

Abstract
Reactive oxygen species (ROS) and nitric oxide (NO) participate in NMDA receptor signaling. However, the source(s) of the ROS and their role in the increase in cerebral blood flow (CBF) induced by NMDA receptor activation have not been firmly established. NADPH oxidase generates ROS in neurons, but there is no direct evidence that this enzyme is present in neurons containing NMDA receptors, or that is involved in NMDA receptor-dependent ROS production and CBF increase. We addressed these questions using a combination of in vivo and in vitro approaches. We found that the CBF and ROS increases elicited by topical application of NMDA to the mouse neocortex were both dependent on neuronal NO synthase (nNOS), cGMP, and the cGMP effector kinase protein kinase G (PKG). In mice lacking the NADPH oxidase subunit NOX2, the ROS increase was not observed, but the CBF increase was still present. Electron microscopy of the neocortex revealed NOX2 immunolabeling in postsynaptic somata and dendrites that also expressed the NMDA receptor NR1 subunit and nNOS. In neuronal cultures, the NMDA-induced increase in ROS was mediated by NADPH oxidase through NO, cGMP and PKG. We conclude that NADPH oxidase in postsynaptic neurons generates ROS during NMDA receptor activation. However, NMDA receptor-derived ROS do not contribute to the CBF increase. The findings establish a NOX2-containing NADPH oxidase as a major source of ROS produced by NMDA receptor activation, and identify NO as the critical link between NMDA receptor activity and NOX2-dependent ROS production.
Keywords: cerebral blood flow, neurovascular coupling, NADPH oxidase, NO electrode, dihydroethidium, iNOS
Introduction
Nitric oxide (NO) and reactive oxygen species (ROS) are key mediators of NMDA receptor signaling (Kishida and Klann, 2007; Garthwaite, 2008). NO is produced by neuronal NO synthase (nNOS), an enzyme tethered to the NMDA receptor complex by the postsynaptic density protein-95 (PSD95) (Christopherson et al., 1999; Garthwaite, 2008). NO is involved in critical NMDA receptor functions, including neuroplasticity, and contributes to the increase in cerebral blood flow (CBF) evoked by neural activity (Iadecola and Nedergaard, 2007; Garthwaite, 2008). However, the ROS produced by NMDA receptor activation have also been implicated in NMDA receptor function (Kishida and Klann, 2007; Pacher et al., 2007), and, because of their vasoactivity (Faraci, 2006), could participate in the increase in CBF. The latter possibility is suggested by the finding that NMDA increases ROS production in cerebral arterioles (Girouard et al., 2006). However, CBF and ROS have not been measured simultaneously to establish whether ROS contribute to the hemodynamic response induced by relatively low NMDA concentrations. Furthermore, the sources of ROS during NMDA receptor activation have not been identified. Several sources have been proposed to mediate the ROS increase (Dugan et al., 1995a; Patel et al., 1996; Atlante et al., 1997), but direct ultrastructural and functional evidence linking NMDA receptors to a specific ROS generating system is lacking (Kishida and Klann, 2007).
NADPH oxidase, a multiunit enzyme comprised of membrane bound and cytoplasmic components. has emerged as an important source of ROS in neurons (Infanger et al., 2006; Bedard and Krause, 2007). The membrane associated components include p22phox and the catalytic subunit of the enzyme NOX, of which 5 homologues have been identified (NOX1–NOX5) (Bedard and Krause, 2007). The cytoplasmic subunit include p40phox, p67phox, and p47phox (Bedard and Krause, 2007). On activation, p47phox is phosphorylated leading to the assembly of the enzyme and superoxide production, a process that also requires the small GTPase Rac1 (Hordijk, 2006). ROS derived from a NOX2-containing NADPH oxidase have been implicated in hippocampal synaptic plasticity and in learning and memory (Kishida and Klann, 2007). However, it remains unclear whether NMDA receptors are present in neurons that also contain NADPH oxidase and whether NMDA receptor activation leads to NADPH oxidase-dependent ROS production.
We sought to determine whether NADPH oxidase is the source of the ROS produced by NMDA receptor activation, and whether ROS contribute to the NMDA-induced increase in CBF. We found that the source of the ROS is a NOX2-containing NADPH oxidase, which is present postsynaptically in neurons of the somatosensory cortex also expressing the NMDA receptor subunit NR1 and nNOS. nNOS-derived NO is required for both the increase in ROS and CBF evoked by NMDA receptor activation. However, ROS do not contribute to the increase in CBF, which can be entirely attributed to NO via cGMP and protein kinase G (PKG). The findings establish postsynaptic NADPH oxidase as a source of ROS during NMDA receptor activation, and identify NO as the critical link between NMDA receptors and ROS production.
Materials and Methods
Mice.
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Experiments were performed in 2- to 3-month-old male nNOS−/− or NOX2−/− mice on a stable C57BL/6 genetic background (Girouard et al., 2007). C57BL/6 mice were used as wild-type controls.
Monitoring of CBF.
As previously described (Girouard et al., 2007; Park et al., 2008), mice were anesthetized with isoflurane and the femoral artery was cannulated for recording of arterial pressure and blood sampling. Mice were artificially ventilated with an oxygen-nitrogen mixture adjusted to provide an arterial pO2 (paO2) of 120–140 mmHg (supplemental Table 1, available at www.jneurosci.org as supplemental material). Rectal temperature was maintained at 37°C using a thermostatically controlled heating device. After surgery, anesthesia was maintained with urethane (750 mg/kg; i.p.; Sigma-Aldrich) and α-chloralose (50 mg/kg; i.p.; Sigma-Aldrich). Corneal reflexes and motor responses to tail pinch were used to test the level of anesthesia. The somatosensory cortex was exposed through a small craniotomy (2 × 2 mm). The dura was removed, and the site was superfused with a modified Ringer's solution (37°C; pH: 7.3–7.4) [see Iadecola (1992) for composition]. CBF was monitored at the site of superfusion with a laser-Doppler probe (Vasamedic) positioned stereotaxically. CBF changes were expressed as percentage increase relative to the resting level (Niwa et al., 2001).
ROS detection in vivo.
