

Abstract
Cytochrome c oxidase (COX) is one of only four bigenomic proteins in mammalian cells, having ten subunits encoded in the nuclear genome and three in the mitochondrial DNA. The mechanism of its bigenomic control is not well understood. The ten nuclear subunits are on different chromosomes, and the possibility of their coordinate regulation by the same transcription factor(s) deserves serious consideration. The present study tested our hypothesis that nuclear respiratory factor 1 (NRF-1) serves such a role in subunit coordination. Following in silico analysis of murine nuclear-encoded COX subunit promoters, electrophoretic mobility shift and supershift assays indicated NRF-1 binding to all ten promoters. In vivo chromatin immunoprecipitation assays also showed NRF-1 binding to all ten promoters in murine neuroblastoma cells. Site-directed mutagenesis of putative NRF-1 binding sites confirmed the functionality of NRF-1 binding on all ten COX promoters. These sites are highly conserved among mice, rats, and humans. Silencing of NRF-1 with RNA interference reduced all ten COX subunit mRNAs and mRNAs of other genes involved in mitochondrial biogenesis. We conclude that NRF-1 plays a significant role in coordinating the transcriptional regulation of all ten nuclear-encoded COX subunits in neurons. Moreover, NRF-1 is known to activate mitochondrial transcription factors A and B, thereby indirectly regulating the expressions of the three mitochondrial-encoded COX subunits. Thus, NRF-1 and our previously described NRF-2 prove to be the two key bigenomic coordinators for transcriptional regulation of all cytochrome c oxidase subunits in neurons. Possible interactions between the NRFs will be investigated in the future.
Cytochrome c oxidase or complex IV is a large transmembrane protein located in the inner mitochondrial membrane of eukaryotes and plasma membrane of prokaryotes. It is the terminal enzyme of the electron transport chain, catalyzing the transfer of electrons from reduced cytochrome c to molecular oxygen to form water. The important outcome of this reaction is the generation of ATP through the coupled process of oxidative phosphorylation. Neurons are highly dependent upon ATP for their activity and functions (1). Approximately 90% of ATP generated in the brain is synthesized in the mitochondria via oxidative phosphorylation (2). The activity of this enzyme is reduced in neurodegenerative diseases, such as Alzheimer disease (3, 4). Among respiratory chain deficiencies presented in infancy and early childhood in humans, cytochrome c oxidase (COX)2 deficiency is the most commonly diagnosed (5). COX deficiency is found with different clinical phenotypes primarily affecting organs with high energy demand, such as the brain, skeletal muscle, heart, and kidney (6).
COX is a complex of 13 different subunits, 3 of which (I, II, and III) are encoded in the mitochondrial DNA, and the remaining 10 are nuclear-encoded (7). To form a functional holoenzyme with 1:1 stoichiometry, exact coordination is essential between the two genomes. All regulatory factors directing the expression of nuclear and mitochondrial respiratory genes are of nuclear origin. Nuclear-encoded factors such as mitochondrial RNA polymerase, a transcription and mitochondrial DNA maintenance factor (TFAM), transcription specificity factors (TFB1M and TFB2M), and a transcription termination factor govern mitochondrial gene expression (8-10). The second type of nuclear-encoded factors encompasses transcription factors and coactivators that control nuclear respiratory gene expression. These factors serve to integrate respiratory gene expression with a wide range of cellular functions (8, 11). Two redox-responsive transcription factors, nuclear respiratory factors 1 and 2, or NRF-1 and NRF-2, have been proposed to mediate such bigenomic coordination in non-neuronal cells (12-13). Both NRF-1 and NRF-2 reportedly regulate the expression of a few nuclear-encoded COX subunit genes and indirectly regulate the three mitochondrial-encoded COX subunit genes by activating mitochondrial transcription factors A and B (TFAM, TFB1M, and TFB2M) (9, 10). In addition, both NRF-1 and NRF-2 regulate a number of genes required for mitochondrial respiratory functions (11).
Previously, we showed that NRF-2 transcriptionally regulates all ten nuclear-encoded COX subunits in neurons (14, 15). We also found that the protein and mRNA levels of NRF-2 changed in response to changing neuronal activity and in concert with altered COX activity (16-20). Neuronal NRF-1, NRF-2, as well as an important coactivator, peroxisome proliferator-activated receptor-γ coactivator 1α all responded to impulse blockade in vitro and visual deprivation in vivo by down-regulating their gene expressions and gene product (21). Our recent study indicates that NRF-1 is transcriptionally regulated by neuronal activity, and sustained activity is required for heightened NRF-1 expression in cultured neurons (22). Whether NRF-1 also plays an important role in regulating all ten nuclear-encoded COX subunit genes located on different chromosomes was entirely unknown.
The goal of the present study was to test our hypothesis that NRF-1 regulates all ten nuclear-encoded COX subunit genes. Using multiple approaches, including in silico analysis, electrophoretic mobility shift assays (EMSA), super shift assays, chromatin immunoprecipitation (ChIP), promoter mutation assays, as well as RNA interference, we documented that NRF-1 has functional binding sites on all ten nuclear-encoded COX subunit promoters in murine neurons. Furthermore, the binding sites are conserved among mice, rats, and humans. Thus, NRF-1 and NRF-2 prove to be the two key bigenomic coordinators for transcriptional regulation of all cytochrome c oxidase subunits in neurons.
EXPERIMENTAL PROCEDURES
Cell Culture
Murine Neuro-2a neuroblastoma (N2a) cells (ATCC, CCL-131) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 50 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen).
In Silico Analysis of Murine COX Subunit Promoters
DNA sequences surrounding the transcription start points (TSPs) of all ten nuclear-encoded murine COX subunit genes were derived from the mouse genome data base in GenBank™. These promoter sequences encompassed 1 kb upstream and up to 1 kb downstream (excluding protein-coding sequence) of the TSP of each gene analyzed. Computer-assisted search for putative NRF-1 core binding sequences “GCGCAT/CGC” or “GCGCAG/CGC” was conducted on each promoter sequence.
Alignment of human, mouse, and rat promoter sequences was done as previously described, using the Genome VISTA genome alignment tool (14). Mouse COX promoter sequences were compared with rat and human genomic sequences using a 5-bp calculation window. Regions of high homology and/or contain known NRF-1 binding sites were compared for the conservation of NRF-1 binding.
Electrophoretic Mobility Shift and Supershift Assays
EMSAs to assay NRF-1 interactions with putative binding elements on all ten COX subunit promoters were carried out as previously described (14) with minor modifications. Briefly, oligonucleotide probes with putative NRF-1 binding site on each murine COX subunit promoter (Table 1, based on in silico analysis) were synthesized, annealed, and labeled by a Klenow fragment fill-in reaction with [α-32P]dATP (50 μCi/200 ng). Each labeled probe was incubated with 2 μg of calf thymus DNA and 5 μg of HeLa nuclear extract (Promega, Madison, WI) and processed for EMSA. Supershift assays were also performed and, in each reaction, 1−1.5 μg of NRF-1-specific antibodies (polyclonal goat antibodies, gift of Dr. Richard Scarpulla, Northwestern University, Chicago) were added to the probe/nuclear extract mixture and incubated for 20 min at room temperature. For competition, 100-fold excess of unlabeled oligonucleotide was incubated with nuclear extract before adding labeled or nonspecific oligonucleotide. Shift reactions were loaded onto 4% polyacrylamide gel and run at 200 V for 2.5 h in 0.25× TBE buffer. Results were visualized by autoradiography. Rat cytochrome c with NRF-1 binding site at position −172/−147 was designed as previously described (12) and used as a positive control. NRF-1 mutants with mutated sequences as shown in Table 1 were used as negative controls.
TABLE 1. EMSA probes.
