

Summary
The TraI relaxase-helicase is the central catalytic component of the multi-protein relaxosome complex responsible for conjugative DNA transfer (CDT) between bacterial cells. CDT is a primary mechanism for the lateral propagation of microbial genetic material, including the spread of antibiotic resistance genes. The 2.4 Å resolution crystal structure of the C-terminal domain of the multifunctional Escherichia coli F plasmid TraI protein (TraI-CT) is presented, and specific structural regions essential for CDT are identified. The crystal structure reveals a novel fold composed of a 28-residue N-terminal α-Domain connected by a proline-rich loop to a compact α/β-Domain. Both the globular nature of the α/β-Domain and the presence and rigidity of the proline-rich loop are required for DNA transfer and single-stranded DNA binding. Taken together, these data establish the specific structural features of this non-catalytic domain that are essential to DNA conjugation.
Keywords: Conjugative DNA Transfer, TraI, F plasmid, DNA binding, novel fold
Introduction
Conjugative DNA Transfer (CDT) is the process by which a mobile genetic element is unidirectionally transferred from a donor to a recipient cell. While there is significant diversity among conjugative plasmids, they share common features related to the mechanism of transfer that allow for the movement of genetic information not only between members of a single species, but also from one species to another and between different kingdoms of life1-4. Conjugative plasmids are most prevalent in bacteria5, where they serve as a major conduit for the horizontal transfer of genetic material6. As such, CDT is the primary route by which antibiotic and virulence factors are propagated within and between bacterial populations7.
CDT requires the formation of a stable mating pair with a close cell-cell junction through which DNA is transferred from donor (plasmid-plus) to recipient (plasmid-minus) cells. In the well studied F (fertility) plasmid of Escherichia coli, two supramolecular complexes are essential for CDT: the 500 kDa relaxosome that assembles at the plasmid's origin of transfer (oriT), and a type-4 secretion system that forms the intercellular junction. The relaxosome is composed of three F plasmid-encoded DNA binding proteins, TraM, TraY and TraI, as well as the E. coli host-encoded heterodimeric integration host factor (IHF). These proteins bind DNA in a site- and sequence-specific manner along oriT, a stretch of approximately 150 bp of plasmid DNA8-11. The TraM and TraY binding sites are distal to the site of strand cleavage in the oriT (the nic site), while TraI binds directly at the oriT nic site. IHF binds between the TraM/TraY and TraI sites and introduces a significant bend in the DNA12,13 that may bring TraM and TraY proximal to TraI (Figure 1A). While the specific roles of the small DNA binding proteins TraM and TraY within the context of the relaxosome are not fully elucidated, it is known that both TraY and IHF must be present on the DNA before TraI is able to bind to the oriT nic site and initiate strand transfer14-16.
Figure 1.
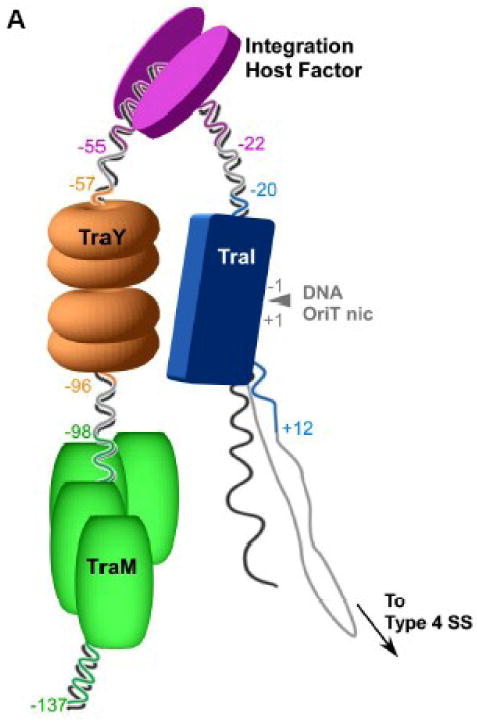
(A) Schematic of the relaxosome. The F plasmid-encoded proteins TraM (green), TraY (orange), TraI (blue), and the E. coli host encoded integration host factor (purple) bind the F plasmid DNA in a site- and sequence-specific manner. The nucleotides most proximal to the nic site (IHFA, SbyA, and SbmC) bound by each protein are indicated by color and numbered relative to the origin of transfer nic site (oriT nic). The region of ssDNA to be transferred to the recipient bacterial cell via a type IV secretion system (Type 4 SS) is shown. (B) Domain structure of TraI. The relaxase region is found between residues 1 and approximately 309, while the canonical helicase motifs (yellow) span a region between 990 and 1450.
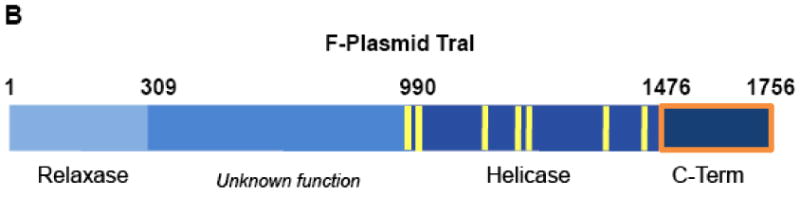
The central catalytic component of the relaxosome is the 1,756-residue multifunctional TraI protein (Figure 1B). TraI performs two enzymatic activities17. The N-terminal 300 residues encode the relaxase that introduces a site- and strand-specific ssDNA break at the oriT nic, forming a covalent phosphotyrosine linkage with the 5′-end of the nicked strand. When DNA transfer is complete, this transient bond is reversed, releasing a single stranded plasmid into the recipient cell2,14,18-21. The second catalytic region is the 5′-3′ helicase located centrally within the polypeptide (∼950 to 1500 based on conserved helicase sequence motifs) that unwinds the DNA strands of the F plasmid and provides the motive force to drive conjugative DNA transfer22,23.
The detailed roles that the residues C-terminal to the helicase domain of TraI (1476-1756; TraI-CT) play in DNA transfer are uncertain. Recent reports provide indirect evidence that TraI-CT interacts with the TraM protein24,25. TraM has been shown to be dispensable for many of steps of CDT but is required for full transfer efficiency24. It has also been hypothesized that TraI-CT couples the relaxosome to the type IV secretion system (T4SS) via the protein TraD26-28. Here, we report the X-ray crystal structure of residues 1476-1629 of the TraI C-terminal domain, which exhibits a novel protein fold. A combination of deletion and site-directed mutants provide detailed information about the TraI-CT features necessary for conjugative DNA transfer. In addition, it is demonstrated that the C-terminal domain of TraI is capable of binding to single-stranded DNA.
Results
The TraI C-Terminus Exhibits a Novel Fold
Crystals of the TraI C-terminus (TraI-CT) were grown by hanging drop vapor diffusion, and the structure was determined to 2.4 Å resolution using multiple-wavelength anomalous dispersion (MAD) phasing and selenomethionine-substituted protein (Table I). The experimental maps produced following phasing and solvent flattening (Figure 2A, B) allowed the building of a dimer of residues 1476-1629, approximately two-thirds, of the TraI-CT in the asymmetric unit. Residues in the far C-terminus (1630-1756) could not be placed in the model. The Matthew's parameter (VM) for two molecules of the 1476-1629 region in the asymmetric unit was 2.8; in contrast, two molecules of the complete TraI C-terminal domain (residues 1476-1756) would exhibit a VM of 1.5. Thus, it is unlikely that the far C-terminal region is present in the crystal. Indeed, SDS-PAGE of washed and dissolved crystal specimen supports this conclusion (data not shown). It is possible that a peptide cleavage event occurred between amino acids Asn-1631 and Ser-1632, a dipeptide known to be susceptible to succinimide-based degradation29,30.