ROS production in brain was determined using dihydroethidium (DHE) microfluorography (Bindokas et al., 1996; Murakami et al., 1998; Girouard et al., 2007). DHE is a cell permeable dye that is oxidized to ethidium and related products by superoxide (Robinson et al., 2006). Ethidium is trapped intracellularly by intercalating with DNA (Dikalov et al., 2007). DHE (Invitrogen; 2 μmol/L) was superfused on the somatosensory cortex for 60 min, as previously described (Girouard et al., 2006, 2007). The brains were removed from the skull, frozen, and then sectioned at a thickness of 20 μm on a cryostat. The sections were collected on slides at 100 μm intervals throughout the region exposed by the cranial window (Girouard et al., 2006, 2007). The mounted sections were examined using a fluorescence microscope (Nikon) equipped with a custom filter set for detection of DHE oxidation products (Girouard et al., 2006, 2007). Images were acquired with a digital camera (Coolsnap; Roper Scientific). Fluorescence intensity was assessed in the brain area of interest. The analysis of ROS production in the different conditions studied was performed in a blinded manner using the IPLab software (Scanalytics). Fluorescence intensities of all sections (30 per animal) were averaged and expressed as relative fluorescence units (RFU) (Girouard et al., 2006, 2007).
Experimental protocol for CBF and ROS measurement in vivo.
Reagents were dissolved in Ringer's solution unless otherwise indicated. The cranial window was first superfused with Ringer's solution. After CBF was in a steady state, the superfusion solution was switched to DHE. Thirty min later, NMDA (40 μm; Sigma) or kainate (10 μm; Sigma) was superfused, and the evoked changes in CBF were recorded after a stable increase in CBF was reached, usually after 10 min. The concentration of NMDA used does not induce spreading depression, as verified by simultaneous recordings of CBF and electrical activity (Zhang et al., 1995; Ayata and Moskowitz, 2006), and does not damage the brain, as determined histologically (Girouard et al., 2006). The experiment was terminated 30 min after the start NMDA superfusion and ROS production was assessed as described above.
In separate mice, we tested the effect on CBF and ROS of the NMDA receptor inhibitor MK-801 (200 μm; Sigma), the free radical scavenger Mn(III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP; 100 μmol) (Niwa et al., 2000), the soluble guanylyl cyclase (sGC) inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ 100 μm; Cayman Chemicals) (Sobey and Faraci, 1997), the cGMP analog 8-(4-chlorophenylthio)-guanosine 3–5-cyclic monophosphate sodium salt (pCPT-cGMP; 10, 100 μm; Sigma) or the PKG inhibitor KT-5823 (5 μm; Sigma) (Xu et al., 2004). The increase in CBF evoked by NMDA was first examined during superfusion with Ringer's solution. Then, the superfusion was switched to Ringer containing DHE plus MK-801, MnTBAP, ODQ, pCPT-cGMP or KT-5823, and the CBF response to NMDA was tested again 30 min later. The stability of the CBF response to NMDA was tested in time control experiments (data not shown). Mice were killed 30 min after NMDA for ROS determination. Mice superfused only with DHE served as vehicle-treated controls.
In some mice, after testing the baseline CBF response to NMDA, the nNOS inhibitor 7-nitroindazole (7-NI; 50 mg/kg, in peanut oil; i.p.; Cayman Chemicals) (Girouard et al., 2007) was administered and the DHE superfusion was started. The CBF response to NMDA was tested 30 min after 7-NI administration, and mice were killed after 60 min of DHE superfusion for ROS determination. We also used a peptide that disrupts the association between the NMDA receptor complex and nNOS through the scaffolding protein PSD95 (Sattler et al., 1999; Park et al., 2008). The peptide comprises the nine COOH-terminal residues of the NMDA receptor subunit NR2B and the HIV tat sequence to facilitate cell penetration (tat-NR2B9c) (Lys-Leu-Ser-Ser-Ile-Glu-Ser-Asp-Val; NR2B9c; Bio·Synthesis) (Sattler et al., 1999; Park et al., 2008). A peptide in which the active sequence was scrambled (s-tat-NR2B9c) served as control (Sattler et al., 1999; Park et al., 2008). The effect of NMDA on CBF or ROS production was tested before and after superfusion with tat-NR2B9c (20 μm) or s-tat-NR2B9c (20 μm) for 30 min. In other studies, we examined the effect of tat-NR2B9c, s-tat-NR2B9c or KT-5823 on CBF responses evoked by neural activity (functional hyperemia) or endothelium-dependent and -independent vasodilators. To study functional hyperemia, the somatosensory cortex was activated by gently stimulating the contralateral whiskers at 3–5 Hz for 60 s and the evoked increase in CBF was recorded. Endothelium-dependent vasodilation was tested by examining the CBF increase evoked by topical superfusion of acetylcholine (ACh; 10 μm; Sigma-Aldrich) or the Ca2+ ionophore A23187 (3 μm) for 3–5 min (Girouard et al., 2006). Endothelium-independent vasodilation was examined by testing the CBF response to adenosine (400 μm; Sigma-Aldrich), an agent that produces vasodilation by acting on vascular smooth muscles (Phillis, 2004). CBF responses to whisker stimulation, ACh, A23187 and adenosine were examined before and 30 min after topical application of the tat-NR2B9c peptide, s-tat-NR2B9c, and KT-5823.
ROS detection in neuronal cultures.
Mixed neocortical cultures [days in vitro (DIV) 13–15) from 16- to 17-d-old mouse embryos were seeded on poly-ornithine-coated glass coverslips were mounted in a perfusion chamber on an inverted microscope (Nikon E300) stage. Cells were loaded with DHE (2 μm) in HEPES buffer composed of (in mm): NaCl 121, KCl 5, CaCl2 1.8, glycine 0.01, Na-pyruvate 1, glucose 20, HEPES 17, KHCO3 3, pH = 7.35. As described previously (Kawano et al., 2006; Park et al., 2008), the composition of the cultures assessed immunocytochemically using specific cell markers at DIV 14 was: neurons 80–85% (microtubule associated protein-2), astrocytes 15–20% (glial fibrillary acidic protein), and microglia <1% (CD11). Cells were then pretreated with vehicle (buffer), MK-801 (5 μm), MnTBAP (100 μm), l-NNA (NG-nitro-l-arginine) (100 μm), the NADPH oxidase peptide inhibitor gp91ds-tat (1 μm), its scrambled control (s-gp91-ds-tat), ODQ (100 μm), or KT-5823 (5 μm) for 10 min and then challenged with NMDA (40 μm). We chose l-NNA as a NOS inhibitor because this inhibitor or its derivatives have been widely used in neuronal cultures (Strijbos et al., 1996; Adamson et al., 1999; Sattler et al., 1999). ROS dependent fluorescence was recorded at room temperature every 30 s using the IPLab software and a CCD digital camera until a stable change was achieved, usually 10–15 min. The relative intensity of ROS imaging was expressed as the ratio Ft/Fo, where Ft is fluorescence in a given cell after application of NMDA, and Fo is the baseline fluorescence of the same cell immediately before NMDA (Wang et al., 2006, 2008).
NO detection in neuronal cultures.