Positions of probes are given relative to TSP. Putative NRF-1 binding sites are in boldface. Mutated nucleotide sequences are underlined.
| Gene | Position | Sequence |
|---|---|---|
| MCOX4i1 | −342/−313 | F 5′-TTTTGCTTCACGCCGGGCATCGCGACCCACGC-3′ |
| R 3′-CGAAGTGCGGCCCGTAGCGCTGGGTGCGTTTT-5′ | ||
| MCOX5a | −155/−133 | F 5′-TTTTGCCGGCGACCGCATGGCGCCCC-3′ |
| R 3′-CGGCCGCTGGCGTACCGCGGGGTTTT-5′ | ||
| MCOX5b | −110/−86 | F 5′-TTTTGCAGAACTGCGCATGTGCGGCGTC-3′ |
| R 3′-CGTCTTGACGCGTACGCGCCGCAGTTTT-5′ | ||
| MCOX6a1 | −166/−142 | F 5′-TTTTGGAACGGGGCGCATGCGCGCTCTC-3′ |
| R 3′-CCTTGCCCCGCGTACGCGCGAGAG′TTTT-5′ | ||
| MCOX6b | −116/−94 | F 5′-TTTTTTAGGCAGAGGCATGCGGATTTCT-3′ |
| R 3′-AATCCGTCTCCGTACGCCTAAAGATTTT-5′ | ||
| MCOX6c | +40/+64 | F 5′-TTTTCGACTCTTGCGCATGCGTGCTGCT-3′ |
| R 3′-GCTGAGAACGCGTACGCACGACGATTTT-5′ | ||
| MCOX7a2 | −66/−42 | F 5′-TTTTGGGTCCGGCGGCAGGCGCGGCAGG-3′ |
| R 3′-CCCAGGCCGCCGTCCGCGCCGTCCTTTT-5′ | ||
| MCOX7b | +87/+111 | F 5′-TTTTCAAGGCAGCAGCATAGTCGCCGCA-3′ |
| R 3′-GTTCCGTCGTCGTATCAGCGGCGTTTTT-5′ | ||
| MCOX7c | +141/+167 | F 5′-TTTTATCATGCTGCGCATAGGAGTTTCT-3′ |
| R 3′-TAGTACGACGCGTATCCTCAAAGATTTT-3′ | ||
| MCOX8a | −161/−137 | F 5′-TTTTGCGTGGTGGCGCATGCCTTTAATC-3′ |
| R 3′-CGCACCACCGCGTACGGAAATTAGTTTT-5′ | ||
| Rat Cyt C | −172/−147 | F 5′-TTTTCTGCTAGCCCGCATGCGCGCGCACCTTA-3′ |
| R 3′-GACGATCGGGCGTACGCGCGCGTGGAATTTTT-5′ | ||
| NRF-1 mutant (MCOX4i1) | −342/−313 | F 5′-TTTTGCTTCATTACGGTTTTCGCGACCCACGC-3′ |
| R 3′-CGAAGTAATGCCCAAAAGCGCTGGGTGCGTTTT-5′ | ||
| NRF-1 mutant (MCOX5a) | −155/−133 | F 5′-TTTTGCCGTTAACCTTTTGGCGCCCC-3′ |
| R 3′-CGGCAATTGGAAAACCGCGGGGTTTT-5′ |
ChIP Assays
ChIP assays were performed similar to those previously described (23, 24). Briefly, ∼750,000 N2a cells were used for each immunoprecipitation and were fixed with 1% formaldehyde for 10 min at room temperature. ChIP assay kit (Upstate, Charlottesville, VA) was used with minor modifications. Following formaldehyde fixation, cells were resuspended in a swelling buffer (5 mm PIPES, pH 8.0, 85 mm KCl, and 1% Nonidet P-40, and protease inhibitors added right before use) and homogenized 10 times in small pestle Dounce tissue homogenizer (7 ml). Nuclei were then isolated by centrifugation before being subjected to sonication. The sonicated lysate was immunoprecipitated with either 0.2 μg of NRF-1 polyclonal rabbit antibodies or 2 μg of anti-nerve growth factor receptor (NGFR) p75 polyclonal goat antibodies (C20 from Santa Cruz Biotechnology, Santa Cruz, CA).
Semi-quantitative PCR was performed using 1/20th of precipitated chromatin. Primers targeting promoter sequences near TSP of COX nuclear subunit genes were designed (Table 2) as previously described (14). TFB2M promoter was used as a positive control, and exon 5 of β-actin gene was used as a negative control (Table 2). PCR reactions were carried out with the EX Taq hot-start polymerase (Takara Mirus Bio, Madison, WI) with the following cycling parameters: 30-s melting at 94 °C, 30-s annealing at 59.5 °C, and 20-s extension at 72 °C (32-36 cycles per reaction). All reactions were hot-started by heating to 94 °C for 120 s. Because the proximal promoters of nuclear COX genes tend to be very GC-rich, reaction conditions for some amplicons were optimized by adding magnesium and/or 0.5 m betaine (Sigma) (reaction conditions are available upon request). Use of hot-start polymerase and PCR additives significantly improved the quality and reproducibility of ChIP. PCR products were visualized on 2% agarose gels stained with ethidium bromide.
TABLE 2. Primers for ChIP assays.
Positions of amplicons are given relative to TSP.
| Gene | Sequence | Amplicon length | Position |
|---|---|---|---|
| bp | |||
| MCOX4i1 promoter | F 5′-GTCTTGGTCTTCCGGTTGC-3′ | 110 | −43 to +49 |
| R 5′-GTCCAAGGCCGTCACCTG-3′ | |||
| MCOX5a promoter | F 5′-CCACGCAGGAATGTTCACTA-3′ | 237 | −246 to −27 |
| R 5′-ACGAGAAGCCGGTGTGAG-3′ | |||
| MCOX5b promoter | F 5′-GTGCGGCGTCTACTTTTAGC-3′ | 171 | −247 to −96 |
| R 5′-ACTCCGCGAAGTAACCTTGA-3′ | |||
| MCOX6a1 promoter | F 5′-CAGGTCTGAGAAGGCGTTG-3′ | 141 | −90 to +51 |
| R 5′-GTCAGCGTCTCGGGTCTCTC-3′ | |||
| MCOX6b promoter | F 5′-ATAGGAGGTCGGGCTTCTTC-3′ | 162 | −102 to +60 |
| R 5′-TAGCAAAGACGCCAATGTCA-3′ | |||
| MCOX6c promoter | F 5′-ATGCGCGCTTGAATACTTTT-3′ | 168 | −4 to +164 |
| R 5′-TGTCAAACCCCAGAATCTCC-3′ | |||
| MCOX7a2 promoter | F 5′-GCTAGGAGGGAGTTCCGTTT-3′ | 139 | +130 to +249 |
| R 5′-ACTCCAGTACCAGCCAAACC-3′ | |||
| MCOX7b promoter | F 5′-AAATCTCGCGACATCTCACC-3′ | 162 | −12 to +150 |
| R 5′-TTTTGGCTAAGGGCAACATC-3′ | |||
| MCOX7c promoter | F 5′-ACAAAGCCCACAAACCTCAG-3′ | 140 | −74 to +66 |
| R 5′-GAAATGGCCGTACCACCTAA-3′ | |||
| MCOX8a promoter | F 5′-TGATGTCGGTTGGTTGTTTC-3′ | 213 | −89 to +124 |
| R 5′-AGTGGCGTCAGGACAGACAT-3′ | |||
| MTFB2M promoter | F 5′-GAAGCGAGTGAGCAAAGGAC-3′ | 179 | −64 to +115 |
| R 5′-GGTCCCCTCATCCTCCTCTA-3′ | |||
| β-Actin exon 5 | F 5′-GCTCTTTTCCAGCCTTCCTT-3′ | 187 | −134 to +53 |
| R 5′-CGGATGTCAACGTCACACTT-3′ |
Construction and Transfection of Luciferase Reporter Vectors for Promoter Mutagenesis Study
Luciferase reporter constructs of all 10 murine COX promoters (mCOX 4i1, 5a, 5b, 6a1, 6b, 6c, 7a2, 7b, 7c, and 8a) were made by PCR cloning the proximal promoter sequences using genomic DNA prepared from mouse N2a cells as template, digesting with KpnI and HindIII, and ligating the product directionally into pGL3 basic (Promega). Sequences of primers used for PCR cloning and mutagenesis primers are provided in Tables 3 and 4. Subunits COX5a and COX6a1 were cloned into EcoR1 site in pGEM-T Easy vector (TA cloning kit, Promega) to generate full-length construct. These pGEM-T vectors with inserts for both subunits were then subcloned into HindIII and KpnI sites and ligated to pGL3-basic vector. Site-directed mutagenesis of putative NRF-1 binding site on each promoter were generated using QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). All constructs were verified by sequencing.