Table I. Data Collection, Phasing, and Refinement Statistics.
| Data collection | |||
|---|---|---|---|
| X-ray source | APS SER-CAT BM-22 | ||
| Space Group | C2221 | ||
| Unit cell: a,b,c (Å); α, β, γ (°) | 40.8, 139.8, 126.5; 90, 90, 90 | ||
| Peak | Inflection | Remote | |
| Wavelength (Å) | 0.97835 | 0.97873 | 0.97126 |
| Resolution (Å) (highest shell) | 50.0-2.39 (2.49-2.39) | 50.0-2.40 (2.49-2.40) | 50.0-2.50 (2.59-2.50) |
| Rsym | 8.8 (27.6) | 9.6 (42.5) | 9.6 (41.9) |
| I/σ | 28.6 (5.4) | 22.9 (3.7) | 13.9 (2.3) |
| Completeness (%) | 97.5 (89.4) | 98.8 (92.9) | 96.6 (81.3) |
| Redundancy | 6.9 (6.10) | 3.7 (3.2) | 6.8 (6.0) |
| Phasing | |||
| Mean Figure of Merit | |||
| Sharp-Centric | 0.301 | ||
| Sharp-Acentric | 0.184 | ||
| Solomon & DM | 0.804 | ||
| Refinement | |||
| Resolution (Å) | 50.0-2.4 | ||
| No. reflections | 24509 | ||
| Rwork | 0.220 | ||
| Rfree | 0.263 | ||
| Molecules per asymmetric unit (AU) | 2 | ||
| No. of amino acids per AU | 307 | ||
| No. of waters per AU | 130 | ||
| No. of sulfates per AU | 10 | ||
| B-factors | |||
| Protein | 36.0 | ||
| Sulfates | 90.6 | ||
| Water | 38.1 | ||
| R.M.S. deviations | |||
| Bond lengths (Å) | 0.008 | ||
| Bond angles (°) | 1.4 | ||
| Ramachandran (%) | |||
| Favored | 96.4 | ||
| Outliers | 0 | ||
Rsym=Σ|I-Imean|/ΣI where I is the observed intensity and Imean is the average intensity of several symmetry related observations.
Rwork=Σ|Fo-Fc|/ΣFo where Fo and Fc are the observed and calculated structure factors, respectively.
Rfree=calculated as above for 5% of data not used in any step of refinement.
Figure 2.
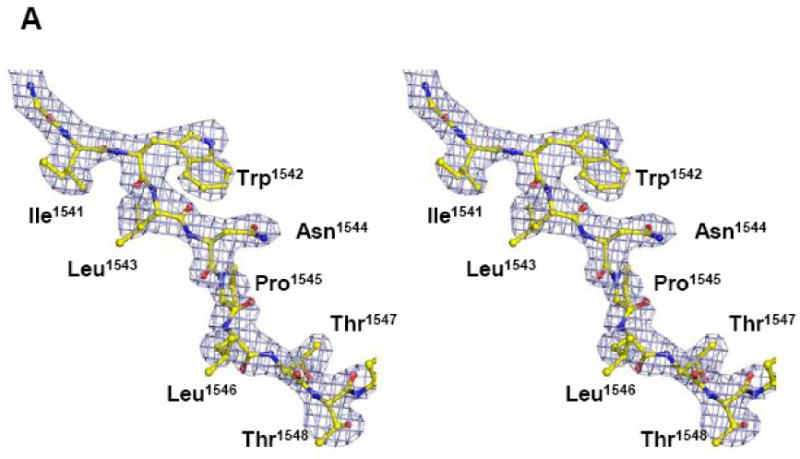
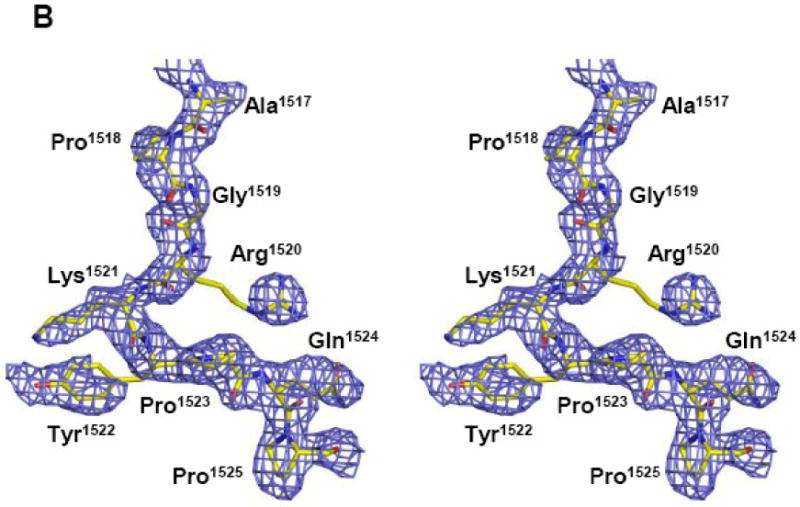

(A, B) Stereoview of two portions of the original 2.4 Å resolution solvent-flattened experimental electron density map after SHARP, SOLOMON and DM (contoured at 1.5 σ) with the final refined protein model. (C) Crystal structure of a monomer of the F plasmid TraI-CT. Secondary structure elements are indicated in green (helices), blue (β-strands), grey (loops) and red (proline-rich loop).
The structured 1476-1629 region of the TraI-CT is composed of an α-Domain containing two α-helices at the N-terminus, a proline-rich loop, and a compact α/β-Domain at the C-terminus (Figure 2C). The α/β-Domain contains two three-stranded β-sheets, a small β-hairpin loop and two helices. Antiparallel β-sheet 1 (β-strands 1-3) contains a β-hairpin loop inserted between β-strands 2 and 3. In β-sheet 2, β-strands 4 and 5 are antiparallel, while the insertion of α-helix 3 allows β-strand 6 to run parallel to β-strand 5.
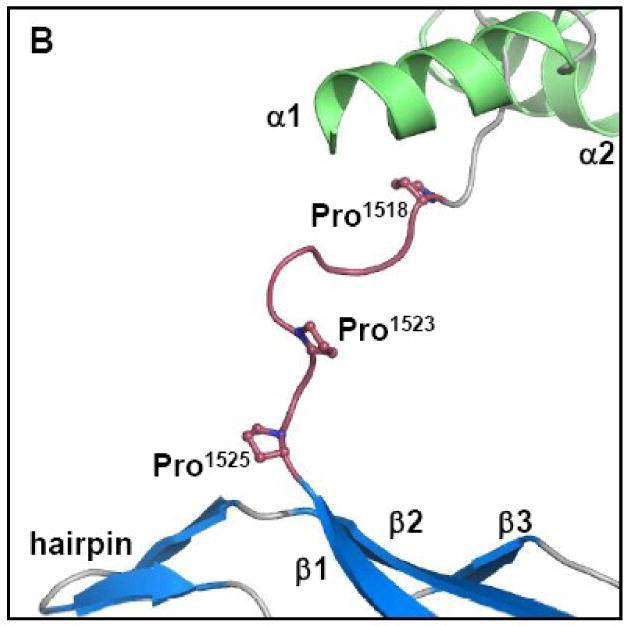
TraI-CT crystallized as a domain-swapped dimer in the asymmetric unit. The N-terminal α-Domain of one monomer docks into the core domain of the second monomer (Figure 3A). β-sheet 1 forms a binding surface for helix 1 of the second monomer in the asymmetric unit, while the second β-sheet (β-strands 4-6) packs against helix 3. The α– and α/β-Domains are connected by a proline-rich loop, which is rigid relative to the remainder of the protein. The mean thermal displacement parameter (B-factor) for all protein atoms is 36.0 Å2 (Table I), while prolines 1523, 1525 and 1530 in this loop exhibit B-factors of 26.1, 25.8 and 26.1 Å2, respectively. A crystallographic 2-fold axis of symmetry in the C2221 space group further generates an intimately associated tetramer formed by two domain-swapped dimers (Figure 3B).
Figure 3.