Amperometric detection of NO released from neuronal cultures was performed using an Apollo 4000 Analyzer equipped with a NO-sensitive electrode (tip diameter: 0.1 μm; ISO-NOPNM; World Precision Instruments), as described previously (Park et al., 2008). After calibration of the electrode with the NO donor SNAP, NO release triggered by NMDA in cultured cortical neurons was measured. After stabilization of the baseline signal, NMDA (40 μm) was applied to the extracellular buffer and NO release was monitored until a stable increase was achieved, usually 10–15 min. After washing NMDA, the NO signal returned to baseline, and neurons were treated with MK-801, l-NNA, gp91ds-tat, s-gp91-ds-tat, ODQ, or KT-5823 and the effects of NMDA application were re-tested 10 min later. Data were digitally acquired and stored for off-line analysis.
NADPH oxidase activity.
NADPH-dependent superoxide production was measured in cell-membrane fractions by an enhanced luminol-based assay as described previously (Anrather et al., 2006). Murine RAW264.7 cells (American Type Culture Collection) were washed twice in Hanks' balanced salt solution and collected by centrifugation. After resuspension in lysis buffer (10 mm Tris-HCl, pH 7.1, 300 mm sucrose, 1 mm MgCl2, 1 mm EGTA, 1 mm phenylmethylsulfonyl fluoride, 5 m leupeptin, 1 m pepstatin A, and 10 g/ml aprotinin), cells were disrupted on ice by sonication and centrifuged (10,000 × g for 10 min at 4°C). The supernatant was centrifuged and the pellet, which constitutes the membrane fraction, was dissolved in assay buffer (100 mm potassium phosphate, pH 7.0, 1 mm MgCl2, 1 mm EGTA, 1 mm sodium azide, 10 μm FAD). 50 μg of protein were incubated with 50 μl of Diogenes reagent (National Diagnostics). Reactions were started by automated injection of ß-NADPH (0.2 mm) and light emission was integrated over the first 30 s of the reaction. Parallel reactions included 100 U of superoxide dismutase and results are reported as superoxide dismutase inhibitable light emission (RLU). Luminol is sensitive to both superoxide and peroxynitrite (Radi et al., 1993). Therefore, increases in peroxynitrite produced by the reaction of NO with superoxide could also be detected. The specificity of the assay in detecting NADPH derived ROS was tested as previously described (Anrather et al., 2006).
Electron microscopy.
C57BL/6 mice (n = 4) were anesthetized (150 mg/kg sodium pentobarbital; i.p.) and their brain tissue fixed by vascular perfusion with 3.75% acrolein and 2% paraformaldehyde in phosphate buffer (PB) (Milner and Drake, 2001). The acrolein-fixed brains were removed, and sectioned coronally (40 μm thickness) through the region of the somatosensory cortex (0.4–0.5 mm rostral to bregma)(Kazama et al., 2004) using a Vibratome. Sections were collected in 0.1 m phosphate buffer, and processed for the dual electron microscopic immunogold and immunoperoxidase labeling of NOX2 and nNOS, or NOX2 and the NMDA receptor subunit NR1, as described previously (Milner and Drake, 2001). Briefly, sections were incubated 24 h at 4°C in a solution containing goat anti-NOX2 antibody (1:200 dilution; Santa Cruz Biotechnology; for specificity and characterization see (Wang et al., 2004) and rabbit nNOS (1:100 dilution) in TS containing 0.1% BSA (Wang et al., 2005). For NOX2 immunoreactivity, sections were incubated sequentially in: (1) anti-goat IgG conjugated to biotin (Vector) diluted 1/400 in TS containing 0.1% BSA, (2) peroxidase-avidin complex (Vectastain Elite kit) in TS, and (3) 3,3′-diaminobenzidine (Aldrich Chemical) and hydrogen peroxide in TS. For nNOS immunogold labeling, sections were incubated in anti-rabbit IgG conjugated to 1 nm gold particles (AuroProbe One; GE Healthcare Bio-Sciences). Sections were rinsed in PBS, postfixed for 10 min in 2% glutaraldehyde and rinsed again. Gold particles were intensified by incubation for 6 min in a silver solution [Electron Microscopy Sciences (EMS)]. Sections then were postfixed for 1 h in 2% osmium tetroxide, dehydrated, and embedded in EMBed (EMS). Ultrathin sections (70 nm thick) were taken from layers 3–4 of the somatosensory cortex (Kazama et al., 2004). These sections were collected on grids and counter-stained with Reynold's lead citrate and uranyl acetate before examination using a Philips Tecnai Biotwin transmission electron microscope connected with a digital camera (Advanced Microscopy Techniques, software version 3.2). Electron microscopic images used for analysis were obtained only in portions of the ultrathin sections at the Epon-tissue interface where the tissue antigens had greatest access to immunoreagents thereby minimizing false negative results. The ultrastructural classification of identified cellular elements was based on the descriptions of Peters et al. (1991).
Data analysis.
Data are expressed as mean ± SEM. Two-group comparisons were analyzed by the two-tailed t test for independent samples. Multiple comparisons were evaluated by ANOVA and Tukey's tests, as appropriate. Statistical significance was considered for p < 0.05.
Results
NMDA receptor activation increases CBF and ROS through nNOS-derived NO
Superfusion of the somatosensory cortex with NMDA (40 μm) increased both CBF and ROS (Fig. 1A–D) (p < 0.05; ANOVA and Tukey's test; n = 5/group). The CBF increases were stable throughout the ROS measurement period (Fig. 1B). The increases in CBF and ROS were abolished by treatment with the NMDA receptor antagonist MK-801 (200 μm) (Fig. 1C,D) (p < 0.05 from NMDA; n = 5/group). In contrast to NMDA, superfusion with kainate (10 μm) increased CBF, but not ROS production (Fig. 1C,D; n = 5/group). NMDA receptor activation increases production of neuronal NO, which contributes to the increase in CBF and to NMDA receptor signaling (Faraci and Breese, 1993; Bhardwaj et al., 1998). Therefore, we investigated whether nNOS-derived NO is responsible for the increase in ROS evoked by NMDA. The nNOS inhibitor 7-NI (50 mg/kg; i.p.) markedly attenuated the increase in CBF evoked by NMDA (Fig. 2A; p < 0.05). 7-NI also attenuated the ROS production evoked by NMDA (Fig. 2B), but not by angiotensin II (Fig. 2B; n = 5/group), an octapeptide that increases ROS via activation of angiotensin II type 1 receptors and NOX2 (Kazama et al., 2004). In agreement with the 7-NI results, NMDA did not increase CBF and ROS in nNOS-null mice (Fig. 2C,D; p > 0.05 from vehicle; n = 5/group).
Figure 1.