TABLE 3.
Primers for PCR cloning
| Cloning primers | Primer sequence |
|---|---|
| MCOX4i1 | F 5′-AAGGTACCCGGGTCTTGCAAATGTCTCT-3′ |
| R 5′-AAAAGCTTGACAGCAAACACCTTTTAGCC-3′ | |
| MCOX5a | F 5′-GCACAGCACAATGTGGGTA-3′ |
| R 5′-GGAGTCTCCTACACGACTCCAG-3′ | |
| MCOX5a subclone | F 5′-AAACGCGTGCACAGCACAATGTGGGTA-3′ |
| R 5′-AAAGATCTACCACAGCGCACTGGCT-3′ | |
| MCOX5b | F 5′-AAGGTACCCAACACTGAGCAATGGAGGA-3′ |
| R 5′-AAAAGCTTGGAACAGGCTGAGCAAGATG-3′ | |
| MCOX6a1 | F 5′-GTTCCATCTGCGTCTTCCTC-3′ |
| R 5′-AGAGACCCGAGACGCTGAC-3′ | |
| MCOX6a1 subclone | F 5′-AAGGTACCGCGTCTTCCTCGCAGATACT-3′ |
| R 5′-AAAAGCTTGGAACTACACCGGCGCGC-3′ | |
| MCOX6b | F 5′-AAGGTACCGCCAGCCCTTAATTGTTTTC-3′ |
| R 5′-AAAAGCTTTCGCAACTAAAAGCTCCACA-3′ | |
| MCOX6c | F 5′-AAACGCGTGTGTGATGGTGCATGTGTCA-3′ |
| R 5′-AACTCGAGTAACCAGCAACAAACCGATG-3′ | |
| MCOX7a2 | F 5′-AAACGCGTTTGCAAAAGGGTGGAAAGAC-3′ |
| R 5′-AACTCGAGAAAAATCGAAAGCCACCAGA-3′ | |
| MCOX7b | F 5′-AAACGCGTAGGGAGACCTGGCTTACACA-3′ |
| R 5′-AACTCGAGGGAGACGAAGATGGAACTGC-3′ | |
| MCOX7c | F 5′-AAGGTACCCACACAGTGACCCCCTTTTC-3′ |
| R 5′-AAAAGCTTGAAATGGCCGTACCACCTAA-3′ | |
| MCOX8a | F 5′-AAGGTACCCCTGGGCTACTGGAGACCTT-3′ |
| R 5′-AAAAGCTTACCACGGACGAGGACCAGTT-3′ |
TABLE 4. Mutagenesis primers.
Boldface letters denote mutated nucleotide sequences.
| MCOX4I1NRF-1MUT | F 5′-CAGCTTCACGCCGGTTTTCGCGACCCACGCAC-3′ |
| R 5′-GTGCGTGGGTCGCGAAAACCGGCGTGAAGCTG-3′ | |
| MCOX5aNRF-1MUT | F 5′-AGCCGCCGGCGACCTTTTGGCGCCCCACC-3′ |
| R 5′-GGTGGGGCGCCAAAAGGTCGCCGGCGGCT-3′ | |
| MCOX5bNRF-1MUT | F 5′-CCACGAAGAGGCAGAACTGCTTTTGTGCGGCGTCTACT-3′ |
| R 5′-AGTAGACGCCGCACAAAAGCAGTTCTGCCTCTTCGTGG-3′ | |
| MCOX6a1NRF-1MUT | F 5′-CACAGGAACGGGGCTTTTGCGCGCTCTCGCTC-3′ |
| R 5′-GAGCGAGAGCGCGCAAAAGCCCCGTTCCTGTG-3′ | |
| MCOX6bNRF-1MUT | F 5′-CAGCACTAGTTAGGCAGAGTTTGGCGGATTTCTGAGTCTAC-3′ |
| R 5′-GTAGACTCAGAAATCCGCCAAACTCTGCCTAACTAGTGCTGG-3′ | |
| MCOX6cNRF-1MUT | F 5′-CCTCCATCGACTCTTGCTTTTGCGTGCTGCTGGAAGG-3′ |
| R 5′-CCTTCCAGCAGCACGCAAAAGCAAGAGTCGATGGAGG-3′ | |
| MCOX7a2NRF-1MUT | F 5′-TTCTGGGTCCGGCGTTTGGCGCGGCAGGG-3′ |
| R 5′-CCCTGCCGCGCCAAACGCCGGACCCAGAA-3′ | |
| MCOX7bNRF-1MUT | F 5′-GAATTTGCACCAAGGCAGCATTTTAGTCGCCGCAGTTCCATC-3′ |
| R 5′-GATGGAACTGCGGCGACTAAAATGCTGCCTTGGTGCAAATTC-3′ | |
| MCOX7cNRF-1MUT | F 5′-CTCCAACCTAATCATGCTGCTTTTAGGAGTTTCTATCTGTATGTCCTC-3′ |
| R 5′-GAGGACATACAGATAGAAACTCCTAAAAGCAGCATGATTAGGTTGGAG-3′ | |
| MCOX8aNRF-1MUT | F 5′-GCCGGGCGTGGTGGCTTTTGCCTTTAATCCCAGC-3′ |
| R 5′-GCTGGGATTAAAGGCAAAAGCCACCACGCCCGGC-3′ |
Each promoter construct was transfected into N2a cells in a 24-well plate using Lipofectamine 2000. Each well received 0.6 μg of reporter construct and 0.03 μg of pCMVβgal, which constitutively expressed β-galactosidase. Cells were harvested 48 h post-transfection and lysed with reporter lysis buffer (Promega). Luciferase activity in the lysate was measured using luciferase assay reagent (Promega), and β-galactosidase activity was measured with Galacto-star reagent (Tropix, Bedford, MA). For each sample, luciferase activity was normalized with β- galactosidase activity. Data from six independent transfections were averaged for each promoter construct. p values were calculated from two-tailed t-tests using normalized data from individual transfections, and p ≤ 0.05 was considered significant.
Plasmid Construction of NRF-1 shRNA
The pLL3.7/U6 promoter vector with puromycin resistance was used to express murine NRF-1 shRNA (GenBank™ accession no. for NRF-1: NM_010938). Four shRNA sequences were selected: 5′-GAAAGCTGCAAGCCTATCT-3′; 5′-GCCACAGGAGGTTAATTCA-3′; 5′-GCATTACGGACCATAGTTA-3′; and 5′-AGAGCATGATCCTGGAAGA-3′. Empty vectors served as negative controls. A green fluorescent protein-containing reporter vector PLVTHM (gift of Dr. P. Aebischer, Swiss Federal Institute of Technology) was used to identify transfected N2a cells. The basic gene clone method was followed and as described previously (15). To determine the effect of silencing NRF-1 expression on endogenous targets, N2a cells were plated in 35-mm dishes at a density of 5 to 8 × 106 cells/dish. Cells were co-transfected 3 days post-plating with either 4 μg of the shRNA plasmids (four sequences at equal amounts) or 4 μg of the empty vector and 1.5 μg of pLL3.7 Puro vector, using Lipofectamine 2000 as described previously (15). Puromycin at a final concentration of 0.5 μg/ml was added to the culture medium on the second day after transfection to select for purely transfected cells. Transfection efficiency was monitored by observing green fluorescence. N2a cells were harvested after 48 h for RNA isolation.