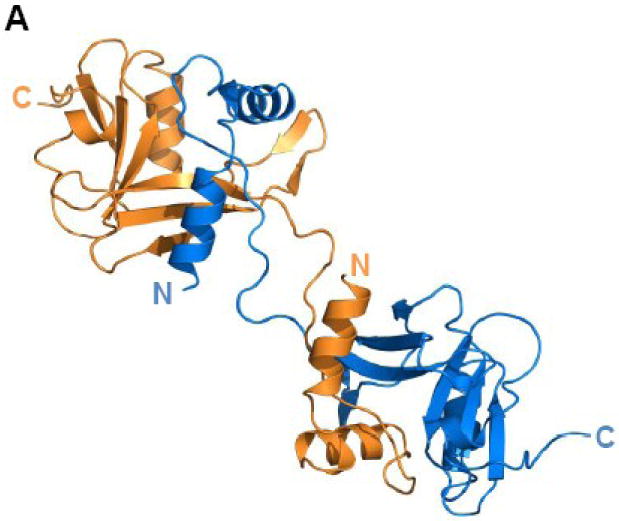
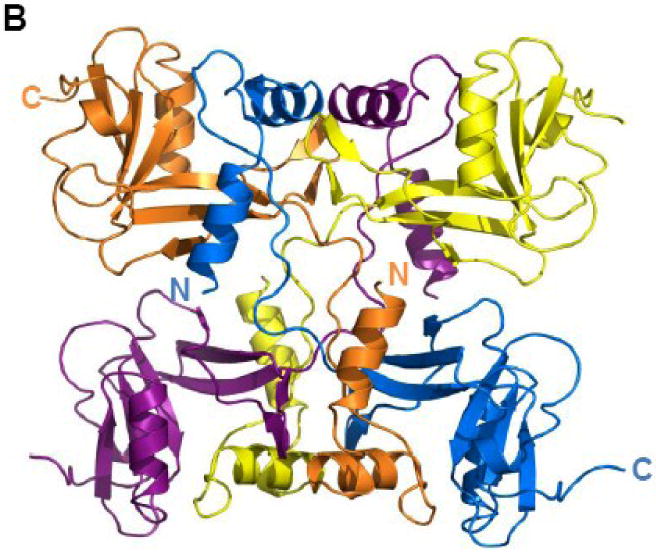
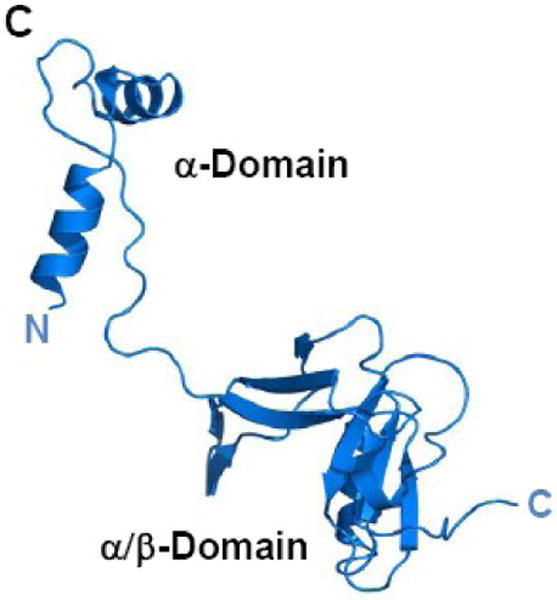
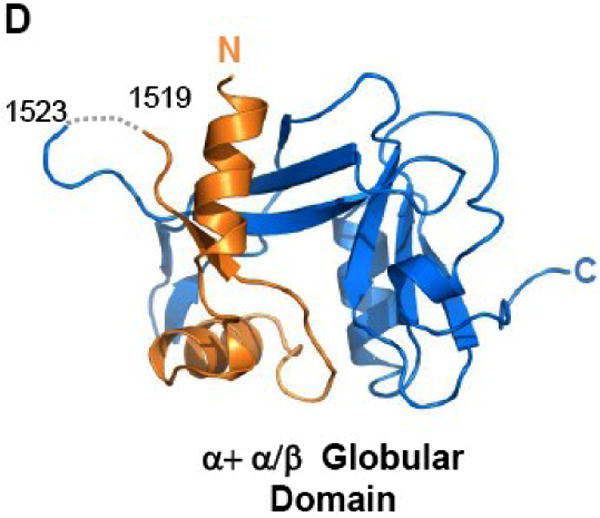
(A) TraI-CT forms a domain-swapped dimer in the asymmetric unit. Each monomer is oriented with the N-terminus (α-Domain) of one monomer contacting the C-terminus (α/β-Domain) of the other monomer. (B) An domain-swapped tetramer is also generated between symmetry-related dimers. Each monomer is oriented with the N-terminus towards the core of the tetramer and the C-terminus at the edge (orange interacts with blue, and purple with yellow). Two monomeric forms of the TraI C-terminus can be modeled: extended (C) and globular (D).
Two potential conformations of 1476-1629 region of the TraI-CT monomer can be hypothesized: an extended arrangement, in which one monomer is simply removed from the dimer (Figure 3C; see also Fig. 2B), and an α+α/β globular arrangement, in which a single domain is generated by pairing the α-Domain of one monomer with the α/β-Domain of its domain-swapping partner (Figure 3D). Both the extended and the globular conformations were examined in DALI searches of the Protein Data Bank31 for structurally-similar folds. Significant homology to TraI-CT was not detected in protein structure similarity searches. Using DALI, two hits were observed for the extended form of TraI-CT, and three for the globular form. For both the extended and the globular conformations, the best match (with a DALI Z-score of 2.5 and 3.1 respectively) was a portion of Pseudomonas putida 2-oxoisovalerate dehydrogenase (PDB ID 1qs0). While this molecule contains a similar degree of α and β character, we discerned no structural similarity with the TraI-CT. Indeed, up to eleven separate regions of structural similarity between the TraI-CT and the oxidoreductase were observed; the aligned segments were 3 to 13 residues in length with root-mean-square deviations of 2.8 to 3.9 Å between Cα positions. After the oxidoreductase, the next best DALI matches had Z-scores of 2.2 and 1.7. Using secondary structure matching server (SSM)32 and either the extended or globular forms of TraI-CT as a search model produced only three hits, all of which exhibited insignificant statistical values. These observations suggest that the 1476-1629 region of the F TraI C-terminal domain exhibits a novel fold.
Oligomerization State of the TraI-C Terminus
The TraI-CT was observed to form an intertwined tetramer composed of two domain-swapped dimers in the crystalline state (Figure 3A, B). To determine the oligomerization state of the TraI C-terminal domain in solution, constructs of the protein were examined by dynamic light scattering. Both the complete C-terminus (1476-1756) and a construct containing the ordered region of the crystal structure (1476-1630) eluted as single peaks on the Superdex 200 column. Coupling size exclusion chromatography with dynamic light scattering (SEC-DLS) and refractometry facilitated the calculation of sample molecular weights. Both C-terminal constructs (1476-1756, 1476-1630) exist as homogeneous monomers with less than 3.5% difference between calculated and theoretical molecular weights (Table II). It was noted that the theoretical molecular weight for the structured region of the TraI-CT (1476-1630), 16.7 kD, was smaller than the measured molecular weights from the SEC-DLS experiment (17.1–17.6 kD; Table II). This observation suggests that the TraI-CT monomer may be in equilibrium between the extended and globular states (see Figures 3C and 3D, respectively). Limited proteolysis using either trypsin or chymotrypsin generated a 3 kD product from the TraI-CT (data not shown). We hypothesize that this 3 kD fragment is the 28-residue α-domain, which is connected to the TraI-CT α/β-domain by the proline-rich loop composed of a sequence susceptible to cleavage by trypsin and chymotrypsin. This result further supports the conclusion that the TraI-CT monomer may shift between the closed and open states, the latter of which providing a substrate more amenable to proteolysis. The “far” C-terminal region (1630-1756), which was not present in our crystal structure, eluted in the void volume (MW > 600 kDa) but does not precipitate in the standard aqueous buffers employed (data not shown), suggesting a soluble aggregate.
Table II. Size Exclusion Chromatography and Dynamic Light Scattering*.
| Protein Construct | TraI 1476-1756 | TraI 1476-1630 |
|---|---|---|
| Theoretical MW (kDa) | 31.180 | 16.690 |
| Molar Mass Moments (kDa) | ||
| Mn (% error) | 32.31 (0.11%) | 17.08 (0.5%) |
| Mw (% error) | 32.32 (0.11%) | 17.35 (0.5%) |
| Mz (% error) | 32.33 (0.25%) | 17.61 (0.5%) |
| Polydispersity | ||
| Mw/Mn (% error) | 1.000 (0.2%) | 1.016 (0.7%) |
| Mz/Mn (% error) | 1.001 (0.3%) | 1.041 91.1%) |
Weight average molar mass defined as Mw=Σ(ci·Mi)/ Σci, Number average molar mass defined as Mn = Σci / Σ(ci/Mi), Z-average molar mass defined as Mz = Σ(ci·Mi)2/ Σ(ci·Mi).
Polydispersity of the sample equals one only when the sample has homogenous molecular mass (i.e. one oligomeric state and independent of averaging method).
Sequence Conservation in the Ordered TraI-CT Region
TraI orthologs in related Yersinia pestis, Klebsiella pneumoniae, Salmonella typhi, Aeromonas salmonicida, Enterobacter sp. 638, Shigella sonnei, and the E. coli R-100 conjugative plasmids were compared to the F plasmid TraI sequence using ClustalX33. A high degree of conservation (27-99% sequence identity) is maintained within the 1476-1629 region ordered in our crystal structure. In contrast, significant sequence divergence is observed in the far C-terminal region (1630-1756) (Figure 4). Only the E. coli R-100 resistance plasmid and a plasmid from Shigella sonnei maintain high sequence identity (96%) through the far C-terminus of their TraI proteins. In contrast, the other plasmids exhibit only 13-44% sequence identify within this region. Thus, residues 1476-1629 in the TraI-CT comprise a conserved core domain present in TraI orthologs in a range of F-like conjugative plasmids.