Neocortical superfusion of NMDA increases local CBF and ROS production. A, Superfusion with NMDA (40 μm) increases CBF. The left y-axis lists CBF as percentage increase (relative to baseline before NMDA application) and the right y-axis lists raw CBF values (perfusion units). B, The increase in CBF induced by NMDA is stable over the 30 min experimental period. C, The increase in CBF induced by NMDA is blocked by topical application of the NMDA receptor antagonist MK-801 (200 μm) (control:16.0 ± 2.0; NMDA+MK-801: 16.3 ± 1.7 perfusion units; p > 0.05 from control) (*p < 0.05 from NMDA; n = 5/group). MK-801 reduced resting CBF (from 19 ± 1 to 16 ± 2 perfusion units; p < 0.05). Superfusion with kainate (10 μm) induces increases in CBF comparable to those elicited by NMDA. D, NMDA increases ROS at the superfusion site, an effect blocked by MK-801. Kainate does not increase ROS production (*p < 0.05; ANOVA and Tukey's test; n = 5/group).
Figure 2.

The increase in CBF and ROS induced by NMDA depends on nNOS-derived NO. A, The nNOS inhibitor 7-NI (50 mg/kg; i.p.) reduces resting CBF from 18.9 ± 1.4 to 16.7 ± 1.2 perfusion units (p < 0.05), and attenuates the increase in CBF induced by NMDA (control: 16.7 ± 1.2; NMDA + 7-NI: 17.6 ± 0.9 perfusion units; p > 0.05 from control) (*p < 0.05 from NMDA; t test; n = 5/group). B, 7-NI attenuates the increase in ROS induced by NMDA, but not that induced by topical application of AngII (50 nm) (*p < 0.05 from Ringer; #p < 0.05 from NMDA and AngII + 7-NI; ANOVA and Tukey's test; n = 5/group). C, NMDA does not increase CBF in nNOS−/− mice (*p < 0.05 from nNOS+/+; t test; n = 5/group). D, NMDA does not increase ROS in nNOS−/− mice (*p < 0.05 from nNOS+/+ and p > 0.05 from Ringer in F; ANOVA and Tukey's test; n = 5/group). E, Tat-NR2B9c, a peptide that disrupts the association between the NMDA receptor complex and nNOS, but not its scrambled control (sTat-NR2B9c), attenuates the increase in CBF induced by NMDA (*p < 0.05 from sTat-NR2B9c; n = 5/group). F, Tat-NR2B9c, but not sTat-NR2B9c, attenuated NMDA induced ROS production (*p < 0.05 from Ringer and Tat-NR2B9c; ANOVA and Tukey's test; n = 5/group).
A peptide disrupting the association between nNOS and NR2 attenuates the increase in CBF and ROS evoked by NMDA
We used the tat-NR2B9c peptide (Sattler et al., 1999; Park et al., 2008) to determine whether the association between the NMDA receptor NR2 subunit and nNOS at postsynaptic sites is required for the effects of NMDA on CBF and ROS. First, we tested the selectivity and specificity of the cerebrovascular effects of the peptide. As anticipated, superfusion of tat-NR2B9c (1 μm) attenuated the increase in CBF induced by whisker stimulation, a response dependent on NMDA receptors (Park et al., 2008), but not CBF responses evoked by ACh, A23187, or adenosine which are not NMDA receptor dependent (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The scrambled peptide did not alter cerebrovascular responses (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Next, we examined the effects of tat-NR2B9c on the increase in CBF and ROS evoked by NMDA. tat-NR2B9c, but not the scrambled control, attenuated both the increase in CBF and ROS induced by NMDA (Fig. 2E,F) (p < 0.05; n = 5/group).
The increases in CBF and ROS are mediated by cGMP and PKG
Because most biological effects of NO are mediated by activation of sGC and cGMP (Hofmann et al., 2006), we investigated whether this signaling mechanism is responsible for the increase in CBF and ROS induced by NMDA. The sGC inhibitor ODQ (100 μm) attenuated (p < 0.05) the increase in both CBF and ROS evoked by NMDA (Fig. 3A,B). The attenuations were counteracted by superfusion with the cGMP analog pCPT-cGMP (Fig. 3A,B; n = 5/group), applied at a concentration (10 μm) that does not increase resting CBF. We then used KT-5823 to determine whether PKG participates in the effects of NMDA on CBF and ROS. KT-5823, at a concentration (5 μm) effective in attenuating the increase in CBF produced by pCPT-cGMP (100 μm; supplemental Fig. 2A, available at www.jneurosci.org as supplemental material), did not affect MAP or resting CBF and attenuated the increase in CBF induced by whisker stimulation or ACh, responses in which NO and cGMP are involved (supplemental Fig. 3A–D, available at www.jneurosci.org as supplemental material). CBF responses evoked by A23187 or adenosine, which are independent of NO and cGMP, were not affected by KT-5823 (supplemental Fig. 3E,F, available at www.jneurosci.org as supplemental material), attesting to the specificity of its cerebrovascular effects. KT5823 attenuated both the increase in CBF and ROS evoked by NMDA (Fig. 3A,B; n = 5/group), indicating the involvement of PKG in these responses. pCPT-cGMP or pCPT-cGMP +KT-5823 did not affect resting ROS production (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material).
Figure 3.

The sGC inhibitor ODQ and the PKG inhibitor KT-5823 attenuate the increase in CBF and ROS induced by NMDA. A, ODQ (100 μm) reduces resting CBF (vehicle 19 ± 2; ODQ: 16 ± 1 perfusion units; p < 0.05; n = 5) and attenuates the increase in CBF evoked by NMDA, an effect reversed by pCPT-cGMP (cGMP; 10 μm). KT-5823 (5 μm) also attenuates the CBF increase (#p < 0.05 from NMDA and NMDA+ODQ+cGMP; ANOVA and Tukey's test; n = 5/group). B, ODQ reduces the increase in ROS evoked by NMDA, an effect reversed by cGMP. KT-5823 also attenuates the ROS increase (*p < 0.05 from Ringer; #p < 0.05 from Ringer; ANOVA and Tukey's test; n = 5/group).
NOX2 is the source of the ROS induced by NMDA receptor activation, but the ROS do not contribute to the CBF increase
We used mice lacking the NOX2 subunit of NADPH oxidase to investigate the role of NADPH oxidase in the ROS and CBF increases induced by NMDA. The increase in ROS induced by NMDA was attenuated in NOX2−/− mice, but the CBF increase was not affected (Fig. 4A,B; p > 0.05 from NOX2+/+; n = 5/group). The increase in CBF induced by NMDA was attenuated by 7-NI (Fig. 4A), attesting to the fact that the vasodilatation was NO-dependent also in NOX2−/− mice. To examine the contribution of ROS not derived from NADPH we used MnTBAP, a compound that scavenges both superoxide and hydrogen peroxide (Day et al., 1997). MnTBAP (100 μm; topical superfusion), blocked the increase in ROS induced by NMDA, but did not alter the associated increase in CBF (Fig. 4C,D) (p > 0.05 from NMDA; n = 5/group). To test whether NO has direct effects on NADPH oxidase activity, we assessed NADPH oxidase catalytic activity using a cell-free system (Anrather et al., 2006). In agreement with previous findings (Selemidis et al., 2007), the NO donor diethylamine NONOate attenuated NADPH oxidase-dependent ROS production in membrane preparations (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), ruling out that the increase in ROS evoked by NMDA is mediated by direct effects of NO on NOX catalytic activity.
