

Abstract
We have recently shown that PPARδ agonism, used clinically to treat insulin resistance, increases fat oxidation and up-regulates mitochondrial PDK4 mRNA and protein expression in resting skeletal muscle. We hypothesized that PDK4 up-regulation, which inhibits pyruvate dehydrogenase complex (PDC)-dependent carbohydrate (CHO) oxidation, would negatively affect muscle function during sustained contraction where the demand on CHO is markedly increased. Three groups of eight male Wistar rats each received either vehicle or a PPARδ agonist (GW610742X) at two doses (5 and 100 mg (kg body mass (bm))−1 orally for 6 days. On the seventh day, the gastrocnemius–soleus–plantaris muscle group was isolated and snap frozen, or underwent 30 min of electrically evoked submaximal intensity isometric contraction using a perfused hindlimb model. During contraction, the rate of muscle PDC activation was significantly lower at 100 mg (kg bm)−1 compared with control (P < 0.01). Furthermore, the rates of muscle PCr hydrolysis and lactate accumulation were significantly increased at 100 mg (kg bm)−1 compared with control, reflecting lower mitochondrial ATP generation. Muscle tension development during contraction was significantly lower at 100 mg (kg bm)−1 compared with control (25%; P < 0.05). The present data demonstrate that PPARδ agonism inhibits muscle CHO oxidation at the level of PDC during prolonged contraction, and is paralleled by the activation of anaerobic metabolism, which collectively impair contractile function.
Pharmacological activation of peroxisome proliferator-activated receptors (PPARs) has been identified as being a viable clinical strategy to treat insulin resistance and dyslipidaemia in humans by decreasing body fat content, most likely by increasing fat oxidation (Berger & Wagner, 2002). PPARs are members of the nuclear receptor superfamily of ligand-activated transcription factors (proteins) that interact with a number of endogenous lipids, thereby controlling many cellular and metabolic processes. Three isotypes (PPARα, PPARβ/δ and PPARγ) have been identified in mammals, which display differential tissue distribution and specific functions (Berger & Wagner, 2002). Nevertheless, all three PPARs affect the regulation of energy homoeostasis and inflammatory responses. In addition to their natural ligands, PPAR activity can be modulated by drug agonists, such as the hypolipidaemic fibrates (PPARα agonists) and the insulin sensitizing thiazolidinediones (PPARγ agonists).
Lately, PPARδ agonists have been identified as also being a viable treatment strategy in hyperlipidaemia, since they can also reduce body fat content and insulin resistance in obese mice by apparently increasing fatty acid oxidation in skeletal muscle (Tanaka et al. 2003). We have recently investigated the effect of 6 day administration of two doses (5 and 100 mg (kg bm)−1) of a high affinity PPARδ agonist, GW610742X (Sznaidman et al. 2003; Abbot et al. 2005), on muscle mitochondrial function and fuel regulation in resting rat skeletal muscle (Constantin et al. 2007). The activity of β-hydroxyacyl-CoA dehydrogenase, which is the rate-limiting step of mitochondrial β-oxidation, increased by 75% in both treatment groups compared with control. Furthermore, muscle pyruvate dehydrogenase kinase 2 and 4 (PDK2 and PDK4) mRNA expression increased compared with control, and was paralleled by a 2-fold increase in mitochondrial PDK4 protein expression. Overall, GW610742X administration switched resting muscle fuel metabolism towards decreased carbohydrate (CHO) use and enhanced lipid utilization.