Figure 4.
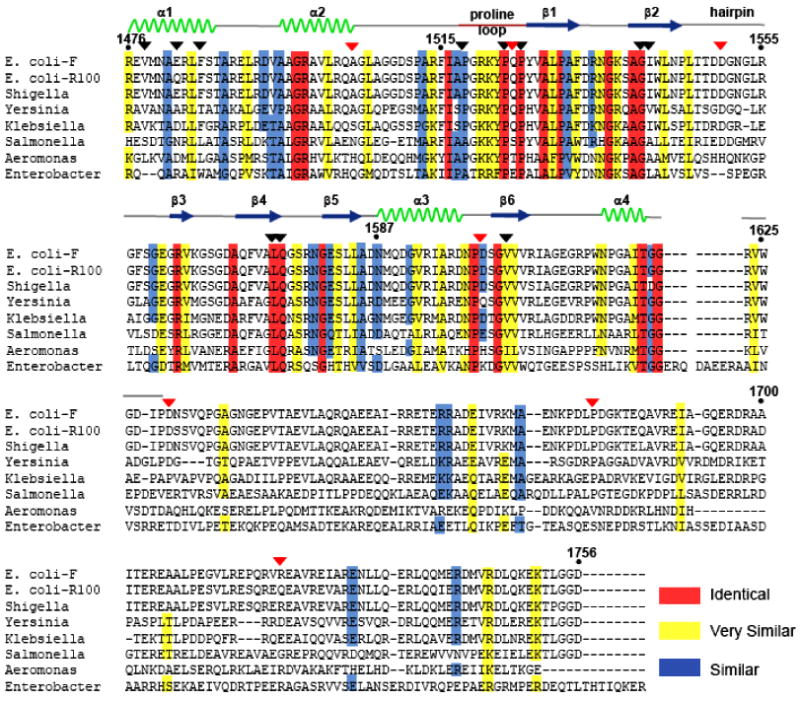
Sequence alignment showing similar (blue), highly similar (yellow) and identical (red) residues. The sequence of the Escherichia coli F plasmid TraI is compared to TraI orthologs from the E. coli plasmid R-100, as well as plasmids from Shigella sonnei (Ss046), Yersinia pestis (MT), Klebsiella pneumoniae (pMGH78587), Salmonella typhi (pED208), Aeromonas salmonicida A449 (p5), and Enterobacter sp.638. The positions of deletion-generating stop codons described in Figure 5 are indicated with red triangles, and the positions of mutations described in Figure 6 are indicated with black triangles.
Functional Analysis of Truncation Mutants
DNA transfer assays were conducted to test the importance of distinct regions of the F TraI C-terminus in conjugation. A panel of C-terminal truncation mutants was generated by introducing a stop codon at positions 1504, 1524, 1550, 1600, 1630, 1680, or 1720. DNA transfer efficiency (the number of transconjugation events per donor cell) was evaluated and normalized to wild-type levels (Figure 5). Previous work establishes that TraI is essential for CDT; when a plasmid with no TraI gene (ΔTraI) is utilized as a control, no conjugation is detected20,23. Deletion of residues 1504-1756 or 1524-1756 resulted in a complete loss of conjugative transfer (Figure 5A). Elimination of residues C-terminal to positions 1550, 1600 or 1720 resulted in a 100-fold decrease in the transfer efficiency, while removal of residues C-terminal to 1630 or 1680 resulted in a 1,000-fold reduction in transfer efficiency. Full activity is observed only with the complete protein; removal of just 36 residues reduces conjugative DNA transfer (CDT) by more than 100-fold (N1720 column in Figure 5A).
Figure 5.
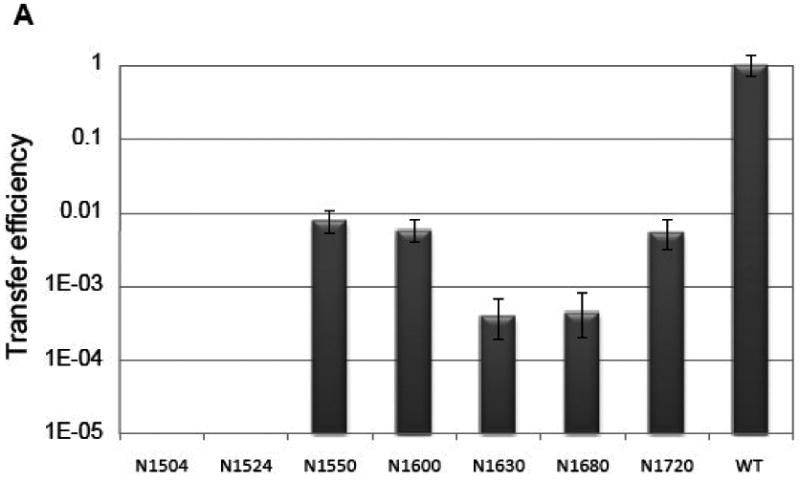
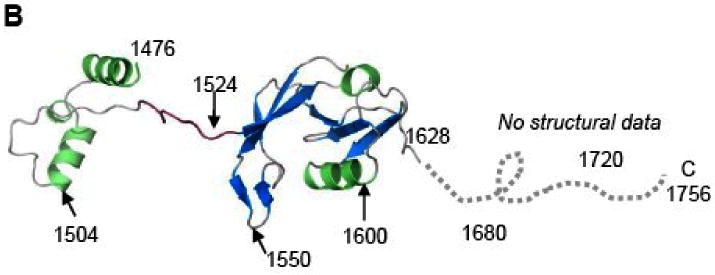
(A) Conjugative DNA transfer efficiencies of TraI deletion mutants. Constructs include residues N-terminal to the indicated residue; for example, N1550 includes residues 1-1550. (B) The TraI-CT structure annotated with the stop codon sites used for this truncation analysis.
Functional Analysis of Specific Structural Features
The importance of specific TraI-CT structural features was next examined using conjugative DNA transfer assays. First, three prolines (1518, 1523 and 1525) within the proline-rich loop were mutated simultaneously to glycine (Figure 6A, B). Prolines 1523 and 1525 are completely conserved, and proline 1518 is highly conserved in related TraI protein sequences (see Figure 4). Together, they may impart rigidity to the 1517-1525 loop (see above for B-factor analysis). Mutation of the three prolines dropped CDT 100,000-fold (Figure 6A). Second, residues 1517-1525, the entire proline-rich loop, were deleted. Elimination of the 1517-1525 loop reduced CDT 10,000-fold (Figure 6A). Thus, the conformationally-restricted proline residues within this loop are essential for CDT.
Figure 6.