RNA Isolation and cDNA Synthesis
Total RNA was isolated by RNeasy kits (Qiagen) according to the manufacturer's instructions. Three micrograms of total RNA was treated with DNase I and purified by phenol-chloroform. cDNA was synthesized using random hexamer primers and SuperScript™ II RNase H-Reverse Transcriptase (Invitrogen) according to the manufacturer's instructions.
Real-time Quantitative PCR
Real-time quantitative PCRs were carried out in a Cepheid Smart Cycler Detection system (Cepheid, Sunnyvale, CA). SyBr Green (BioWhittaker Molecular Application) and EX Taq real-time quantitative PCR hot-start polymerase were used following the manufacturer's protocols and as described previously (21). Primer sequences are shown in Table 5. PCR runs: hot start 2 min at 95 °C, denaturation 10 s at 95 °C, annealing 15 s according to the Tm of each primer, and extension 10 s at 72 °C for 15−30 cycles. Melt curve analyses verified the formation of single desired PCR product. Mouse β-actin was the internal control, and the ΔΔCT method (25) was used to calculate the relative amount of transcripts. The group means were then analyzed for overall statistical significance using analysis of variance and the Student's t test. p values of 0.05 or less were considered significant.
TABLE 5.
Primers for real-time PCR
| Gene | Sequence | Amplicon length | Tm |
|---|---|---|---|
| bp | °C | ||
| β-Actin | F 5′-GGCTGTATTCCCTCCATCG-3′ | 154 | 59.5 |
| R 5′-CCAGTTGGTAACAATGCCATGT-3′ | |||
| MCOX4i1 | F 5′-TCTACTTCGGTGTGCCTTCG-3′ | 253 | 59.5 |
| R 5′-ACTCATTGGTGCCCTTGTTC-3′ | |||
| MCOX5a | F 5′-GGAGTTGCGTAAAGGGATGA-3′ | 247 | 60 |
| R 5′-CACTTTGTCAAGGCCCAGTT-3′ | |||
| MCOX5b | F 5′-GGAGGTGGTGTCCCTACTGA-3′ | 241 | 59.5 |
| R 5′-CAGCCAGAACCAGATGACAG-3′ | |||
| MCOX6a1 | F 5′-TCAACGTGTTCCTCAAGTCGC-3′ | 115 | 60 |
| R 5′-AGGGTATGGTTACCGTCTCCC-3′ | |||
| MCOX6b | F 5′-ATGGCCGAAGACATCAAGAC-3′ | 250 | 60 |
| R 5′-CAGGAAATGTGCCTTCTGCT-3′ | |||
| MCOX6c | F 5′-AGCGTCTGCGGGTTCATA-3′ | 154 | 60 |
| R 5′-GCCTGCCTCATCTCTTCAAA-3′ | |||
| MCOX7a2 | F 5′-GAGGACCATCAGCACCACTT-3′ | 234 | 59.5 |
| R 5′-TGGAGACTGGGATGACGAC-3′ | |||
| MCOX7b | F 5′-CGAAGCATTCAGCAAGTGGT-3′ | 209 | 59.5 |
| R 5′-TGGCATGACTACTGATCTCTCC-3′ | |||
| MCOX7c | F 5′-TCTGCCTTCCGTCTCTGC-3′ | 145 | 60 |
| R 5′-AGAAAGGAGCAGCAAATCCA-3′ | |||
| MCOX8a | F 5′-TCCTGCTTCGTGTGTTGTCT-3′ | 70 | 59 |
| R 5′-TCCCGCTTCTTGTAGCTTTC-3′ | |||
| NRF-2α | F 5′-CTCCCGCTACACCGACTAC-3′ | 145 | 59.5 |
| R 5′-TCTGACCATTGTTTCCTGTTCTG-3′ | |||
| TFAM | F 5′-CCGAAGTGTTTTTCCAGCAT-3′ | 144 | 60 |
| R 5′-CAGGGCTGCAATTTTCCTAA-3′ | |||
| TFB1M | F 5′-AAGATGGCCCTTTCGTTTATGG-3′ | 102 | 60 |
| R 5′-GACTGTGCTGTTTGCTTCCTG-3′ | |||
| TFB2M | F 5′-CCAAAACCCATCCCGTCAAAT-3′ | 135 | 60 |
| R 5′-AAGGGCTCCAAATGTGGAATAAA-3′ | |||
| SURF1 | F 5′-GGTTCCTGCTTTTAATCCCTGC-3′ | 119 | 61 |
| R 5′-GATGGGCTCAGCCATGACT-3′ | |||
| VDAC1 | F 5′-CTGAGTATGGGCTGACGTTTAC-3′ | 189 | 60 |
| R 5′-GGTGAGCTTCAGTCCACGAG-3′ | |||
| TOM20 | F 5′-GCCCTCTTCATCGGGTACTG-3′ | 101 | 61 |
| R 5′-ACCAAGCTGTATCTCTTCAAGGA-3′ |
Western Blot Assay
Control and NRF-1 shRNA samples were loaded onto 10% SDS-PAGE gel and electrophoretically transferred onto polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). Subsequent to blocking, blots were incubated in primary antibodies against NRF-1 (1:500) or monoclonal antibodies against β-actin (Sigma) at 1:3,000 dilution as loading controls, and in secondary goat-anti-rabbit or goat-anti-mouse antibodies (Bio-Rad). Blots were then reacted with ECL and exposed to autoradiographic film (Amersham Biosciences). Quantitative analyses of relative changes were done with an Alpha Imager (Alpha Innotech, San Leandro, CA).
RESULTS
In Silico Promoter Analysis of Nuclear-encoded Mouse COX Subunit Genes
In silico analysis of the proximal promoters with DNA sequence 1 kb 5′ upstream and 100 bps beyond 3′ of TSP of murine nuclear-encoded COX 5b, 6a1, 6c, and 8a promoters showed typical sequence G/TGCGCATG/CC/TG/C/T, whereas COX 4i1, 5a, 6b, 7a2, 7b, and 7c showed atypical sequencesC/(A/T)/G/(C/A)C/(G/A) GCAT/GG/(C/A)C/GG/T/C, with the core GCA being the invariant sequence.
In Vitro Binding of NRF-1 Transcription Factor on All Ten Nuclear-encoded COX Subunit Promoters
Electrophoretic mobility shift assays (EMSAs) for NRF-1 interactions were carried out in vitro, using 32P-labeled oligonucleotide probes that encompassed putative NRF-1 binding sites on each of 10 murine COX promoters (4i1, 5a, 5b, 6a1, 6b, 6c, 7a2, 7b, 7c, and 8a). NRF-1 site from position −172/−147 of the rat cytochrome c promoter was used as a positive control, and it formed specific DNA/NRF-1 shift and supershift complexes (Fig. 1A, lanes 1 and 3, respectively). When an excess of unlabeled probe was added as a competitor, no band was found (Fig. 1A, lane 2). When labeled oligonucleotides were incubated with NRF-1 antibody but without HeLa nuclear extract, neither shift nor supershift band was observed (Fig. 1A, lanes 4 and 9 for COX 4i1- and COX 5a-specific oligonucleotides, respectively), ruling out nonspecific antibody-oligonucleotide interactions. As shown in Fig. 1 (A–C), all ten mouse COX promoters formed specific DNA-protein shift complexes when incubated with purified HeLa nuclear extract (Fig. 1A, lanes 5 and 10; Fig. 1, B and C, lanes 1, 4, 7, and 10, respectively). These complexes were displaced by competition with excess unlabeled probes (Fig. 1A, lanes 6, 11, and 15; Fig. 1, B and C, lanes 2, 5, 8, and 11, respectively), but were not displaced with scrambled unlabeled mutant NRF-1 probes (Fig. 1A, lanes 7 and 12). Shift assays using anti-NRF-1 antibodies produced a super-shift band of DNA/NRF-1/antibody complex for each of the subunits (Fig. 1A, lanes 8 and 13; Fig. 1, B and C, lanes 3, 6, 9, and 12, respectively). NRF-1 mutant showed neither shift nor super shift complexes (Fig. 1A, lanes 14 and 16, respectively).