If PPARδ agonism is to be used as a viable therapy for insulin resistance and dyslipidaemia, it is important to understand the implications of this change in fuel use on muscle function, especially during contraction when CHO becomes an essential fuel, rather than at rest where CHO oxidation is relatively low and function is not an issue. During prolonged steady-state submaximal contraction, muscle contractile function is entirely maintained by oxidation of endogenous and exogenous free fatty acid and CHO. However, at workloads equivalent to ∼75% and above, CHO becomes the predominant fuel utilized. This increase in CHO oxidation is at least partly dependent on the transformation of PDC to its active (dephosphorylated) form (PDCa) in contracting rat skeletal and heart muscle (Hennig et al. 1975; Illingworth & Mullings, 1976), and in human skeletal muscle (Constantin-Teodosiu et al. 1991a; Constantin-Teodosiu et al. 1993). Furthermore, this transformation of PDC occurs in parallel with contraction intensity and CHO oxidation up to an intensity of about 75–90%
and above, CHO becomes the predominant fuel utilized. This increase in CHO oxidation is at least partly dependent on the transformation of PDC to its active (dephosphorylated) form (PDCa) in contracting rat skeletal and heart muscle (Hennig et al. 1975; Illingworth & Mullings, 1976), and in human skeletal muscle (Constantin-Teodosiu et al. 1991a; Constantin-Teodosiu et al. 1993). Furthermore, this transformation of PDC occurs in parallel with contraction intensity and CHO oxidation up to an intensity of about 75–90% , when PDC activation becomes maximal (Constantin-Teodosiu et al. 1991a; Howlett et al. 1998). These findings show that activation of PDC in contracting muscle is an important determinant of cellular energy delivery and therefore to muscle contractile function at these workloads.
, when PDC activation becomes maximal (Constantin-Teodosiu et al. 1991a; Howlett et al. 1998). These findings show that activation of PDC in contracting muscle is an important determinant of cellular energy delivery and therefore to muscle contractile function at these workloads.
In the light of the above considerations, we hypothesized that PDK4-mediated inhibition of muscle PDC activity, and thereby CHO oxidation, following administration of a high affinity PPARδ agonist (GW610742X; Sznaidman et al. 2003; Abbot et al. 2005) would impair mitochondrial ATP production, activate non-oxidative ATP generation and negatively affect tension development during contraction, particularly at exercise intensities where CHO is the predominant fuel utilized, i.e. 75%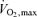 and above.
and above.
Methods
Materials
PPARδ agonist GW610742X was obtained from GlaxoSmithKline Pharmaceuticals (for structure see (Sznaidman et al. 2003). All other chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA), unless noted.
Animals
Twenty-four male Wistar rats (Charles River, Margate, UK), with an average body mass of 310 g, were housed at the University of Nottingham Medical School Animal Facility, given free access to regular chow and water and exposed to a constant ambient temperature with light/dark cycles of 12 h. After acclimation to the holding facility, animals were dosed by oral gavage with either PPARδ agonist GW610742X at 5 (n= 8) or 100 mg (kg bm)−1 (n= 8) (doses derived from Graham et al. (2005)), or an iso-volume of vehicle (0.1% solution (hydroxylpropylmethyl cellulose; n= 8) daily for six consecutive days. On the seventh day, animals were anaesthetized with sodium thiobutabarbital (Inactin, 120 mg (kg bm)−1; i.p.) and surgically prepared for hindlimb perfusion and subsequent contraction as previously described (Baker et al. 2005). All experimental procedures on the animals used in the present study were approved by the UK Home Office and by the University of Nottingham Ethical Review Committee and were conducted in accordance with the laws governing the use of animals in research in the UK (Animals (Scientific Procedures) Act 1986).
Hindlimb perfusion model and muscle contraction
Following anaesthesia, musculature of the left hindlimb was exposed by removal of the skin from this region. The branches of the femoral artery and vein were ligated (using silk ligatures or thermocautery) to the point where these vessels entered the gastrocnemius–plantaris–soleus (GPS) muscle complex. This ensured that blood flow to and from this muscle group occurred solely via the intact femoral artery and vein. The bicep femoris was then removed, a length of thread was tied around the Achilles tendon and the tendon was then cut distal to this ligature, leaving the GPS muscle group fixed to the limb on the dorsal side of the knee joint.
At this stage, the GPS muscle complex from the contralateral limb was exposed, excised and snap frozen in liquid nitrogen. Samples were stored frozen in liquid nitrogen until further analysis was performed to determine resting muscle metabolite content and enzyme activity.