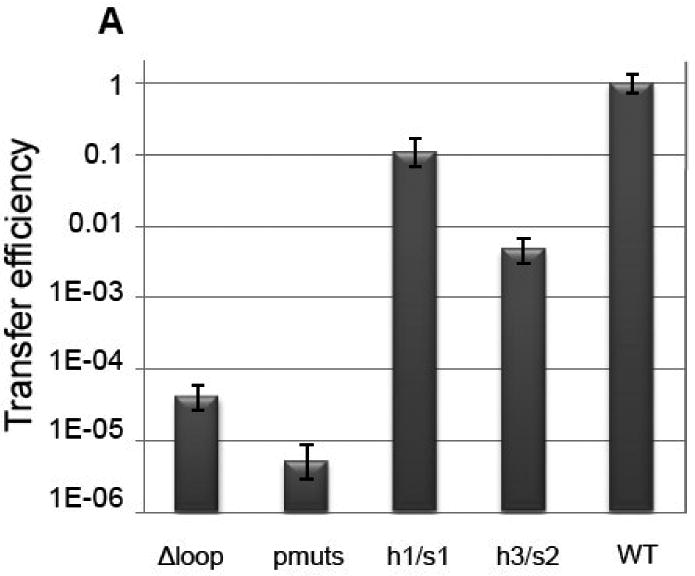
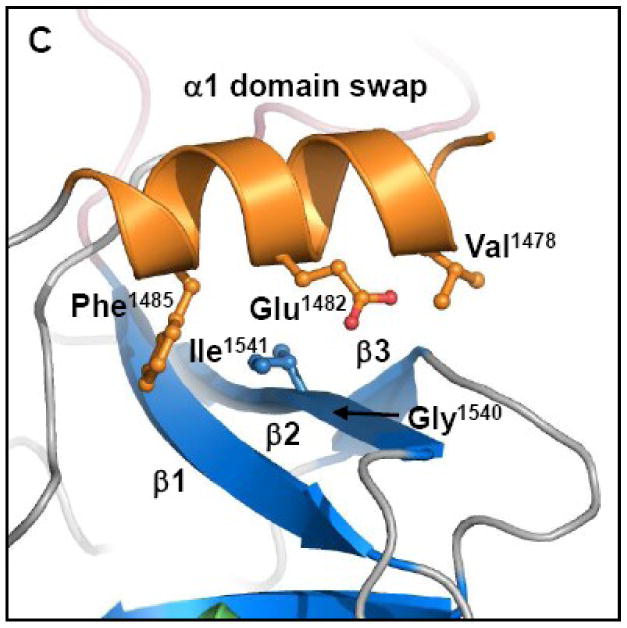
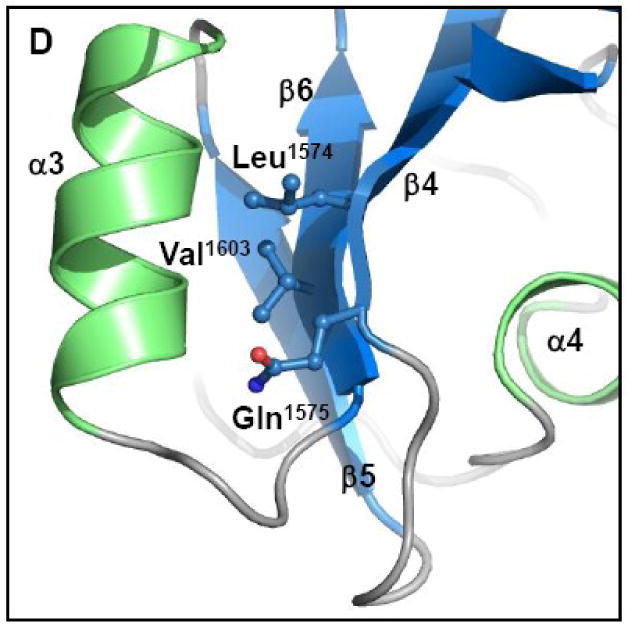
(A) Conjugative DNA transfer efficiencies of bacterial strains containing specifically designed TraI-CT mutations. (B) “Pmuts” indicates the mutation of prolines 1518, 1523 and 1525 simultaneously to glycine, while “Δloop” indicates the removal of the entire proline-rich loop. (C) The helix 1/sheet1 (h1/s1) variant is the mutation of V1478, E1482 and F1485 all to alanine on helix 1 and the mutation of I1541 to alanine and G1540 to glutamic acid on strand 2. (Helix 1 is shown here in orange to indicate the domain swapped interaction.) (D) The helix 3/sheet2 (h3/s2) mutants replace L1574, Q1575 and V1603 all with alanine.
Third, the importance of contacts between α1 and β-sheet 1 (h1/s1) was examined (Figure 6C; see also Figs. 2C and 3). Mutation of residues V1478, E1482 and F1485 to alanine on α1, coupled with mutation of G1540 to glutamic acid and I1541 to alanine on β2 (h1/s1), resulted in only a 10-fold decrease in transfer efficiency relative to wild type TraI (Figure 6A). These mutations were designed to disrupt the interaction between α1 and β-sheet 2. Although we did not establish that this interaction was successfully disrupted (by measuring changes in protein stability, for example), the relatively moderate 10-fold decrease in transfer efficiency lead us to conclude that the α1/β-sheet 1 interaction is not essential to CDT.
Fourth, contacts between α3 and β-strands 4 and 6 of β-sheet 2 (h3/s2) were examined. In contrast to the α1/β-sheet 1 interaction, the mutation of residues L1574, Q1575 and V1603 (Figure 6D) simultaneously to alanine resulted in a >200-fold decrease in transfer efficiency (Figure 6A). Mutation of L1574 and V1603 to alanine is expected to disrupt the hydrophobic interactions between helix 3 and sheet 2. The Q1575 side chain nitrogen on β-strand 4 forms polar contacts with two main-chain oxygen atoms in the loop that packs helix 3 against β-strand 6, and also forms a polar contact with a main-chain oxygen on helix 3. When alanine replaces Q1575, these polar contacts would be eliminated. Thus, the globular nature of the α/β-domain of the TraI C-terminus plays an important role in conjugative DNA transfer. Taken together, these data functionally annotate the crystal structure of the TraI C-terminal domain, and they establish that the α/β-domain and the rigid 1517-1525 loop are important for conjugative DNA transfer.
DNA Binding of the TraI C-Terminus
Because TraI contains domains that perform site-specific DNA nicking and highly processive DNA unwinding activities, the ability of constructs of the TraI C-terminal domain to bind to DNA was examined using fluorescence anisotropy. The full TraI C-terminal domain (1476-1756) did not bind to a double stranded stretch of DNA (data not shown), in accordance with previously published data25. The C-terminal domain binds to a 34 nucleotide single-stranded DNA (ssDNA) oligo with Kds of 2.9 μM and 7.7 μM in 75 mM and 150 mM NaCl, respectively (Figure 7, blue). A protein construct containing only the far C-terminal residues (1630-1756) formed a soluble aggregate in solution and was therefore not suitable for anisotropy studies. Similarly, the 1476-1629 fragment without the far C-terminal residues aggregated at 75 mM NaCl. At 150 mM NaCl, the 1476-1629 fragment exhibited poor binding to ssDNA (Kd>22.6 μM) (Figure 7, black). (A Kd value greater than 22.6 μM indicates a dissociation constant higher than the highest protein concentration tested.)
Figure 7.
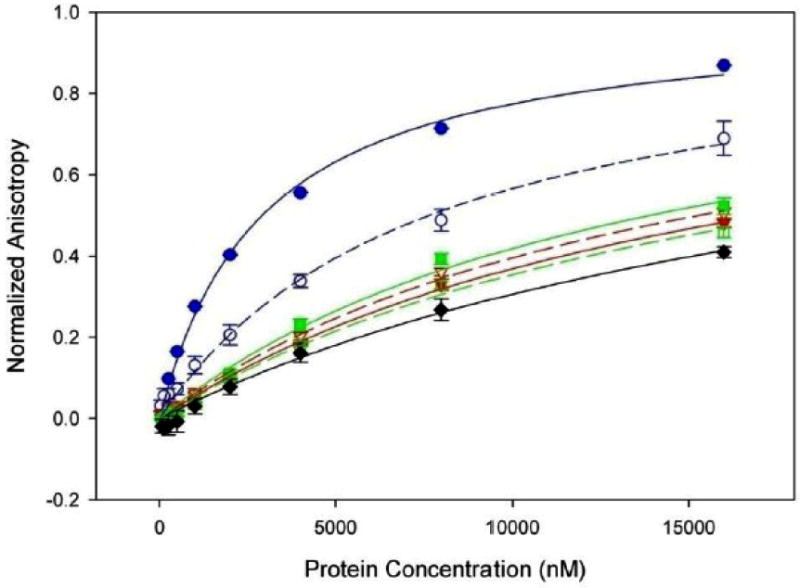
Binding of ssDNA by the TraI C-terminus measured by fluorescence anisotropy. TraI 1476-1756 at 75 and 150 mM NaCl is indicated by solid and dashed blue lines (Kd =2.9 μM and Kd =7.7 μM) respectively. TraI 1476-1756 with a deletion of the proline rich loop at 75 and 150 mM NaCl is indicated by solid and dashed red lines (Kd >17.1 μM and Kd >15.9 μM) respectively. TraI 1476-1756 with mutations of prolines 1518, 1523 and 1525 to glycine at 75 and 150 mM NaCl is indicated by solid and dashed green lines (Kd >13.9 μM and Kd >18.2 μM) respectively, while the binding of 1476-1630 at 150 mM NaCl is indicated in black (Kd >22.6μM).
The proline loop deletion (Δloop) and Pro-to-Gly mutant (pmut) forms of the TraI C-terminal domain (1476-1756) were also examined; recall that these variants significantly disrupted conjugative DNA transfer (see Fig. 6). The Δloop form of TraI 1476-1756 binds ssDNA poorly, with Kds of >17.1 μM and >15.9 μM at 75 mM and 150 mM NaCl, respectively (Figure 7, red). Similarly, mutating prolines 1518, 1523 and 1525 to glycine within this loop region also produced a form of TraI 1476-1756 that binds ssDNA poorly, with Kds of >13.9 μM and >18.2 μM at 75 mM and 150 mM NaCl, respectively (Figure 7, green). As a negative control, we found that bovine serum albumin did not bind to ssDNA at either 75 mM or 150 mM NaCl (data not shown). These results demonstrate that the TraI C-terminal domain binds ssDNA, and indicate that both the 1517-1525 loop and the 1630-1756 region are essential for this activity. Thus, the presence and the relative structural rigidity of the 1517-1525 loop are required for both ssDNA binding and conjugative DNA transfer (see Figs. 6, 7).