FIGURE 1. In vitro binding activity of NRF-1 to putative binding sites on all ten nuclear-encoded COX subunits as measured with EMSA and supershift assays.
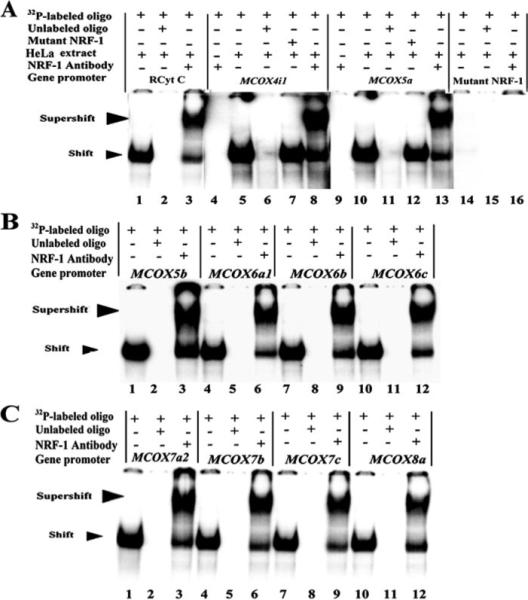
32P-labeled oligonucleotides (oligo), excess unlabeled oligonucleotides as competitors, excess unlabeled mutant NRF-1 as competitors, HeLa extract, and NRF-1 antibodies added to or absent from the reactions are indicated by a + or a − sign, respectively, above each lane. Arrowheads mark the specific NRF-1/probe shift complex and antibody-supershifted complex. Rat cytochrome c served as a positive control, showing shift band (A, lanes 1) and supershift band (A, lane 3) with labeled NRF-1 binding site. Excess unlabeled probe eliminated the shift band (A, lane 2). Labeled oligonucleotides with mutated NRF-1 site on COX 4i1 served as a negative control (A, lanes 14−16). Labeled oligonucleotides with putative NRF-1 binding sites on all ten mouse COX subunits showed specific shift and supershift bands that are eliminated by excess unlabeled competitors (A, lanes 5, 6, 8, 10, 11, and 13; B and C, all lanes). On the other hand, excess unlabeled oligonucleotides with mutated NRF-1 site were unsuccessful in competing for the binding (A, lanes 7 and 12). Labeled oligonucleotides for COX 4i1 and COX5a with NRF-1 antibodies alone did not yield any bands (A, lanes 4 and 9), ruling out nonspecific antibody-oligonucleotide interactions.
In Vivo Occupancy of the Proximal Promoters of COX Subunits by NRF-1
To determine whether NRF-1 actually binds to the promoters of COX subunits in vivo, ChIPs were performed. As a negative control, another immunoprecipitation from the same stock of cell lysate was done using anti-nerve growth factor receptor p75 antibodies (NGFR). PCRs targeting regions of ten COX subunit promoters surrounding putative NRF-1 binding sites were carried out in parallel on chromatin immunoprecipitated from N2a cells. A 0.5 and 0.1% dilution of input chromatin (i.e. prior to immunoprecipitation) was used as a standard to indicate the efficiency of the PCRs.
The proximal promoters of all 10 COX subunits 4i1, 5a, 5b, 6a1, 6b, 6c, 7a2, 7b, 7c, and 8a were co-immunoprecipitated with NRF-1 antibodies and were amplified in semi-quantitative PCRs (Fig. 2). The amount of DNA precipitated by anti-NRF-1 antibodies (NRF-1 lanes) was greater than the amount precipitated by anti-NGFR (a negative control for background, NGFR lanes) for each of the ten COX subunit promoters. TFB2M (positive control) showed a clear band, whereas β-actin (negative control) yielded no band with NRF-1 (Fig. 2).
FIGURE 2. In vivo ChIP assays for NRF-1 interaction with COX nuclear-encoded subunit promoters.
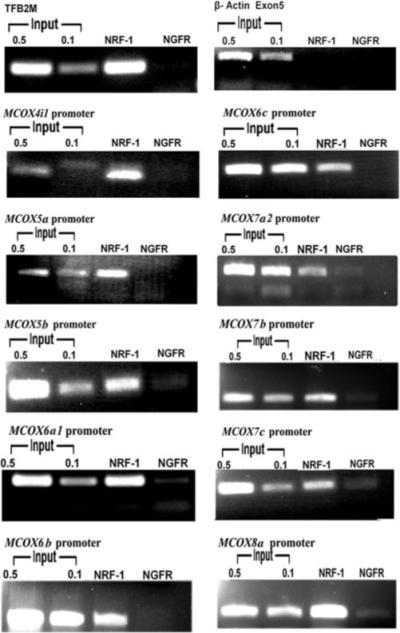
PCR reactions were performed on N2a cell chromatin precipitated with anti-NRF-1 antibodies (NRF-1 lanes) or anti-nerve growth factor receptor p75 antibodies (negative control, NGFR lanes). Control reactions were performed with 0.5% and 0.1% of input chromatin (input lanes). PCR products targeting COX 4i1, 5a, 5b, 6a1, 6b, 6c, 7a2, 7b, 7c, and 8a promoters revealed that all ten COX promoters’ DNA co-immunoprecipitated with NRF-1. Reactions targeting TFB2M promoter was used as a positive control, and β-actin was used as a negative control.
Investigation of Mutated NRF-1 Binding Sites within the Ten COX Subunit Promoters
Site-directed mutagenesis of putative NRF-1 binding sites were constructed in luciferase reporter plasmids, and their effects on promoter function were assayed by gene transfection. NRF-1 binding sites mutated were based on EMSA probes shown to form NRF-1-specific complexes (Fig. 1, A–C). As depicted in Fig. 3, mutated NRF-1 binding sites for each of the ten COX subunit promoters caused a significant reduction of promoter activity. Specifically, mutations of NRF-1 sites at positions −110/−86, +40/+64, and −161/−137 on COX 5b, 6c, and 8a promoters, respectively, reduced 70−80% of luciferase reporter activity. For COX 4i1, 6a1, and 6b promoters, mutations of NRF-1 sites at positions −342/−313, −166/−142, and −116/−94, respectively, led to a 50−60% reduction of promoter activity. For the remaining subunits, COX 5a, 7a2, 7b, and 7c, mutations of NRF-1 sites at positions −155/−133, −66/−42, +87/+111, and +141/+167, respectively, reduced by ∼40% of the original promoter activity.
FIGURE 3. Mutational analysis of the promoter elements of COX genes.