The femoral artery and vein of the left hindlimb were then cannulated, and heparinized saline (10 U ml−1) was slowly flushed through the vasculature of the GPS muscle group. The arterial cannula was then attached to a primed perfusion system (enclosed in a chamber that maintained an ambient temperature of 37°C), and the muscle group was perfused with previously prepared perfusion media (see below for details). The animal was then killed humanely by injecting ∼5 ml of sodium pentobarbital (Sagatal; Rhône Mérieux) directly into the heart (according to UK Home Office Guidelines), and placed ventral surface down to enable the tibia to be secured to a clamp fixed to a stereological frame, after which the thread from the Achilles tendon was attached to an isometric force transducer (Grass Instruments, Warwick, RI, UK). Clamping the tibia facilitated the measurement of muscle force production during contraction by minimizing inertia generated by movement of the animal.
The perfusion media contained isolated porcine red blood cells suspended in a modified Krebs’ buffer containing 5% bovine serum albumin, 100 U ml−1 insulin, and 0.15 mmol l−1 pyruvate (adjusted to pH 7.4; 47% haematocrit and 6 mmol l−1 glucose). The GPS muscle group was perfused with the cell suspension for 60 min at a constant pressure of 120 mmHg using a Marlow-Watson peristaltic pump (210 U, Cornwall, UK) before undergoing repeated single twitch, submaximal isometric contractions (0.3 Hz; 200 ms; 3 V) for 30 min, which was achieved via direct electrical stimulation of the sciatic nerve using a hook electrode (Harvard Apparatus, Holliston, MA, USA). These stimulation parameters aimed to elicit a submaximal contraction intensity necessitating a marked increase in CHO oxidation (Baker et al. 2005, 2006). Isometric tension was recorded throughout (MacLab 400; ADInstruments Pty Ltd, Castle Hill, Australia), and muscle force generation was calculated as the total area under the tension–time curve for the 30 min time of contraction. Immediately following contraction, the GPS muscle group was rapidly excised and snap-frozen in liquid nitrogen. The muscle was then stored in liquid nitrogen until further analysis was performed to determine postcontraction muscle metabolite content and enzyme activity.
Muscle metabolite analysis
Each frozen GPS muscle group was crushed in liquid nitrogen, thoroughly mixed to create a homogeneous mix of fibre types and subsequently divided into two parts. One part was freeze-dried, dissected free from visible connective tissue and blood, and powdered; 5–10 mg of this muscle powder was then extracted with 0.5 mmol l−1 perchloric acid containing 1 mmol l−1 EDTA, and, after centrifugation, the supernatant was neutralized with 2.1 mmol l−1 KHCO3. Muscle acetylcarnitine was measured in the neutralized extract by enzymatic assays that made use of a radioisotopic substrate, as previously described (Cederblad et al. 1990). Muscle ATP, PCr, creatine and lactate concentrations were determined fluorometrically using a modification of the method of Harris et al. (1974).
Muscle PDC activity
A small portion of frozen ‘wet’ muscle was used to determine PDC activity as previously described (Constantin-Teodosiu et al. 1991b). Briefly, the activity of PDC in its dephosphorylated active form (PDCa) was assayed in a buffer containing NaF and dichloroacetate (DCA), and was expressed as a rate of acetyl-CoA formation (mmol min−1 (kg muscle wet tissue (wt))−1) at 37°C.
Statistical analysis
All data are expressed as means ±s.e.m. To investigate the treatment effect two-way repeated measures analysis of variance (ANOVA) was applied. When a significant F-ratio was obtained, a least significant difference (LSD) post hoc test was applied to locate specific differences. Significance was set at the P < 0.05 level of confidence.
Results
Body mass
The increase in body mass over six days of treatment with either vehicle (0.1% hydroxypropylmethylcellulose) or GW610742X was no different among treatment groups (data not shown).