Discussion
The crystal structure of residues 1476-1629 of F plasmid TraI reveals a novel fold with a compact α/β-domain at the C-terminus connected via a proline-rich loop to two α-helices at the N-terminus. By sequence analysis, this fold appears to be conserved in related conjugative relaxase-helicase enzymes (Figure 4). While a domain-swapped dimer was observed in the asymmetric unit of the crystal structure (Figure 3A), biophysical studies confirmed that the purified TraI-CT exists as a monomer in solution. Based on the crystal structure, two conformational models of the protein monomer can be generated – an extended, two-domain form (Figure 3C) and a compact globular form (Figure 3D). The region swapped in our structure is the 28-residue α-domain, which is located at the N-terminus of the TraI-CT construct used for crystallization. Protein domain swaps frequently involve segments at the N- or C-terminus of a polypeptide34-36. Mutagenesis and deletion analyses indicate that interactions between the α-domain and the α/β-domain (Figure 6C) do not dramatically impact transfer; in contrast the proline-rich loop with intact proline residues is required for efficient conjugative DNA transfer mediated by TraI. Taken together with the SEC-DLS results, these data are consistent with the active conformation being an extended form (Figure 3C). It is known that full-length TraI is a monomer17,37,38, and there is no evidence of TraI oligomers playing a role in relaxosome function. In spite of this, the crystal packing arrangement observed may provide clues about protein-protein interactions formed by TraI. For example, the α-domain of the TraI-CT may contact other regions of TraI or other relaxosome proteins such as TraM. Ragonese et al. reported electrophoretic mobility gel shift assay (EMSA) data in which the formation of a supershifted TraM-TraI-DNA complex was observed with full length TraI, but not with TraI 1-150425. This is indirect evidence that elements within 1504-1756 of TraI mediate formation of the supershifted complex. Our attempts to extend these results, however, by fluorescence anisotropy using TraI-CT and TraM purified proteins, peptides and DNA in order to show a direct contact between these elements were unsuccessful.
Detailed deletion analysis of the TraI C-terminus indicates residues 1-1550 are required for any observable conjugative DNA transfer. Deletion of residues between 1550 and 1756 yield an intermediate level of transfer. Wild type transfer is observed only with the complete protein. This result corroborates published data by Ragonese and colleagues that the TraI-CT domain is essential for DNA conjugation25. Traxler et al. observed a < 10-7 transfer complementation ratio with deletion of TraI residues C-terminal to 162839. Their result showed a greater reduction of transfer than in this study; however, their truncation also eliminated TraX, a well conserved protein essential to pilin acetylation40,41. Both reduced and normal levels of conjugation have been observed for TraX deletion mutants42,43. Haft et al. described a series of 31 residue insertion mutants in F plasmid TraI; of relevance is the finding that a 31 amino acid insertion at position 1685 did not decrease transfer44. An analysis of the sequence of the far C-terminal domain (residues 1630-1756) by the program COILS45 predicts three coiled-coil (CC) domains. A gap between coil 1 and coil 2 from residue 1678 to 1687 may therefore permit insertion within that region without any affect on function. Coiled-coil domains are secondary structure elements (found in 2-3% of polypeptides) characterized by heptad repeats of hydrophobic and polar residues along α-helices, which facilitates coiled-coil formation46. Interestingly, despite the poor sequence conservation observed in the ClustalX alignment of the far C-termini, all TraI orthologs in Figure 4 display a high probability of at least one coiled-coil in this region. Coiled-coils have been reported previously to mediate protein-protein interactions particularly within multimeric protein complexes46-48. The predicted coiled-coil domain may indicate that the far C-terminal region is responsible for mediating the contact between TraI and other relaxosome proteins.
In summary, we have established that the TraI C-terminal domain contains structural elements critical for conjugative DNA transfer and ssDNA binding. Specifically, the proline-rich loop is required for proper conjugative function. These results advance our understanding of DNA conjugation, and facilitate the assembly of specific and testable models for TraI domain function in the relaxosome.
Materials and Methods
Expression and Purification
The gene encoding the TraI C-terminal domain (residues 1476-1756) was cloned into the vector pMCGS949 that fuses the expressed protein C-terminal to maltose binding protein (MBP) and a 6-His tag. A TEV cleavage site is located between the target protein and the tags. One liter flasks of LB broth were inoculated at a ratio of 1:100 from a saturated overnight culture of BL21 cells containing this protein-expression plasmid. Cells were grown at 37 °C under antibiotic selection with vigorous shaking until the cell density reached an OD600 of 0.6. IPTG was then added to a final concentration of 0.5 mM and the temperature was dropped to 16 °C for overnight expression. Cells were pelleted and resuspended in Nickel A buffer (20 mM Tris pH 7.4, 5% glycerol, 20 mM imidazole and 300 mM NaCl) at a ratio of 10 mL buffer/L of original culture. Lysis was carried out using a sonicator (Heat Systems) pulsed on ice for approximately three minutes. Following centrifugation at 27,000 g, the cleared lysate was loaded onto a gravity column packed with Nickel Sepharose 6 Fast Flow resin (Amersham) pre-equilibrated with Nickel A Buffer. Following washing to baseline, the His-tagged protein was eluted with a high imidazole Nickel B buffer (20 mM Tris pH 7.4, 5% glycerol, 500 mM imidazole and 300 mM NaCl). Fractions containing the TraI-CT MBP fusion protein (as evaluated by SDS-PAGE) were pooled and cleaved with 1% (w/w) TEV protease during dialysis into Nickel A buffer overnight at 4 °C. Following cleavage, a second run through the nickel column separated TraI-CT from MBP; TraI-CT flowed through the column while the His-tagged MBP remained bound to the column and was later eluted with Nickel B buffer. The purity of TraI-CT was assessed by SDS gel electrophoresis. Fractions containing clean TraI- CT were pooled for a final polishing step. The protein was loaded onto a 26/60 Superdex 75 size exclusion column in 20 mM HEPES pH 7.4, 150 mM NaCl on an Akta Express FPLC (GE Healthcare). TraI-CT peak fractions were concentrated and buffer exchanged into 150 mM ammonium acetate using an Amicon ultracentrifugation filter (Millipore). TraI-CT was concentrated to 25 mg/ml as determined by UV280 nm measurement.
TraI-CT containing selenomethionine was generated using B834 cells, a methionine auxotroph cell line. Cells were grown in selenomethionine specific media (Athena) supplemented with 50 mg/L selenomethionine. Expression and purification were preformed as described above. Additional proteins (TraM, TraI full length, TraI 1476-1630, and TraI 1630-1756) were expressed and purified using the same protocol.
Structure Determination
TraI-CT and selenomethionyl TraI-CT crystals were grown by hanging drop vapor diffusion at 25 °C. Equal volumes of TraI-CT protein solution and well solution (1 M ammonium sulfate, 100 mM MES pH 6.0) were mixed, and crystals appeared in 7-10 days. Data were collected from a single crystal cryoprotected in 1.8 M lithium sulfate and flash cooled in liquid nitrogen. A three wavelength MAD data set was collected at Sector 22-BM (SER-CAT) of the Advanced Photon Source, Argonne National Laboratory. Data were indexed and scaled using HKL200050 (Table I).
The structure was determined in space group C2221, which showed the best statistics during data reduction and scaling. Four of ten possible selenium atoms in the two molecules in the asymmetric unit (five methionines per monomer) were located by the program SHARP51 and used to build an initial model to 2.4 Å. Two methionine residues (1479, 1588) per monomer were located in the ordered 1476-1629 region of the structure, while the remaining three methionines (1672, 1739, and 1743) per monomer were located in the missing 1630-1756 region. The electron density map was improved by a combination of solvent flipping using the program SOLOMON52 and density modification using DM53, both operating under SHARP. Automatic model building using ARP/wARP placed approximately 100 alanine residues in each monomer. The remaining residues and side chains were placed manually using the program Coot54. Model refinement was conducted using the maximum likelihood method in REFMAC 5.255, CNS56, and the free R-factor (with 5% of the data set aside for free R). The final asymmetric unit contains two protein monomers forming a dimer, along with 130 water molecules and 10 sulfate ions. The C-terminal residues 1630-1756 of the protein construct were missing in each protein monomer and are not present in the final refined model.