Figure 4.

NMDA increases CBF, but not ROS production, in NOX2−/− mice. A, The increases in CBF induced by NMDA in NOX2−/− mice are attenuated by 7-NI (*p < 0.05 from NMDA; t test; n = 5/group). B, The increases in ROS induced by NMDA are not observed in NOX2−/− mice. 7-NI attenuates the increase in ROS in NOX2+/+ mice. (*p < 0.05 from NOX2+/+ Ringer and NMDA + 7-NI; ANOVA and Tukey's test; n = 5/group). C, The ROS scavenger MnTBAP (100 μm) does not attenuate NMDA-induced CBF increase. D, MnTBAP blocks the NMDA-induced increase in ROS (*p < 0.05 from Ringer and NMDA+MnTBAP; ANOVA and Tukey's test; n = 5/group).
NOX2 is localized to endomembranes in neocortical somata and dendrites that express NMDA receptors and nNOS
Next, we sought to provide ultrastructural evidence supporting the involvement of NADPH oxidase and nNOS in NMDA receptor-dependent ROS production. Immunogold labeling for NOX2 was associated with endomembranes within the cytoplasm in postsynaptic somatodendritic profiles that also contained immunoperoxidase labeling for nNOS (Fig. 5A). In addition, NOX2 immunoreactivity was also shown to be present in somata and dendrites that expressed the NMDA receptor essential subunit NR1 (Fig. 5B,C). Together, these observations show that NOX2 has a subcellular location consistent with involvement in NMDA-induced ROS production, which may be influenced by NO signaling in somatosensory cortical neurons.
Figure 5.
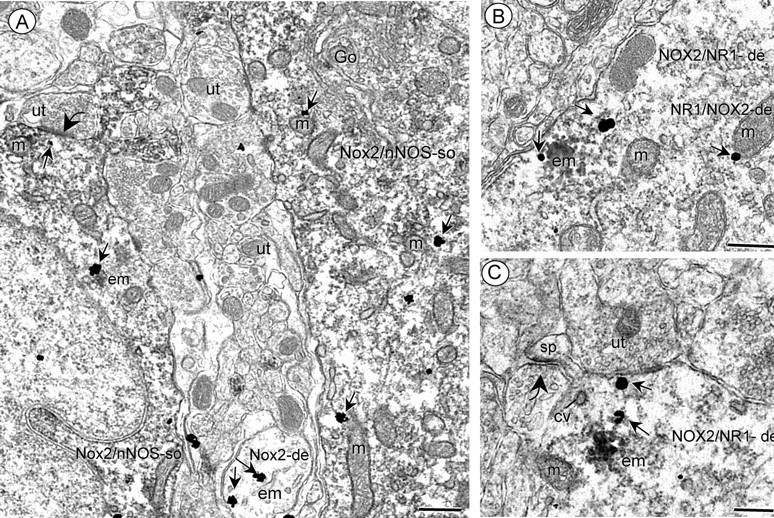
Electron micrographs showing somatodendritic colocalization of NOX2 and nNOS (A) or NR1 (B, C) in somatosensory cortex. A shows immunogold labeling (small arrows) for NOX2 located near endomembranes (em) and mitochondria (m) in two somata that also exhibit diffuse cytoplasmic immunoperoxidase labeling for nNOS (Nox2/nNOS-so). Within the neuropil, NOX2-immunogold (arrows) is seen near endomembranes (em) in a small dendrite (Nox2-de) without nNOS labeling. B, C, immunogold labeling (small arrows for NR1 in the cytoplasmic compartment of dendrites showing dense aggregated endomembrane (em) labeling for NOX2 (NOX2/NR1-de). These dendrites also contain several mitochondria (m) and coated vesicles (cv). Scale bars, 500 nm.
NMDA induces NOX2-dependent ROS production through NO, cGMP and PKG in cortical neuronal cultures
In neuronal cultures, NMDA (40 μm) increased ROS production assessed using DHE. The increase in ROS was attenuated by MK-801 (5 μm) or l-NNA (100 μm), attesting to its dependence on NMDA receptors and NOS activity (Fig. 6A,B). Furthermore, the ROS increase was blocked by the NADPH oxidase peptide inhibitor gp91ds-tat (1 μm), but not its scrambled control (Fig. 6B). Similarly, the increase in ROS was attenuated by ODQ (100 μm) or KT-5823 (5 μm) (Fig. 6C). NO release in the cultures was assessed using a NO electrode (Park et al., 2008). NMDA increased the NO signal, an effect blocked by MK-801 or l-NNA (Fig. 6D). The increase in NO induced by NMDA was not attenuated by gp91ds-tat, ODQ or KT-5823 (Fig. 6D).
Figure 6.

NMDA increases ROS in neuronal cultures via NO, cGMP, PKG, and NADPH oxidase. A, Bright-field images (a, d) and ROS-dependent fluorescence (DHE) (b, c, e, f) in neuronal cultures treated with vehicle (b), NMDA (40 μm) (c), MK-801 (5 μm) (e), or MK-801+NMDA (f). NMDA increases the ROS signal in neurons (arrows in a, b, c), but not after pretreatment with MK-801 (e, f). B, NMDA (40 μm) increases ROS production (n = 7). The increase is blocked by MK-801 (5 μm; n = 5), MnTBAP (100 μm; n = 9), the NOS inhibitor l-NNA (100 μm; n = 14), the peptide inhibitor of NADPH oxidase gp91ds-tat (gp91ds; 1 μm; n = 10), but not a scrambled control peptide (sgp91ds; 1 μm; n = 7) (*p < 0.05 from vehicle; ANOVA and Tukey's test). C, The increase in ROS induced by NMDA is attenuated by ODQ (100 μm; n = 10) or KT5823 (5 μm; n = 6) (*p < 0.05 from vehicle; ANOVA and Tukey's test). D, NMDA (40 μm) increases NO release. The increase is blocked by MK-801 or l-NNA (100 μm). gp91ds-tat (gp91ds; 1 μm), ODQ, and KT5823 do not affect NMDA-induced NO release (*p < 0.05 from NMDA; ANOVA and Tukey's test; n = 4/group). Scale bar, 10 μm.