Muscle function
The rate of muscle perfusion, which was similar across treatment groups, amounted to 18.9 ± 0.6, 18.3 ± 0.6 and 19.5 ± 0.8 ml min 100 (g wet muscle)−1 mass in the control, 5 and 100 mg (kg bm)−1 treatment group, respectively. Peak tension development occurred towards the end of the first minute of stimulation and was similar among vehicle and the 5 and 100 mg (kg bm)−1 treated groups (2.9 ± 0.3, 2.8 ± 0.3 and 2.6 ± 0.2 kg (100 g wet muscle)−1, respectively). An example of the force trace obtained in each of the three experimental groups is presented in Fig. 1. Muscle fatigue (defined as the percentage decline in tension from peak tension) during contraction is presented in Fig. 2A. Muscle tension loss occurred continuously throughout contraction. However, the rate of fatigue in the 100 mg (kg bm)−1 treated group was significantly greater compared with vehicle (P < 0.05). There was also a trend (P= 0.10) for muscle fatigue in the 5 mg (kg bm)−1 treated group to be greater compared to vehicle. Figure 2B shows the total work done by the GPS muscle group during the 30 min of electrically evoked isometric contraction. Work output was 25% less in the 100 mg (kg bm)−1 treated group compared with control (P < 0.05). There was also a trend (P= 0.07) for work output to be lower in the 5 mg (kg bm)−1 treated group compared to vehicle.
Figure 1. An example of a muscle tension development trace obtained over the course of 30 min of submaximal intensity electrically evoked isometric contraction.

Isometric contraction was electrically evoked at 0.3 Hz, 200 ms, 3 V. Rats were fed for 6 days with either vehicle (control; hydroxypropylmethylcellulose; n= 8) or GW610742X at 5 (n= 8) and 100 (n= 8) mg (kg bm)−1.
Figure 2. Percentage decline from peak tension development (A) and total muscle work output (B) during 30 min of submaximal intensity electrically evoked isometric contraction.

Isometric contraction was electrically evoked at 0.3 Hz, 200 ms, 3 V. Rats were fed for 6 days with either vehicle (control; hydroxypropylmethylcellulose; n= 8) or GW610742X at 5 (n= 8) and 100 (n= 8) mg (kg bm)−1. *Significantly different from the corresponding vehicle (control) group (P < 0.05).
Muscle lactate efflux and glucose uptake
Muscle lactate efflux after 5, 10, 15, 20, 25 and 30 min of contraction was calculated as:
where [lactate] is in μmol and blood flow is in ml min−1 (100 g wet muscle)−1. Date for vehicle and treated groups are presented in Fig. 3A. There was a tendency towards lower muscle lactate efflux in the vehicle group compared with the 100 mg (kg bm)−1 treated group, but no significance could be reached (P > 0.05).
Figure 3. Blood–muscle lactate (A) and glucose (B) exchange during 30 min of submaximal intensity electrically evoked isometric contraction.

Isometric contraction was electrically evoked at 3 Hz, 200 ms, 3 V. Rats were fed for 6 days with either vehicle (control; hydroxypropylmethylcellulose; n= 8) or GW610742X at 5 (n= 8) and 100 (n= 8) mg (kg bm)−1.
Muscle glucose uptake after 5, 10, 15, 20, 25 and 30 min of contraction calculated as:
where [glucose] is in μmol and blood flow in ml min−1 (100 g wet muscle)−1. Data for vehicle and treated groups are presented in Fig. 3B. There was a tendency towards higher muscle glucose uptake in the 100 mg (kg bm)−1 treated group compared with the vehicle, but no statistical difference was reached (P > 0.05).