Dynamic Light Scattering
To investigate the polydispersity and oligomerization state of TraI constructs, dynamic light scattering experiments were employed using a Wyatt DAWN EOS light scattering instrument interfaced to an Amersham Biosciences Akta FPLC with Superdex S200 size exclusion column, a Wyatt Optilab refractometer, and Wyatt dynamic light scattering module. Constructs of TraI residues 1476-1630 and 1476-1756 were expressed and purified using the protocol outlined above. The samples were dialyzed into dynamic light scattering (DLS) buffer (10 mM potassium phosphate pH 7.1 and 150 mM NaCl). Data were analyzed using the ASTRA software from Wyatt Technology. Molecular weights were calculated using the following equation:
where K* is an optical parameter, c is the sample concentration, M is molecular weight, R(Θ) is the excess intensity of scattered light at DAWN angle Θ, P(Θ) is a function describing the angular dependence on scattered light, and A2 is the second virial coefficient.
Conjugative Mating Assays
To probe the importance of TraI-CT in conjugative DNA transfer, a mating assay protocol was developed based on previous studies23 and established E. coli variants. Briefly, the donor strain (JS11) contains pOX38TΔTraI, a tetracycline-resistant mini-F-plasmid that has all the genes necessary for transfer except TraI. TraI is supplied on an additional, smaller plasmid pET11c-TraI, which is a more manageable cloning substrate. The mutations described in this work were introduced in pET11c-TraI using the Quikchange mutagenesis kit (Stratagene). The recipient strain (JS4) does not contain the F-plasmid (F-) and is streptomycin-resistant.
Donor and recipient strains from saturated, antibiotic selected overnight cultures were diluted 1:50 in LB and then grown at 37 °C in the absence of selection to an OD600≈0.6. Donors and recipients were then mixed at a ratio of 1:9 and incubated at 37 °C with shaking for 70 min. Mating pairs were disrupted by pipetting up and down, then solutions were diluted a hundred-fold in LB media. From this initial 10-2 dilution, serial 10-fold dilutions were made in a 96-well plate, resulting in a dilution range of 10-3 to 10-10. Appropriate antibiotics were present in the media to select for donors (tetracycline), recipients (streptomycin) and transconjugates (both). The Most Probable Number (MPN) method was then used to determine the number of bacteria in the initial culture57. Twelve independent matings were performed for each mutant each evaluation cycle, with at least 3 cycles averaged and reported in Figures 5 and 6. Transfer efficiency indicates the number of transconjugates relative to the number of available donors; E=T/D, and was 0.12 for wild-type TraI complementation. The transfer efficiency of the wild type TraI plasmid was then normalized to a value of 1 in figures 5 and 6. Previous work establishes that when a plasmid with no TraI gene (ΔTraI) is utilized as a control, no conjugation is detected20,23.
Fluorescence Anisotropy
To investigate DNA binding to TraI-CT, fluorescence anisotropy experiments were employed. Purified proteins were exchanged into the following buffer for anisotropy experiments: 10 mM Tris pH 8.0 and either 75 or 150 mM NaCl. DNA reagents containing a 5′ fluorescein label were commercially synthesized and HPLC purified (IDT DNA). We utilized two oligonucleotides in our studies. The first consisted of a 34-mer poly-N sequence with a 5′ label. The second was a 50 nucleotide section of duplex DNA containing the SbmC site previously reported9. The label was placed on the 5′ end of the coding (top) strand. (Fluorescent labeling of the rod-shaped DNA, instead of the protein, is recommended to generate the largest change in anisotropy58.) To generate the duplex, the complimentary SbmC oligonucleotides were mixed in an equimolar ratio, heated at 95 °C for 5 minutes, and slow cooled to room temperature. All DNA oligonucleotides were resuspended in the anisotropy buffer.
To perform the fluorescence polarization (FP) assay, protein and DNA reagents were mixed in 96 well plates (Costar) in anisotropy buffer. The labeled DNA probe was used at a concentration suitable for signal detection (50 nM) and held constant while protein concentration was varied in two-fold dilutions starting at 16 μM. The plate was read on a Pherastar instrument (BMG Labtech) utilizing the excitation and emission wavelengths suitable for fluorescein (485 nm/520 nm). At least four replicates of data were generated. These data were subjected to a baseline subtraction and were averaged, with error bars representing one standard deviation. Binding (Kd) values were calculated using the one site ligand binding equation in the Sigma Plot program. The data shown in Figure 7 were normalized by dividing the anisotropy values by the Bmax calculated in Sigma Plot. The addition of 0.02% Tween had no effect on observed DNA binding characteristics, indicating that non-specific binding was not being measured. In certain cases the Kd values are given as greater than a certain value because the highest concentration tested in the assay is lower than the predicted Kd value. It was not possible to increase the concentration of protein in the assay due to solubility constraints.
Acknowledgments
We thank C. Zhang for experimental assistance, and S. Matson, S. Lujan, L. Charlton, A. Tripathy and J. Schuermann for helpful advice. Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) beamline 22 at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. Supported by a Kirschstein NIH Postdoctoral Fellowship (L.M.G.).
Footnotes
Structural coordinates have been deposited with the PDB as 3FLD.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frost LS, Leplae R, Summers AO, Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol. 2005;3:722–732. doi: 10.1038/nrmicro1235. [DOI] [PubMed] [Google Scholar]
- 2.Lanka E, Wilkins BM. DNA processing reactions in bacterial conjugation. Annu Rev Biochem. 1995;64:141–169. doi: 10.1146/annurev.bi.64.070195.001041. [DOI] [PubMed] [Google Scholar]
- 3.Lessl M, Lanka E. Common mechanisms in bacterial conjugation and Ti-mediated T-DNA transfer to plant cells. Cell. 1994;77:321–324. doi: 10.1016/0092-8674(94)90146-5. [DOI] [PubMed] [Google Scholar]
- 4.Zupan JR, Zambryski P. Transfer of T-DNA from Agrobacterium to the plant cell. Plant Physiol. 1995;107:1041–1047. doi: 10.1104/pp.107.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazel D, Davies J. Antibiotic resistance in microbes. Cell Mol Life Sci. 1999;56:742–754. doi: 10.1007/s000180050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burrus V, Waldor MK. Shaping bacterial genomes with integrative and conjugative elements. Res Microbiol. 2004;155:376–386. doi: 10.1016/j.resmic.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 7.de la Cruz F, Davies J. Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol. 2000;8:128–133. doi: 10.1016/s0966-842x(00)01703-0. [DOI] [PubMed] [Google Scholar]
- 8.Di Laurenzio L, Frost LS, Paranchych W. The TraM protein of the conjugative plasmid F binds to the origin of transfer of the F and ColE1 plasmids. Mol Microbiol. 1992;6:2951–2959. doi: 10.1111/j.1365-2958.1992.tb01754.x. [DOI] [PubMed] [Google Scholar]
- 9.Fekete RA, Frost LS. Characterizing the DNA contacts and cooperative binding of F plasmid TraM to its cognate sites at oriT. J Biol Chem. 2002;277:16705–16711. doi: 10.1074/jbc.M111682200. [DOI] [PubMed] [Google Scholar]
- 10.Lahue EE, Matson SW. Purified Escherichia coli F-factor TraY protein binds oriT. J Bacteriol. 1990;172:1385–1391. doi: 10.1128/jb.172.3.1385-1391.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lum PL, Rodgers ME, Schildbach JF. TraY DNA recognition of its two F factor binding sites. J Mol Biol. 2002;321:563–578. doi: 10.1016/s0022-2836(02)00680-0. [DOI] [PubMed] [Google Scholar]
- 12.Rice PA, Yang S, Mizuuchi K, Nash HA. Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell. 1996;87:1295–1306. doi: 10.1016/s0092-8674(00)81824-3. [DOI] [PubMed] [Google Scholar]
- 13.Tsai MM, Fu YH, Deonier RC. Intrinsic bends and integration host factor binding at F plasmid oriT. J Bacteriol. 1990;172:4603–4609. doi: 10.1128/jb.172.8.4603-4609.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrd DR, Matson SW. Nicking by transesterification: the reaction catalysed by a relaxase. Mol Microbiol. 1997;25:1011–1022. doi: 10.1046/j.1365-2958.1997.5241885.x. [DOI] [PubMed] [Google Scholar]