Discussion
We sought to determine whether ROS participate in the increase in CBF induced by NMDA receptor activation, and to identify the source and signaling mechanisms responsible for the ROS production. One of the main functions of the NMDA receptor is synaptic plasticity, a phenomenon thought to underlie learning and memory (Lau and Zukin, 2007). Both NO and ROS are involved in synaptic plasticity, but whereas the sources of NO are relatively well defined (Garthwaite, 2008), the sources of ROS remain controversial (Kishida and Klann, 2007). Recent data in NOX2-null mice have implicated NADPH oxidase-derived ROS (Kishida et al., 2006), but functional evidence linking NMDA receptor activity to NOX2-dependent ROS production was lacking. Furthermore, the signaling pathway and the ultrastructural bases for NOX2-dependent ROS production during NMDA receptor activation remained unclear. Our findings fill several knowledge gaps in the mechanisms by which NMDA receptors trigger ROS production. First, the data provide direct evidence that NOX2 is the major source of ROS during activation of NMDA receptors. Second, they demonstrate that nNOS-derived NO is the signaling molecule linking NMDA receptors to NOX2-dependent ROS production. Third, they demonstrate that the association between NMDA and nNOS via PSD95 is required for the ROS production, pointing to the involvement of synaptic NMDA receptors and excluding NMDA receptors on non-neuronal cells (Stys and Lipton, 2007). Fourth, they demonstrate that nNOS and NOX2 are colocalized with the NMDA receptor subunit NR1 at postsynaptic sites, providing the ultrastructural bases for their interaction. Fifth, they demonstrate that NO leads to NOX2 activation via cGMP and PKG, the same signaling pathway that mediates the hemodynamic effects of NMDA. Sixth, they suggest that in the setting of NMDA receptor activation NO and ROS may not act by distinct signaling mechanisms, but that NO is upstream of ROS in the same signaling pathway. These novel observations provide a more complete understanding of the mechanisms by which NMDA receptor activation leads to ROS production.
Exclusion of potential sources of artifacts
The findings of the present study cannot be attributed to instability of the preparation or artifacts of the pharmacological agents used. Arterial blood pressure and blood gases were carefully monitored and maintained in the physiological range. The attenuation by 7-NI of NMDA-induced ROS production cannot be due to a ROS scavenging effect or to NADPH oxidase inhibition, because 7-NI did not attenuate the increase in ROS evoked by angiotensin II, a response also mediated by NOX2-derived ROS (Kazama et al., 2004). Similarly, the cerebrovascular effects of ODQ, KT-5823 and tat-NR2B9c cannot be attributed to attenuation of all vascular responses independent of their mechanism: ODQ and KT-5823 inhibited only NO-dependent responses whereas tat-NR2B9c inhibited only responses dependent on NMDA receptor activation. The effect of ODQ and KT-5823 cannot be attributed to inhibition of nNOS rather that sGC and PKG because these inhibitors did not alter the NO production induced by NMDA in neuronal cultures. Finally, in some systems NO and ROS can rapidly interact and inactivate each other, thereby reducing their respective concentrations (Pacher et al., 2007). However, there was little evidence of interaction between NO and ROS in our cultures, because inhibition of NO synthesis did not enhance ROS production and ROS scavenging did not enhance NO production.
NO links NMDA receptor activation with ROS production
We found that nNOS-derived NO is an absolute requirement for the ROS production evoked through NMDA receptors. Although NO can act as antioxidant by reacting with superoxide (Pacher et al., 2007), our data demonstrate that NO can activate a signaling pathway that leads to ROS production. NO failed to increase NADPH oxidase-dependent ROS production in a cell free system, ruling out an enhancing effect of NO on NOX2 catalytic activity. Rather, the effect of NO on ROS production requires sGC, cGMP and PKG activation. The pro-oxidant effects of NO and cGMP have also been described in the heart (Xu et al., 2004). The specificity of the involvement of NO in NMDA-induced ROS production is demonstrated by our observation that the ROS increase produced by angiotensin II, which is also mediated by NOX2 (Kazama et al., 2004), is not affected by nNOS inhibition. These data provide evidence for a previously unrecognized signaling mechanism linking NMDA receptor activation with NOX2-derived ROS production through NO.
NOX2 is a major source of ROS during NMDA receptor activation
Recent data have implicated NADPH oxidase derived ROS in NMDA receptor signaling. For example, NADPH oxidase derived ROS are needed for the NMDA receptor-dependent activation of extracellular regulate kinase (ERK) in the hippocampus, a key signaling step in synaptic plasticity, and are involved in hippocampal long term potentiation (Kishida et al., 2005). Mice lacking the NADPH oxidase subunits NOX2 or p47phox have mild deficits in learning and memory (Kishida et al., 2006), as has been suggested to occur in patients with chronic granulomatous disease, a genetic condition caused by mutations of the genes encoding for NADPH oxidase complexes (Pao et al., 2004). However, it remained unclear whether NMDA receptor activation evokes ROS production from NADPH oxidase. Here, we provided in vivo and in vitro evidence that NMDA receptor activation induces ROS production via a NOX2 containing NADPH oxidase. The functional interaction between NOX2, nNOS and NMDA receptors was substantiated by ultrastructural studies indicating a postsynaptic somatodendritic distribution of NOX2, NMDA receptors, as well as nNOS. Therefore, our data support the following scenario. NMDA receptor activation leads to increased NO production by activating nNOS, a process that depends on the association of nNOS with postsynaptic NMDA receptors. NO, in turn, activates sGC increasing cGMP production, which leads to activation of PKG and increased NOX2 activity. The mechanisms by which PKG activates NADPH oxidase have not been elucidated. However, the observations that NMDA activates Rac1 in mouse hippocampal neurons (Tejada-Simon et al., 2006) and that PKG activates Rac1 in HEK293 cells (Hou et al., 2004), support the involvement of this GTPase. Regardless of the mechanisms of PKG activation, our findings identify NO-cGMP-PKG signaling as the critical link between NMDA receptors and NOX2-dependent ROS production.
Concentrations of NMDA (100–300 μm) higher than those used in the present study (40 μm) can trigger ROS production from other sources, especially mitochondria (Dugan et al., 1995b; Reynolds and Hastings, 1995; Bindokas et al., 1996). Therefore, under conditions of excessive NMDA receptor activation other source of ROS could also be involved. Alternatively, NADPH oxidase-derived ROS could prime the mitochondria to produce additional radicals, as reported to occur in the heart (Kimura et al., 2005) and suggested by our own finding that NOX2 is located near mitochondria in neurons. Therefore, additional ROS sources downstream of NADPH oxidase cannot be excluded, especially with NMDA receptor overactivation.