Muscle metabolites
Muscle metabolite concentrations at rest and following 30 min of electrically evoked isometric contraction are presented in Table 1. Muscle PCr hydrolysis occurred in all treatment groups during contraction. However, the magnitude of hydrolysis (rest minus post contraction value) in the 100 mg (kg bm)−1 group was twofold greater compared with control (Δ48.2 ± 2.1 versus 21.1 ± 3.4 mmol (kg dry muscle (dm))−1, respectively; P < 0.01). Contraction markedly increased muscle lactate concentration in all treatment groups, but the magnitude of accumulation was significantly greater in the 100 mg (kg bm)−1 treated group compared with control (Δ55.9 ± 3.8 versus 42.3.0 ± 3.1 mg (kg dm)−1, respectively; P < 0.05). Muscle acetylcarnitine accumulation, which is indicative of flux through the PDC reaction (Constantin-Teodosiu et al. 1993), was significantly lower in the 100 mg (kg bm)−1 group compared with control (Δ0.89 ± 0.04 versus 1.06 ± 0.04 mmol (kg dm)−1, respectively; P < 0.05). Muscle glycogen hydrolysis (glycogenolysis) occurred in all treatment groups during contraction. However, the magnitude of hydrolysis (rest minus post contraction value) in the 100 mg (kg bm)−1 group was significantly greater compared with control (Δ71.7 ± 5.8 versus 52.5 ± 4.4 mmol (kg dm)−1, respectively; P < 0.05).
Table 1.
Muscle metabolite concentrations in gastrocnemius–plantaris–soleus muscle group after 6 days of oral gavage feeding of GW610742X
| Control | 5 mg kg−1 | 100 mg kg−1 | ||||
|---|---|---|---|---|---|---|
| Pre | Post | Pre | Post | Pre | Post | |
| ATP | 30.8 ± 1.1 | 30.4 ± 1.2 | 28.2 ± 0.5 | 29.3 ± 1.5 | 30.1 ± 0.8 | 29.3 ± 1.7 |
| PCr | 70.9 ± 1.4 | 49.8 ± 7.9 | 70.2 ± 1.1 | 43.7 ± 5.2 | 73.8 ± 2.0 | 25.6 ± 2.4* |
| Lactate | 3.3 ± 0.9 | 45.6 ± 4.1 | 2.0 ± 0.3 | 50.6 ± 5.4 | 2.8 ± 0.4 | 58.7 ± 5.0* |
| Acetylcarnitine | 0.28 ± 0.04 | 1.34 ± 0.06 | 0.22 ± 0.02 | 1.26 ± 0.05 | 0.19 ± 0.02 | 1.08 ± 0.05* |
| Glycogen | 129.2 ± 4.0 | 76.7 ± 3.5 | 130.9. ± 6.3 | 68.5 ± 7.9 | 131.4 ± 6.0 | 59.7 ± 3.0* |
Oral gavage feeding of GW610742X at 5 (n= 8), and 100 mg (kg bm)−1 (n= 8) or with a 0.1% vehicle solution (hydroxypropylmethylcellulose; n= 8) and after 30 min of submaximal intensity electrically evoked isometric contraction (0.3 Hz; 200 ms; 3 V). Values expressed as means ±s.e.m. (n= 8). Concentrations are expressed as mmol (kg dm)−1*Significantly different from the corresponding control (vehicle) group (P < 0.05).
Anaerobic ATP production
ATP production derived from substrate level phosphorylation during contraction was calculated from ΔPCr + (1.5 ×Δlactate) + (2 ×ΔATP) (Fig. 4), where Δis the difference between pre- and post-exercise concentrations (Table 1). Anaerobic ATP production was 49% greater in the 100 mg (kg bm)−1 treated group compared with control (Fig. 4, P < 0.05).
Figure 4. Muscle anaerobic ATP production (mmol (kg dm)−1) during 30 min of submaximal intensity electrically evoked isometric contraction.

Isometric contraction was electrically evoked at 3 Hz, 200 ms, 3 V. Rats were fed for 6 days with either vehicle (control; hydroxypropylmethylcellulose; n= 8) or GW610742X at 5 (n= 8) and 100 (n= 8) mg (kg bm)−1. *Significantly different from the corresponding control (vehicle) group (P < 0.05).