Relative luciferase activity of wild-type and site-directed mutations of NRF-1 binding sites on all ten nuclear-encoded COX promoters indicates significant reductions in luciferase activity in all mutants. (n = 6 for each construct). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
RNA Interference-mediated Silencing of NRF-1 Decreased mRNAs of COX Subunit and Other Nuclear-encoded Transcripts
To study the cellular effect caused by the silencing of NRF-1 transcripts, we used plasmid vectors expressing shRNA against four target sequences in the NRF-1 mRNA. Transfection of N2a cells with shRNA vectors resulted in ∼80−90% reduction of NRF-1 mRNA as measured by real-time quantitative PCR relative to empty vector controls (Fig. 4A). This reduction of NRF-1 mRNA was matched by a ∼75−85% (p < 0.05−0.01) decrease in NRF-1 protein as measured by Western blots (Fig. 4B).
FIGURE 4. RNA interference-mediated silencing of NRF-1 suppresses mRNAs in all 10 nuclear-encoded COX subunit genes and another six nuclear genes important in mitochondrial biogenesis.
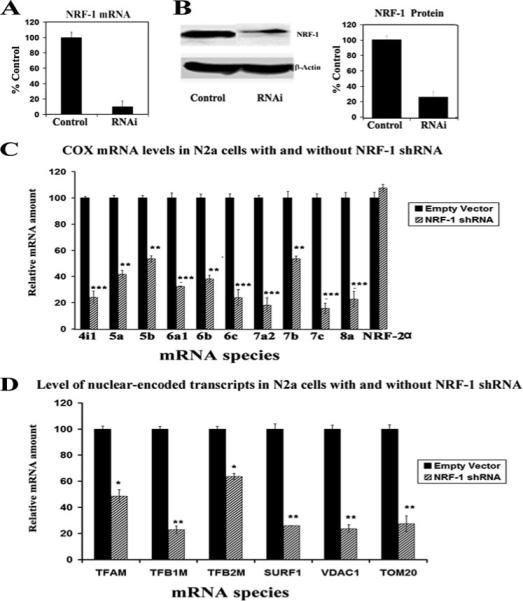
A, N2a cells transfected with NRF-1 shRNA expressed significantly less NRF-1 mRNA than those with empty vectors. B, Western blot reveals a down-regulation of NRF-1 protein in shRNA-transfected neurons. β-Actin served as a loading control. C, N2a cells were transfected with shRNA against NRF-1 (hatched bars) or with empty vectors (solid bars). NRF-2α served as a negative control. All ten COX subunit mRNAs show significant decreases in shRNA-treated samples as compared with those with empty vectors, whereas NRF-2α mRNA remained unchanged. n = 5−6 for each data point; *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, mRNAs were quantified for six other known target genes of NRF-1: TFAM, TFB1M, TFB2M, SURF1, VDAC1, and TOM20 in N2a cells. All six genes showed significant reductions in mRNA expression in NRF-1 shRNA-transfected cells (hatched bars) as compared with empty vector controls (solid bars). n = 5−6 for each data point; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
cDNAs from control (empty vector) and shRNA-transfected N2a cells were analyzed for the effect of NRF-1 silencing on transcriptional expression of nuclear-encoded COX subunit genes. Levels of all ten nuclear-encoded COX subunit mRNAs were significantly reduced in cells transfected with shRNA as compared with those transfected with empty vectors (Fig. 4C). The amount of reduction in COX mRNAs ranged from ∼40% to 75% (p < 0.05−0.001). The expression of NRF-2α, which served as a negative control, remained unchanged.
In addition, we measured mRNAs of other nuclear-encoded target genes of NRF-1: TFAM, TFB1M, TFB2M, surfeit 1 (SURF1), voltage-dependent anion channel (VDAC1), and transporter of outer mitochondrial membrane 20 (TOM20). All of these genes had significantly reduced levels of transcripts in NRF-1-silenced N2a cells (Fig. 4D). The extent of decrease ranged from ∼35% to 70% with p < 0.05−0.01.
Conservation of NRF-1 Binding Sites among Mouse, Rat, and Human COX Promoters
We aligned the sequences of all ten mouse COX subunit genes to the homologous regions in the rat and human genomes to verify whether the NRF-1 binding sites tested in the present study (EMSA and mutational analyses) on the mouse COX subunit promoters are conserved in other species. Alignment of these sequences showed a high degree of homology (60−100%) in the three species (Fig. 5). Additional NRF-1 binding sites were also noted (data not shown). Thus, NRF-1 sites are highly conserved among mice, rats, and humans.
FIGURE 5. All ten nuclear-encoded COX subunit promoters from mouse, rat, and human contained conserved putative NRF-1 binding sites.

Aligned partial sequences of COX promoters from human (H), mouse (M), and rat (R) genomes show conservation of typical and atypical NRF-1 binding sites. Conserved binding site sequences are in boldface. Solid boxes highlight NRF-1 sites that are highly conserved in all three or at least two species.
DISCUSSION
Using multiple approaches, including in silico analysis, electrophoretic mobility shift and supershift assays, chromatin immunoprecipitation assays, promoter mutational analysis, as well as RNA interference, the present study documents for the first time the significance of NRF-1 in regulating all ten nuclear-encoded COX subunit genes located on different chromosomes in murine neurons. Furthermore, the binding sites are conserved among mice, rats, and humans. In addition, NRF-1 is known to indirectly activate the three mitochondrial-encoded COX subunit genes by regulating TFAM, TFB1M, and TFB2M (9, 10). Thus, NRF-1, as well as our previously described NRF-2 (14, 15), are the only two transcription factors known thus far to be responsible for bigenomically coordinating the transcriptional activities of all 13 COX subunits in neurons. These results shed light on the mechanism of mitochondrial biogenesis, which requires precise bigenomic coordination (26).
NRF-1 was discovered during mutational and DNA binding analysis of the cytochrome c promoter, indicating that it is involved in the transcriptional regulation of cytochrome c, the substrate for cytochrome c oxidase (27). Additional target genes of NRF-1 include TFAM, TFB1M, TFB2M, SURF1, VDAC, and TOM20 genes (10, 23, 26, 28). Mitochondrial transcription factors A and B are required for the coupled transcription and replication of mitochondrial DNA, SURF1 is important for the assembly of COX subunits, while VDAC and TOM20 are involved in ion and protein transport in the mitochondria. The present study confirms that all of these genes are down-regulated when NRF-1 is silenced. Thus, NRF-1's control over these functions has the potential to link the expressions and functions of respiratory proteins encoded in both genomes (11, 28).
Thus far, only a limited number of COX subunit promoters have been analyzed (5b and 6a in humans, 5b, 6a, and 6c in rats, and 7a in cows) (11-14, 29-32). Human COX5b and TFAM genes have both NRF-1 and NRF-2 sites that are evolutionarily conserved in the proximal promoter regions (9, 29). COX 8−3 gene has been identified in human, lemur, rat, and mouse, and its promoter contains a consensus sequence for NRF-1 near the origin of transcription (34). Our present in silico analysis of all ten mouse COX genes revealed both typical and atypical NRF-1 binding sites. Promoter sequence alignment indicates high homology for NRF-1 binding sites in all three species examined (mouse, rat, and human), consistent with general species conservation reported previously (12, 14, 24).