- 15.Howard MT, Nelson WC, Matson SW. Stepwise assembly of a relaxosome at the F plasmid origin of transfer. J Biol Chem. 1995;270:28381–28386. [PubMed] [Google Scholar]
- 16.Nelson WC, Howard MT, Sherman JA, Matson SW. The traY gene product and integration host factor stimulate Escherichia coli DNA helicase I-catalyzed nicking at the F plasmid oriT. J Biol Chem. 1995;270:28374–28380. [PubMed] [Google Scholar]
- 17.Byrd DR, Sampson JK, Ragonese HM, Matson SW. Structure-function analysis of Escherichia coli DNA helicase I reveals non-overlapping transesterase and helicase domains. J Biol Chem. 2002;277:42645–42653. doi: 10.1074/jbc.M205984200. [DOI] [PubMed] [Google Scholar]
- 18.Datta S, Larkin C, Schildbach JF. Structural insights into single-stranded DNA binding and cleavage by F factor TraI. Structure (Camb) 2003;11:1369–1379. doi: 10.1016/j.str.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Larkin C, Datta S, Harley MJ, Anderson BJ, Ebie A, Hargreaves V, Schildbach JF. Inter- and intramolecular determinants of the specificity of single-stranded DNA binding and cleavage by the F factor relaxase. Structure. 2005;13:1533–1544. doi: 10.1016/j.str.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 20.Lujan SA, Guogas LM, Ragonese H, Matson SW, Redinbo MR. Disrupting antibiotic resistance propagation by inhibiting the conjugative DNA relaxase. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0702760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pansegrau W, Lanka E. Enzymology of DNA transfer by conjugative mechanisms. Prog Nucleic Acid Res Mol Biol. 1996;54:197–251. doi: 10.1016/s0079-6603(08)60364-5. [DOI] [PubMed] [Google Scholar]
- 22.Lahue EE, Matson SW. Escherichia coli DNA helicase I catalyzes a unidirectional and highly processive unwinding reaction. J Biol Chem. 1988;263:3208–3215. [PubMed] [Google Scholar]
- 23.Matson SW, Sampson JK, Byrd DR. F plasmid conjugative DNA transfer: the TraI helicase activity is essential for DNA strand transfer. J Biol Chem. 2001;276:2372–2379. doi: 10.1074/jbc.M008728200. [DOI] [PubMed] [Google Scholar]
- 24.Matson SW, Ragonese H. The F-plasmid TraI protein contains three functional domains required for conjugative DNA strand transfer. J Bacteriol. 2005;187:697–706. doi: 10.1128/JB.187.2.697-706.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ragonese H, Haisch D, Villareal E, Choi JH, Matson SW. The F plasmid-encoded TraM protein stimulates relaxosome-mediated cleavage at oriT through an interaction with TraI. Mol Microbiol. 2007;63:1173–1184. doi: 10.1111/j.1365-2958.2006.05576.x. [DOI] [PubMed] [Google Scholar]
- 26.Beranek A, Zettl M, Lorenzoni K, Schauer A, Manhart M, Koraimann G. Thirty-eight C-terminal amino acids of the coupling protein TraD of the F-like conjugative resistance plasmid R1 are required and sufficient to confer binding to the substrate selector protein TraM. J Bacteriol. 2004;186:6999–7006. doi: 10.1128/JB.186.20.6999-7006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Disque-Kochem C, Dreiseikelmann B. The cytoplasmic DNA-binding protein TraM binds to the inner membrane protein TraD in vitro. J Bacteriol. 1997;179:6133–6137. doi: 10.1128/jb.179.19.6133-6137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu J, Frost LS. Mutations in the C-terminal region of TraM provide evidence for in vivo TraM-TraD interactions during F-plasmid conjugation. J Bacteriol. 2005;187:4767–4773. doi: 10.1128/JB.187.14.4767-4773.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mills KV, Perler FB. The mechanism of intein-mediated protein splicing: variations on a theme. Protein Pept Lett. 2005;12:751–755. doi: 10.2174/0929866054864337. [DOI] [PubMed] [Google Scholar]
- 30.Stephenson RC, Clarke S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J Biol Chem. 1989;264:6164–6170. [PubMed] [Google Scholar]
- 31.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 32.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 33.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 1995;4:2455–2468. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Eisenberg D. 3D domain swapping: as domains continue to swap. Protein Sci. 2002;11:1285–1299. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Gotte G, Libonati M, Eisenberg D. Structures of the two 3D domain-swapped RNase A trimers. Protein Sci. 2002;11:371–380. doi: 10.1110/ps.36602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdel-Monem M, Hoffmann-Berling H. Enzymic unwinding of DNA. 1. Purification and characterization of a DNA-dependent ATPase from Escherichia coli. Eur J Biochem. 1976;65:431–440. doi: 10.1111/j.1432-1033.1976.tb10358.x. [DOI] [PubMed] [Google Scholar]
- 38.Abdel-Monem M, Taucher-Scholz G, Klinkert MQ. Identification of Escherichia coli DNA helicase I as the traI gene product of the F sex factor. Proc Natl Acad Sci U S A. 1983;80:4659–4663. doi: 10.1073/pnas.80.15.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Traxler BA, Minkley EG., Jr Evidence that DNA helicase I and oriT site-specific nicking are both functions of the F TraI protein. J Mol Biol. 1988;204:205–209. doi: 10.1016/0022-2836(88)90609-2. [DOI] [PubMed] [Google Scholar]
- 40.Cram DS, Loh SM, Cheah KC, Skurray RA. Sequence and conservation of genes at the distal end of the transfer region on plasmids F and R6-5. Gene. 1991;104:85–90. doi: 10.1016/0378-1119(91)90469-r. [DOI] [PubMed] [Google Scholar]
- 41.Moore D, Hamilton CM, Maneewannakul K, Mintz Y, Frost LS, Ippen-Ihler K. The Escherichia coli K-12 F plasmid gene traX is required for acetylation of F pilin. J Bacteriol. 1993;175:1375–1383. doi: 10.1128/jb.175.5.1375-1383.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cole SP, Guiney DG. Site-specific mutations in the traI relaxase and upstream region of plasmid RP4. Plasmid. 1995;34:236–239. doi: 10.1006/plas.1995.0011. [DOI] [PubMed] [Google Scholar]
- 43.Maneewannakul K, Maneewannakul S, Ippen-Ihler K. Characterization of traX, the F plasmid locus required for acetylation of F-pilin subunits. J Bacteriol. 1995;177:2957–2964. doi: 10.1128/jb.177.11.2957-2964.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haft RJ, Palacios G, Nguyen T, Mally M, Gachelet EG, Zechner EL, Traxler B. General mutagenesis of F plasmid TraI reveals its role in conjugative regulation. J Bacteriol. 2006;188:6346–6353. doi: 10.1128/JB.00462-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 46.Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 2001;11:82–88. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- 47.Ciferri C, De Luca J, Monzani S, Ferrari KJ, Ristic D, Wyman C, Stark H, Kilmartin J, Salmon ED, Musacchio A. Architecture of the human ndc80-hec1 complex, a critical constituent of the outer kinetochore. J Biol Chem. 2005;280:29088–29095. doi: 10.1074/jbc.M504070200. [DOI] [PubMed] [Google Scholar]
- 48.Cusack S, Berthet-Colominas C, Hartlein M, Nassar N, Leberman R. A second class of synthetase structure revealed by X-ray analysis of Escherichia coli seryl-tRNA synthetase at 2.5 A. Nature. 1990;347:249–255. doi: 10.1038/347249a0. [DOI] [PubMed] [Google Scholar]
- 49.Donnelly MI, Zhou M, Millard CS, Clancy S, Stols L, Eschenfeldt WH, Collart FR, Joachimiak A. An expression vector tailored for large-scale, high-throughput purification of recombinant proteins. Protein Expr Purif. 2006;47:446–454. doi: 10.1016/j.pep.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 51.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 52.Abrahams JP, Leslie AG. Methods used in the structure determination of bovine mitochondrial F-1 ATPase. Acta Cryst D. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 53.Cowtan K, Main P. Miscellaneous algorithms for density modification. Acta Crystallogr D Biol Crystallogr. 1998;54:487–493. doi: 10.1107/s0907444997011980. [DOI] [PubMed] [Google Scholar]
- 54.Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallographica Section D - Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 55.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 56.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 57.Garthright WE, B RJ. FDA's preferred MPN methods for Standard, large or unusual tests, with a spreadsheet. Food Microbiology. 2003:439–445. [Google Scholar]
- 58.Lundblad JR, Laurance M, Goodman RH. Fluorescence polarization analysis of protein-DNA and protein-protein interactions. Mol Endocrinol. 1996;10:607–612. doi: 10.1210/mend.10.6.8776720. [DOI] [PubMed] [Google Scholar]