitzpatrick E, Wilcox S, Delatycki M, Williamson R, Efron D, Lynch M, Forrest S. Disruption of a novel member of a sodium/hydrogen exchanger family and DOCK3 is associated with an attention deficit hyperactivity disorder-like phenotype. J Med Genet. 2003;40:733–740. doi: 10.1136/jmg.40.10.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JE, Goldstein LS. The genetics of axonal transport and axonal transport disorders. PLoS Genet. 2006;2:e124. doi: 10.1371/journal.pgen.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Deconstructing the axon: Wallerian degeneration and the ubiquitin-proteasome system. Trends Neurosci. 2004;27:3–6. doi: 10.1016/j.tins.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Gandy S, Zhang YW, Ikin A, Schmidt SD, Bogush A, Levy E, Sheffield R, Nixon RA, Liao FF, Mathews PM, Xu H, Ehrlich ME. Alzheimer's presenilin 1 modulates sorting of APP and its carboxyl-terminal fragments in cerebral neurons in vivo. J Neurochem. 2007;102:619–626. doi: 10.1111/j.1471-4159.2007.04587.x. [DOI] [PubMed] [Google Scholar]
- Glaumann H, Ericsson JL, Marzella L. Mechanisms of intralysosomal degradation with special reference to autophagocytosis and heterophagocytosis of cell organelles. Int Rev Cytol. 1981;73:149–182. doi: 10.1016/s0074-7696(08)61288-7. [DOI] [PubMed] [Google Scholar]
- Gohla A, Bokoch GM. 14-3-3 regulates actin dynamics by stabilizing phosphorylated cofilin. Curr Biol. 2002;12:1704–1710. doi: 10.1016/s0960-9822(02)01184-3. [DOI] [PubMed] [Google Scholar]
- Grimsley CM, Kinchen JM, Tosello-Trampont AC, Brugnera E, Haney LB, Lu M, Chen Q, Klingele D, Hengartner MO, Ravichandran KS. Dock180 and ELMO1 proteins cooperate to promote evolutionarily conserved Rac-dependent cell migration. J Biol Chem. 2004;279:6087–6097. doi: 10.1074/jbc.M307087200. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Herrera F, Martin V, Carrera P, García-Santos G, Rodriguez-Blanco J, Rodriguez C, Antolin I. Tryptamine induces cell death with ultrastructural features of autophagy in neurons and glia: possible relevance for neurodegenerative disorders. Anat Rec A Discov Mol Cell Evol Biol. 2006;288A:1026–1030. doi: 10.1002/ar.a.20368. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Glicksman MA, Willard MB. Organization of mammalian neurofilament polypeptides within the neuronal cytoskeleton. J Cell Biol. 1984;98:1523–1536. doi: 10.1083/jcb.98.4.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PN, Lasek RJ. The slow component of axonal transport: identification of major structural polypeptides of the axon and their generality among mammalian neurons. J Cell Biol. 1975;66:351–366. doi: 10.1083/jcb.66.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol. 1993;121:306–315. doi: 10.1083/jcb.121.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- Jung C, Chylinski TM, Pimenta A, Ortiz D, Shea TB. Neurofilament transport is dependent on actin and myosin. J Neurosci. 2004;24:9486–9496. doi: 10.1523/JNEUROSCI.1665-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28:449–459. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- Kashiwa A, Yoshida H, Lee S, Paladino T, Liu Y, Chen Q, Dargusch R, Schubert D, Kimura H. Isolation and characterization of novel presenilin binding protein. J Neurochem. 2000;75:109–116. doi: 10.1046/j.1471-4159.2000.0750109.x. [DOI] [PubMed] [Google Scholar]
- Kim SH, Lah JJ, Thinakaran G, Levey A, Sisodia SS. Subcellular localization of presenilins: association with a unique membrane pool in cultured cells. Neurobiol Dis. 2000;7:99–117. doi: 10.1006/nbdi.1999.0280. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lah JJ, Levey AI. Endogenous presenilin-1 targets to endocytic rather than biosynthetic compartments. Mol Cell Neurosci. 2000;16:111–126. doi: 10.1006/mcne.2000.0861. [DOI] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascaño J, Tokito M, Van Winkle T, Howland DS, Holzbaur ELF. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR, Koliatsos VE, Kins S, Lee VM, Wong PC, Price DL, Brady ST, Sisodia SS. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci. 2005;25:2386–2395. doi: 10.1523/JNEUROSCI.3089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, Morfini GA, Pigino G, Gadadhar A, Chen X, Robinson J, Ho H, Brady ST, Sisodia SS. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer's disease-linked mutant presenilin 1. J Neurosci. 2007;27:7011–7020. doi: 10.1523/JNEUROSCI.4272-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Cleveland DW. Neuronal intermediate filaments. Annu Rev Neurosci. 1996;19:187–217. doi: 10.1146/annurev.ne.19.030196.001155. [DOI] [PubMed] [Google Scholar]