Functions of the ten nuclear-encoded cytochrome c oxidase subunits are beginning to be explored. They are reportedly involved in the regulation of the catalytic activity, assembly, or stability of the enzyme (34-36). Three of the nuclear subunit proteins (VIa, VIIa, and VIII) have muscle and non-muscle-specific isoforms, whereas subunit IV has a lung-specific isoform, and VIb has a testes-specific isoform (34-43). Different isoforms presumably regulate COX activity in different tissues. COX IV binds ATP at the matrical side, leading to an allosteric inhibition of enzyme activity at high intramitochondrial ATP/ADP ratios (44, 45). Recent study suggests that suppression of COX IV expression leads to a loss of respiratory function and assembly of cytochrome c oxidase complex. It is thus important for COX activity in higher organisms and yeast (46, 47). On the other hand, subunit Va binds the thyroid hormone T2, releasing this allosteric ATP inhibition and allowing a high turnover at an elevated ATP/ADP ratio (47). Phosphorylation of subunit Vb, located at the matrical side of the enzyme complex, is accompanied by an increase in the allosteric ATP inhibition of the soluble enzyme (35). Subunit VIa isoforms have also been shown to respond to high ATP/ADP ratios. Binding of ATP to the heart and skeletal muscle isoform, VIaH, reduces the proton to electron stoichiometric ratio (48), and binding of palmitate to the liver isoform, VIaL, has a comparable effect (49). Subunit VIb, in addition to subunits III, Vb, and VIa, provides unique contact sites between the two monomers of COX. Subunit VIb has also been proposed to be involved in the cooperativity between the two substrate binding sites within the dimeric enzyme complex at high intramitochondrial ATP/ADP ratios, causing sigmoidal kinetics (50). The cytosolic domain of VIc transmembranous subunit participates with three of its acidic amino acids (Asp-49, Asp-55, and Asp-59) in a ring of negatively charged amino acids, which could represent a low affinity binding site for a second molecule of cytochrome c (47). Subunit VIIaH is expressed in bovine heart and skeletal muscles but not in smooth muscles. All non-skeletal and non-heart muscle tissues contain subunit VIIaL (51, 52). In mice, all three COX heart isoform genes (6ah, 7ah, and 8ah) are localized to chromosome 7 to a conserved region syntenic with that in humans (52). The transmembranous subunit VIII is located in close vicinity to subunit IV and has tissue-specific isoforms that are differentially expressed in different species. In cows, dogs, and rats, the heart isoform (VIIIH) differs from the liver isoform (VIIIL), whereas in human, sheep, and rabbit, one and the same isoform is expressed in all tissues examined (32, 34, 41). COX VIII-3 is mitochondrially targeted, and its sequence conservation implies a functional role in the COX holoenzyme with possible tissue specificity (34). Thus, control of energy metabolism in eukaryotes is based on variable efficiency of energy transduction in cytochrome c oxidase. It is also dependent on the turning on and off of respiratory control via the intramitochondrial ATP/ADP ratio by reversible and cAMP-dependent phosphorylation of the enzyme. This regulation is mediated by ten nuclear-encoded subunits of cytochrome c oxidase.
Our in vitro and in vivo studies showed that all ten nuclear-encoded subunit genes respond, by and large, in unison to increasing or decreasing functional demands, indicating that they are coordinately regulated (53). The coordinated regulation among the ten subunits may be multifactorial, requiring various processes. In addition to transcription factors, a transcriptional coactivator, peroxisome proliferator-activated receptor-γ coactivator 1α, has been shown to induce mitochondrial biogenesis by interacting with NRF-1, NRF-2, peroxisome proliferator-activated receptor α, and other nuclear factors (10, 11, 33, 54, 55). Peroxisome proliferator-activated receptor-γ coactivator 1α is markedly up-regulated in brown fat during adaptive thermogenesis and can induce mitochondrial biogenesis when expressed ectopically in cultured cells or in transgenic mice (26, 54).
The current study supports our earlier reports describing the importance of NRF transcription factors for activating COX gene promoters in neurons (14, 15, 30). Indeed, our results allow us to extend what is known about the participation of NRF transcription factors (NRF-1 and NRF-2) in the coordination of nuclear and mitochondrial genetic systems to the level of COX subunit genes. All ten nuclear-encoded COX subunits, whose genes are in different chromosomal loci, are transcribed and translated independently. NRFs and associated regulatory proteins may serve to integrate all ten nuclear subunits as well as the mitochondrial genetic systems to accommodate cellular demands for respiratory energy. The interplay of these nuclear factors is likely to be a major determinant in regulating the biogenesis of mitochondria.
In summary, the promoters of all ten COX ubiquitous nuclear genes have been found in this study to have functional binding sites for NRF-1, a feature not shared by a number of known respiratory-related factors, such as Sp-1 or YY1 (37). Thus, NRF-1, along with NRF-2, can serve as an effective master coordinator of transcriptional activities of all COX subunit genes in neurons. Our findings further support a bigenomic mechanism of energy regulation in neurons. Future studies will be directed at possible transcriptional interactions between NRF-1 and NRF-2. Co-recruitment of COX genes into a transcription factory is one such possible mechanism.
Acknowledgments
It gives us great pleasure to thank Dr. Richard Scarpulla for his generous gift of NRF-1 antibodies and for critically reviewing our report. We thank Drs. H. Liang and H. Meng for assisting in the construction of shRNA vectors.
Footnotes
This work was supported by Grant R01 EY05439 from the National Institutes of Health. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: COX, cytochrome c oxidase; ChIP, chromatin immunoprecipitation; EMSA, electrophoretic mobility shift assay; NRF-1, nuclear respiratory factor-1; NRF-2, nuclear respiratory factor-2; Sp-1, specificity protein 1; TFAM, transcription factor A of mitochondria; TFB1M, transcription factor B1 of mitochondria; TFB2M, transcription factor B2 of mitochondria; TSP, transcription start point; YY1, ying-yang protein 1; NGFR, nerve growth factor receptor; CMV, cytomegalovirus; shRNA, short hairpin RNA.
REFERENCES
- 1.Wong-Riley MTT. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- 2.Gunter TE, Gunter KK, Sheu SS, Gavin CE. Am. J. Physiol. 1994;267:C313–C339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- 3.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Hum. Mol. Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 4.Wong-Riley M, Antuono P, Ho KC, Egan R, Hevner R, Liebl W, Huang Z, Rachel R, Jones J. Vision Res. 1997;37:3593–3608. doi: 10.1016/S0042-6989(96)00210-6. [DOI] [PubMed] [Google Scholar]