NMDA receptor-dependent hyperemia does not require ROS
Activation of NMDA receptors is a critical step in the coupling between neural activity and CBF (Iadecola and Nedergaard, 2007). ROS derived from NMDA receptor activation are vasoactive and could contribute to the increases in CBF. ROS are potent cerebrovasodilators (Wei et al., 1985, 1996), and have been implicated in flow-mediated vasodilatation and in the vasodilatation produced by bradykinin, the Ca2+ ionophore A23187 and arachidonic acid (Kontos et al., 1984; Niwa et al., 2001; Faraci, 2006; Paravicini et al., 2006). Previous studies did not measure CBF and ROS simultaneously, and used concentrations of NMDA (100, 300 μm) that cause spreading depression (Arrick et al., 2007), a phenomenon well known to confound the hemodynamic effects of NMDA (Zhang et al., 1995; Ayata and Moskowitz, 2006). Therefore, we evaluated the cerebrovascular effects of NMDA by measuring CBF and ROS in the same animal and using concentrations of NMDA that do not induce spreading depression. We found that ROS are not involved in the hemodynamic response induced by NMDA, which is mediated by NO and sGC through the cGMP effector kinase PKG. Although these observations do not support a role of ROS in the hemodynamic response to NMDA, they identify PKG as a key target of the NO signaling mediating functional hyperemia.
Conclusions
We have provided in vivo and in vitro evidence that NMDA receptor activation elicits ROS production from a NOX2-containing NADPH oxidase through signaling pathway involving NO, cGMP and PKG. Ultrastructural evidence indicates that NMDA receptors, nNOS and NOX2 are located postsynaptically in the same neurons and in close proximity. However, NMDA-induced ROS do not play a role in the increase in CBF evoked by NMDA receptor activation, which is entirely dependent on NO, cGMP and PKG. Although the mechanisms by which PKG activates NADPH oxidase have not been elucidated, increasing evidence points to PKG-dependent activation of the small GTPase Rac1, an essential step in NADPH oxidase activation. These novel findings demonstrate that NADPH oxidase, colocalized postsynaptically with nNOS and NR1, is a major source of ROS generated by NMDA receptor activation, and that NO is the essential link between NMDA receptor activity and NOX2-dependent ROS production.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS34179 and HL18974. C.I. is the recipient of a Javits award from NIH–National Institute of Neurological Disorders and Stroke. H.G. held a fellowship from the Canadian Institutes of Health Research. We thank Dr. Charles Inturrisi for his comments on an earlier version of this manuscript.
References
- Adamson DC, Kopnisky KL, Dawson TM, Dawson VL. Mechanisms and structural determinants of HIV-1 coat protein, gp41-induced neurotoxicity. J Neurosci. 1999;19:64–71. doi: 10.1523/JNEUROSCI.19-01-00064.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anrather J, Racchumi G, Iadecola C. NF-kappa B regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- Arrick DM, Sharpe GM, Sun H, Mayhan WG. nNOS-dependent reactivity of cerebral arterioles in Type 1 diabetes. Brain Res. 2007;1184:365–371. doi: 10.1016/j.brainres.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlante A, Gagliardi S, Minervini GM, Ciotti MT, Marra E, Calissano P. Glutamate neurotoxicity in rat cerebellar granule cells: a major role for xanthine oxidase in oxygen radical formation. J Neurochem. 1997;68:2038–2045. doi: 10.1046/j.1471-4159.1997.68052038.x. [DOI] [PubMed] [Google Scholar]
- Ayata C, Moskowitz MA. Cortical spreading depression confounds concentration-dependent pial arteriolar dilation during N-methyl-d-aspartate superfusion. Am J Physiol Heart Circ Physiol. 2006;290:H1837–H1841. doi: 10.1152/ajpheart.01102.2005. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bhardwaj A, Sawada M, London ED, Koehler RC, Traystman RJ, Kirsch JR. Potent sigma1-receptor ligand 4-phenyl-1-(4-phenylbutyl) piperidine modulates basal and N-methyl-d-aspartate-evoked nitric oxide production in vivo. Stroke. 1998;29:2404–2410. [PubMed] [Google Scholar]
- Bindokas VP, Jordán J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-d-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–27473. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- Day BJ, Fridovich I, Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide-mediated injury. Arch Biochem Biophys. 1997;347:256–262. doi: 10.1006/abbi.1997.0341. [DOI] [PubMed] [Google Scholar]
- Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan LL, Lin TS, He YY, Hsu CY, Choi DW. Detection of free radicals by microdialysis/spin trapping EPR following focal cerebral ischemia-reperfusion and a cautionary note on the stability of 5,5-dimethyl-1-pyrroline N-oxide (DMPO) Free Radic Res. 1995a;23:27–32. doi: 10.3109/10715769509064016. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J Neurosci. 1995b;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraci FM. Reactive oxygen species: influence on cerebral vascular tone. J Appl Physiol. 2006;100:739–743. doi: 10.1152/japplphysiol.01044.2005. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Breese KR. Nitric oxide mediates vasodilation in response to activation of N-methyl-d-aspartate receptors in brain. Circ Res. 1993;72:476–480. doi: 10.1161/01.res.72.2.476. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2-derived radicals. Arterioscler Thromb Vasc Biol. 2006;26:826–832. doi: 10.1161/01.ATV.0000205849.22807.6e. [DOI] [PubMed] [Google Scholar]
- Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler Thromb Vasc Biol. 2007;27:303–309. doi: 10.1161/01.ATV.0000253885.41509.25. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- Hou Y, Ye RD, Browning DD. Activation of the small GTPase Rac1 by cGMP-dependent protein kinase. Cell Signal. 2004;16:1061–1069. doi: 10.1016/j.cellsig.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Nitric oxide participates in the cerebrovasodilation elicited from cerebellar fastigial nucleus. Am J Physiol. 1992;263:R1156–R1161. doi: 10.1152/ajpregu.1992.263.5.R1156. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006;8:1583–1596. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E(2) EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, Iadecola C. Angiotensin II impairs neurovascular coupling in neocortex through NADPH-oxidase-derived radicals. Circ Res. 2004;95:1019–1026. doi: 10.1161/01.RES.0000148637.85595.c5. [DOI] [PubMed] [Google Scholar]
- Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Suzuki T, Maeta H, Abe Y. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 2005;45:860–866. doi: 10.1161/01.HYP.0000163462.98381.7f. [DOI] [PubMed] [Google Scholar]
- Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid Redox Signal. 2007;9:233–244. doi: 10.1089/ars.2007.9.ft-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Pao M, Holland SM, Klann E. NADPH oxidase is required for NMDA receptor-dependent activation of ERK in hippocampal area CA1. J Neurochem. 2005;94:299–306. doi: 10.1111/j.1471-4159.2005.03189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Hoeffer CA, Hu D, Pao M, Holland SM, Klann E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol Cell Biol. 2006;26:5908–5920. doi: 10.1128/MCB.00269-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontos HA, Wei EP, Povlishock JT, Christman CW. Oxygen radicals mediate the cerebral arteriolar dilation from arachidonate and bradykinin in cats. Circ Res. 1984;55:295–303. doi: 10.1161/01.res.55.3.295. [DOI] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Milner TA, Drake CT. Ultrastructural evidence for presynaptic mu opioid receptor modulation of synaptic plasticity in NMDA-receptor-containing dendrites in the dentate gyrus. Brain Res Bull. 2001;54:131–140. doi: 10.1016/s0361-9230(00)00415-9. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa K, Carlson GA, Iadecola C. Exogenous A beta1–40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J Cereb Blood Flow Metab. 2000;20:1659–1668. doi: 10.1097/00004647-200012000-00005. [DOI] [PubMed] [Google Scholar]
- Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res. 2001;88:600–608. doi: 10.1161/01.res.88.6.600. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao M, Wiggs EA, Anastacio MM, Hyun J, DeCarlo ES, Miller JT, Anderson VL, Malech HL, Gallin JI, Holland SM. Cognitive function in patients with chronic granulomatous disease: a preliminary report. Psychosomatics. 2004;45:230–234. doi: 10.1176/appi.psy.45.3.230. [DOI] [PubMed] [Google Scholar]
- Paravicini TM, Miller AA, Drummond GR, Sobey CG. Flow-induced cerebral vasodilatation in vivo involves activation of phosphatidylinositol-3 kinase, NADPH-oxidase, and nitric oxide synthase. J Cereb Blood Flow Metab. 2006;26:836–845. doi: 10.1038/sj.jcbfm.9600235. [DOI] [PubMed] [Google Scholar]
- Park L, Gallo EF, Anrather J, Wang G, Norris EH, Paul J, Strickland S, Iadecola C. Key role of tissue plasminogen activator in neurovascular coupling. Proc Natl Acad Sci U S A. 2008;105:1073–1078. doi: 10.1073/pnas.0708823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Day BJ, Crapo JD, Fridovich I, McNamara JO. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345–355. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay S, Webster HD. The fine structure of the nervous system. New York: Oxford UP; 1991. [Google Scholar]
- Phillis JW. Adenosine and adenine nucleotides as regulators of cerebral blood flow: roles of acidosis, cell swelling, and KATP channels. Crit Rev Neurobiol. 2004;16:237–270. doi: 10.1615/critrevneurobiol.v16.i4.20. [DOI] [PubMed] [Google Scholar]
- Radi R, Cosgrove TP, Beckman JS, Freeman BA. Peroxynitrite-induced luminol chemiluminescence. Biochem J. 1993;290:51–57. doi: 10.1042/bj2900051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Selemidis S, Dusting GJ, Peshavariya H, Kemp-Harper BK, Drummond GR. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc Res. 2007;75:349–358. doi: 10.1016/j.cardiores.2007.03.030. [DOI] [PubMed] [Google Scholar]
- Sobey CG, Faraci FM. Effects of a novel inhibitor of guanylyl cyclase on dilator responses of mouse cerebral arterioles. Stroke. 1997;28:837–842. doi: 10.1161/01.str.28.4.837. [DOI] [PubMed] [Google Scholar]
- Strijbos PJ, Leach MJ, Garthwaite J. Vicious cycle involving Na+ channels, glutamate release, and NMDA receptors mediates delayed neurodegeneration through nitric oxide formation. J Neurosci. 1996;16:5004–5013. doi: 10.1523/JNEUROSCI.16-16-05004.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Lipton SA. White matter NMDA receptors: an unexpected new therapeutic target? Trends Pharmacol Sci. 2007;28:561–566. doi: 10.1016/j.tips.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Tejada-Simon MV, Villasana LE, Serrano F, Klann E. NMDA receptor activation induces translocation and activation of Rac in mouse hippocampal area CA1. Biochem Biophys Res Commun. 2006;343:504–512. doi: 10.1016/j.bbrc.2006.02.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Anrather J, Huang J, Speth RC, Pickel VM, Iadecola C. NADPH oxidase contributes to Angiotensin II signaling in the nucleus tractus solitarius. J Neurosci. 2004;24:5516–5524. doi: 10.1523/JNEUROSCI.1176-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Anrather J, Glass MJ, Tarsitano MJ, Zhou P, Frys KA, Pickel VM, Iadecola C. Nox2, Ca2+, and protein kinase C play a role in angiotensin II-induced free radical production in nucleus tractus solitarius. Hypertension. 2006;48:482–489. doi: 10.1161/01.HYP.0000236647.55200.07. [DOI] [PubMed] [Google Scholar]
- Wang G, Milner TA, Speth RC, Gore AC, Wu D, Iadecola C, Pierce JP. Sex differences in angiotensin signaling in bulbospinal neurons in the rat rostral ventrolateral medulla. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1149–R1157. doi: 10.1152/ajpregu.90485.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hitron IM, Iadecola C, Pickel VM. Synaptic and vascular associations of neurons containing cyclooxygenase-2 and nitric oxide synthase in rat somatosensory cortex. Cereb Cortex. 2005;15:1250–1260. doi: 10.1093/cercor/bhi008. [DOI] [PubMed] [Google Scholar]
- Wei EP, Christman CW, Kontos HA, Povlishock JT. Effects of oxygen radicals on cerebral arterioles. Am J Physiol. 1985;248:H157–H162. doi: 10.1152/ajpheart.1985.248.2.H157. [DOI] [PubMed] [Google Scholar]
- Wei EP, Kontos HA, Beckman JS. Mechanisms of cerebral vasodilation by superoxide, hydrogen peroxide, and peroxynitrite. Am J Physiol. 1996;271:H1262–H1266. doi: 10.1152/ajpheart.1996.271.3.H1262. [DOI] [PubMed] [Google Scholar]
- Xu Z, Ji X, Boysen PG. Exogenous nitric oxide generates ROS and induces cardioprotection: involvement of PKG, mitochondrial KATP channels, and ERK. Am J Physiol Heart Circ Physiol. 2004;286:H1433–H1440. doi: 10.1152/ajpheart.00882.2003. [DOI] [PubMed] [Google Scholar]
- Zhang F, Xu S, Iadecola C. Role of nitric oxide and acetylcholine in neocortical hyperemia elicited by basal forebrain stimulation: evidence for an involvement of endothelial nitric oxide. Neuroscience. 1995;69:1195–1204. doi: 10.1016/0306-4522(95)00302-y. [DOI] [PubMed] [Google Scholar]