- Leem JY, Vijayan S, Han P, Cai D, Machura M, Lopes KO, Veselits M, Xu H, Thinakaran G. Presenilin 1 is required for maturation and cell surface accumulation of nicastrin. J Biol Chem. 2002;277:19236–19240. doi: 10.1074/jbc.C200148200. [DOI] [PubMed] [Google Scholar]
- Maloney MT, Bamburg JR. Cofilin-mediated neurodegeneration in Alzheimer's disease and other amyloidopathies. Mol Neurobiol. 2007;35:21–44. doi: 10.1007/BF02700622. [DOI] [PubMed] [Google Scholar]
- Masliah E, Crews L, Hansen L. Synaptic remodeling during aging and in Alzheimer's disease. J Alzheimers Dis. 2006;9(Suppl 3):91–99. doi: 10.3233/jad-2006-9s311. [DOI] [PubMed] [Google Scholar]
- Meberg PJ, Bamburg JR. Increase in neurite outgrowth mediated by overexpression of actin depolymerizing factor. J Neurosci. 2000;20:2459–2469. doi: 10.1523/JNEUROSCI.20-07-02459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain. 2003;126:515–530. doi: 10.1093/brain/awg061. [DOI] [PubMed] [Google Scholar]
- Medori R, Jenich H, Autilio-Gambetti L, Gambetti P. Experimental diabetic neuropathy: similar changes of slow axonal transport and axonal size in different animal models. J Neurosci. 1988;8:1814–1821. doi: 10.1523/JNEUROSCI.08-05-01814.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller N, Merlot S, Guda C. CZH proteins: a new family of Rho-GEFs. J Cell Sci. 2005;118:4937–4946. doi: 10.1242/jcs.02671. [DOI] [PubMed] [Google Scholar]
- Minamide LS, Striegl AM, Boyle JA, Meberg PJ, Bamburg JR. Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat Cell Biol. 2000;2:628–636. doi: 10.1038/35023579. [DOI] [PubMed] [Google Scholar]
- Mizisin AP, Bache M, DiStefano PS, Acheson A, Lindsay RM, Calcutt NA. BDNF attenuates functional and structural disorders in nerves of galactose-fed rats. J Neuropathol Exp Neurol. 1997;56:1290–1301. doi: 10.1097/00005072-199712000-00004. [DOI] [PubMed] [Google Scholar]
- Namekata K, Enokido Y, Iwasawa K, Kimura H. MOCA induces membrane spreading by activating Rac1. J Biol Chem. 2004;279:14331–14337. doi: 10.1074/jbc.M311275200. [DOI] [PubMed] [Google Scholar]
- Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci. 2006;29:528–535. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Ono S. Regulation of actin filament dynamics by actin depolymerizing factor/cofilin and actin-interacting protein 1: new blades for twisted filaments. Biochemistry. 2003;42:13363–13370. doi: 10.1021/bi034600x. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Petzold A. Neurofilament phosphoforms: Surrogate markers for axonal injury, degeneration and loss. J Neurol Sci. 2005;233:183–198. doi: 10.1016/j.jns.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Sarmiere PD, Bamburg JR. Regulation of the neuronal actin cytoskeleton by ADF/cofilin. J Neurobiol. 2004;58:103–117. doi: 10.1002/neu.10267. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Stowers CC, Scallet AC, Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen A, Cumming RC, Finley KD. Linking lysosomal trafficking defects with changes in aging and stress response in Drosophila. Autophagy. 2007;3:499–501. doi: 10.4161/auto.4604. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Wirths O, Weis J, Kayed R, Saido TC, Bayer TA. Age-dependent axonal degeneration in an Alzheimer mouse model. Neurobiol Aging. 2007;28:1689–1699. doi: 10.1016/j.neurobiolaging.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Yang YC, Bauer C, Strasser G, Wollman R, Julien JP, Fuchs E. Integrators of the cytoskeleton that stabilize microtubules. Cell. 1999;98:229–238. doi: 10.1016/s0092-8674(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Näslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]