- 5.Rahman S, Brown RM, Chong WK, Wilson CJ, Brown GK. Ann. Neurol. 2001;49:797–800. doi: 10.1002/ana.1060. [DOI] [PubMed] [Google Scholar]
- 6.Lerman-Sagie T, Leshinsky-Silver E, Watemberg N, Luckman Y, Lev D. Mol. Genet. Metab. 2005;2:127–136. doi: 10.1016/j.ymgme.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Kadenbach B, Jarausch J, Hartmann R, Merle P. Anal. Biochem. 1983;129:517–521. doi: 10.1016/0003-2697(83)90586-9. [DOI] [PubMed] [Google Scholar]
- 8.Scarpulla RC. J. Bioenerg. Biomembr. 1997;29:109–119. doi: 10.1023/a:1022681828846. [DOI] [PubMed] [Google Scholar]
- 9.Virbasius JV, Scarpulla RC. Proc. Natl. Acad. Sci. U. S. A. 1994;91:1309–1313. doi: 10.1073/pnas.91.4.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gleyzer N, Vercauteren K, Scarpulla RC. Mol. Cell Biol. 2005;25:1354–1366. doi: 10.1128/MCB.25.4.1354-1366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scarpulla RC. J. Cell Biochem. 2006;97:673–683. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- 12.Evans MJ, Scarpulla RC. Genes Dev. 1990;4:1023–1034. doi: 10.1101/gad.4.6.1023. [DOI] [PubMed] [Google Scholar]
- 13.Virbasius JV, Scarpulla RC. Mol. Cell Biol. 1991;11:5631–5638. doi: 10.1128/mcb.11.11.5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ongwijitwat S, Wong-Riley MTT. Gene (Amst.) 2005;360:65–77. doi: 10.1016/j.gene.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 15.Ongwijitwat S, Liang HL, Graboyes EM, Wong-Riley MTT. Gene (Amst.) 2006;374:39–49. doi: 10.1016/j.gene.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Nie F, Wong-Riley MTT. J. Comp. Neurol. 1999;404:310–320. doi: 10.1002/(sici)1096-9861(19990215)404:3<310::aid-cne3>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 17.Guo A, Nie F, Wong-Riley MTT. J. Comp. Neurol. 2000;417:221–232. doi: 10.1002/(sici)1096-9861(20000207)417:2<221::aid-cne7>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 18.Zhang C, Wong-Riley MTT. Eur. J. Neurosci. 2000;12:1013–1023. doi: 10.1046/j.1460-9568.2000.00997.x. [DOI] [PubMed] [Google Scholar]
- 19.Yang SJ, Liang HL, Ning G, Wong-Riley MTT. Eur. J. Neurosci. 2004;19:1153–1162. doi: 10.1111/j.1460-9568.2004.03250.x. [DOI] [PubMed] [Google Scholar]
- 20.Wong-Riley MTT, Yang SJ, Liang HL, Ning G, Jacobs P. Vis. Neurosci. 2005;22:1–18. doi: 10.1017/S0952523805221016. [DOI] [PubMed] [Google Scholar]
- 21.Liang HL, Wong-Riley MT. Neuroreport. 2006;17:401–405. doi: 10.1097/01.wnr.0000204980.98876.11. [DOI] [PubMed] [Google Scholar]
- 22.Yang SJ, Liang HL, Wong-Riley MTT. Neurosci. 2006;141:1181–1192. doi: 10.1016/j.neuroscience.2006.04.063. [DOI] [PubMed] [Google Scholar]
- 23.Scarpulla RC. Biochim. Biophys. Acta. 2002;1576:1–14. doi: 10.1016/s0167-4781(02)00343-3. [DOI] [PubMed] [Google Scholar]
- 24.Ongwijitwat S, Wong-Riley MTT. Gene (Amst.) 2004;337:163–171. doi: 10.1016/j.gene.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Kelly DP, Scarpulla RC. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 27.Evans MJ, Scarpulla RC. J. Biol. Chem. 1989;264:14361–14368. [PubMed] [Google Scholar]
- 28.Scarpulla RC. Gene (Amst.) 2002;286:81–89. doi: 10.1016/s0378-1119(01)00809-5. [DOI] [PubMed] [Google Scholar]
- 29.Bachman NJ, Yang TL, Dasen JS, Ernst RE, Lomax MI. Arch. Biochem. Biophy. 1996;333:152–162. doi: 10.1006/abbi.1996.0376. [DOI] [PubMed] [Google Scholar]
- 30.Wong-Riley MTT, Guo A, Bachman NJ, Lomax MI. Gene (Amst.) 2000;247:63–75. doi: 10.1016/s0378-1119(00)00121-9. [DOI] [PubMed] [Google Scholar]
- 31.Hüttemann M, Mühlenbein N, Schmidt TR, Grossman LI, Kadenbach B. Biochim. Biophys. Acta. 2000;1492:252–258. doi: 10.1016/s0167-4781(00)00087-7. [DOI] [PubMed] [Google Scholar]
- 32.Seelan RS, Grossman LI. J. Biol. Chem. 1997;272:10175–10181. doi: 10.1074/jbc.272.15.10175. [DOI] [PubMed] [Google Scholar]
- 33.Vercauteren K, Pasko RA, Gleyzer N, Marino VM, Scarpulla RC. Mol. Cell Biol. 2006;20:7409–7419. doi: 10.1128/MCB.00585-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hüttemann M, Schmidt TR, Grossman LI. Gene (Amst.) 2003;312:95–102. doi: 10.1016/s0378-1119(03)00604-8. [DOI] [PubMed] [Google Scholar]
- 35.Kadenbach B, Hüttemann M, Arnold S, Lee I, Bender E. Free Radic. Biol. Med. 2000;29:211–221. doi: 10.1016/s0891-5849(00)00305-1. [DOI] [PubMed] [Google Scholar]
- 36.Hüttemann M, Kadenbach B, Grossman LI. Gene (Amst.) 2001;267:111–123. doi: 10.1016/s0378-1119(01)00385-7. [DOI] [PubMed] [Google Scholar]
- 37.Grossman LI, Lomax MI. Biochim. Biophys. Acta. 1997;1352:174–192. doi: 10.1016/s0167-4781(97)00025-0. [DOI] [PubMed] [Google Scholar]
- 38.Fabrizi GM, Sadlock J, Hirano M, Mita S, Koga Y, Rizzuto R, Zeviani M, Schon EA. Gene. 1992;119:307–312. doi: 10.1016/0378-1119(92)90288-z. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt TR, Jaradat SA, Goodman M, Lomax MI, Grossman LI. Mol. Biol. Evol. 1997;14:595–601. doi: 10.1093/oxfordjournals.molbev.a025798. [DOI] [PubMed] [Google Scholar]
- 40.Arnaudo E, Hirano M, Seelan RS, Milatovich A, Hsieh CL, Fabrizi GM, Grossman LI, Francke U, Schon EA. Gene (Amst.) 1992;119:299–305. doi: 10.1016/0378-1119(92)90287-y. [DOI] [PubMed] [Google Scholar]
- 41.Hüttemann M, Jaradat S, Grossman LI. Mol. Reprod. Dev. 2003;66:8–16. doi: 10.1002/mrd.10327. [DOI] [PubMed] [Google Scholar]
- 42.Rizzuto R, Nakase H, Darras B, Francke U, Fabrizi GM, Mengel T, Walsh F, Kadenbach B, DiMauro S, Schon EA. J. Biol. Chem. 1989;264:10595–10600. [PubMed] [Google Scholar]
- 43.Napiwotzki J, Shinzawa-Itoh K, Yoshikawa S, Kadenbach B. Biol. Chem. 1997;378:1013–1021. doi: 10.1515/bchm.1997.378.9.1013. [DOI] [PubMed] [Google Scholar]
- 44.Arnold S, Kadenbach B. FEBS Lett. 1999;443:105–108. doi: 10.1016/s0014-5793(98)01694-9. [DOI] [PubMed] [Google Scholar]
- 45.Li Y, Park J, Deng J, Bai Y. J. Bioenerg. Biomembr. 2006;38:283–291. doi: 10.1007/s10863-006-9052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ludwig B, Bender E, Arnold S, Hüttemann M, Lee I, Kadenbach B. Chembiochem. 2001;2:392–403. doi: 10.1002/1439-7633(20010601)2:6<392::AID-CBIC392>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 47.Arnold S, Goglia F, Kadenbach B. Eur. J. Biochem. 1998;252:325–330. doi: 10.1046/j.1432-1327.1998.2520325.x. [DOI] [PubMed] [Google Scholar]
- 48.Frank V, Kadenbach B. FEBS Lett. 1996;382:121–124. doi: 10.1016/0014-5793(96)00096-8. [DOI] [PubMed] [Google Scholar]
- 49.Lee I, Kadenbach B. Eur. J. Biochem. 2001;268:6329–6334. doi: 10.1046/j.0014-2956.2001.02602.x. [DOI] [PubMed] [Google Scholar]
- 50.Arnold S, Kadenbach B. Eur. J. Biochem. 1997;249:350–354. doi: 10.1111/j.1432-1033.1997.t01-1-00350.x. [DOI] [PubMed] [Google Scholar]
- 51.Yu M, Jaradat SA, Grossman LI. Biochim. Biophys. Acta. 2002;1574:345–353. doi: 10.1016/s0167-4781(02)00228-2. [DOI] [PubMed] [Google Scholar]
- 52.Jaradat SA, Ko MS, Grossman LI. Genomics. 1998;49:363–370. doi: 10.1006/geno.1998.5279. [DOI] [PubMed] [Google Scholar]
- 53.Liang HL, Ongwijitwat S, Wong-Riley MTT. Neuroscience. 2006;140:177–190. doi: 10.1016/j.neuroscience.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 54.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 55.Vega RB, Huss JM, Kelly DP. Mol. Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]