Muscle PDC activity
Skeletal muscle PDCa at rest and after 30 min of electrically evoked muscle contraction in the control and the GW610742X treated groups is presented in Fig. 5. Resting muscle PDCa in the 100 mg (kg bm)−1 treated group was significantly lower compared with control (Fig. 5, P < 0.05). As would be expected, muscle PDCa increased following contraction in all groups. However, the PDC activation in the 100 mg (kg bm)−1 treated group was significantly lower than in the control group (Fig. 5, P < 0.01). Furthermore, the magnitude of change in PDC activity during contraction (rest minus post contraction value) was 34% lower in the 100 mg (kg bm)−1 treated group compared with control (Δ0.56 ± 0.09 versus 0.89 ± 0.09 mmol acetyl-CoA formed min−1 (kg wt)−1, respectively, P < 0.05).
Figure 5. Muscle PDCa activity (mmol min−1 (kg wm)−1) at rest and after 30 min of submaximal intensity electrically evoked isometric contraction.

Isometric contraction was electrically evoked at 3 Hz, 200 ms, 3 V. Rats were fed for 6 days with either vehicle (control; hydroxypropylmethylcellulose; n= 8) or GW610742X at 5 (n= 8) and 100 (n= 8) mg (kg bm)−1. **Significantly different from the corresponding control (vehicle) group (P < 0.01).
Discussion
The main finding of the present study was that 6 days of treatment with a specific PPARδ agonist, GW610742X, at 100 mg day−1 (kg bm)−1, reduced the rate of mitochondrial PDC activation and flux during 30 min of submaximal electrically evoked muscle contraction at an intensity where carbohydrate is an obligate fuel. Furthermore, muscle PCr hydrolysis and lactate accumulation during contraction were markedly increased in this group compared with control, and this was reflected by a 49% greater contribution from anaerobic energy delivery during contraction. These observations, which were mostly likely to be the consequence of the inhibition of PDC controlled CHO oxidation, take on an even greater significance when it is acknowledged that the acceleration of anaerobic ATP generation occurred under conditions where muscle tension development was 25% less compared with control. PPARα agonists in general have been associated with tiredness or weakness in patients (Franc et al. 2003; Carvalho et al. 2004; Davidson et al. 2007). Since administration of PPARα and δ agonists have been reported to increase muscle free fatty acid (FFA) utilization in vivo (Motojima, 2002; Tanaka et al. 2003), and PPARδ agonists are known to impair carbohydrate oxidation at the level of PDC (Constantin et al. 2007) in vivo in resting muscle, it raises the question as to whether some of these clinical manifestations may be the consequence of an exacerbated PPAR-mediated reduction in PDC activation and CHO oxidation during physical activity limiting the mitochondrial ATP delivery.
Previous in vivo human studies have shown complete activation of muscle PDC to PDCa during submaximal voluntary contraction (Constantin-Teodosiu et al. 1993). Nevertheless, the present results clearly show that transformation of PDC to its active form (PDCa) can be markedly attenuated during submaximal contraction following PPARδ agonism, which is likely to be mediated by an increase in PDK4 protein expression (Constantin et al. 2007). The increase in FFA oxidation induced by PPARδ agonism (Tanaka et al. 2003), an increase in circulating FFA following starvation in rodents (Wu et al. 2001) and humans (Tsintzas et al. 2006), or following high dietary fat intake in humans (Peters et al. 2001) have also been shown to result in up-regulation of muscle PDK4 mRNA and protein expression. In this way, the long-term impairment of CHO oxidation has been implicated as being a causative factor in insulin resistance and the metabolic syndrome (central obesity, high fasting glucose, hypertriglyceridaemia, low HDL-cholesterol and hypertension (Gorter et al. 2004). The mechanism of PDK4-mediated PDC inhibition in starvation has been attributed to either increased FFA-mediated activation of the forkhead transcription factor FOXO1, followed by direct binding to the promoter region of the PDK4 gene (Furuyama et al. 2003), or through FFA-mediated activation of the PPARα receptor (Wu et al. 2001). Alternatively, recent data suggest that the increase in muscle PDK4 expression with starvation and diabetes may be due to either decreased insulin availability (Lee et al. 2004) or insulin deficiency (Kim et al. 2006), rather than to an increase in circulating FFAs. A reduction/impairment of the insulin-signalling pathway would reduce insulin-mediated phosphorylation of Akt1, and therefore increase FOXO1 dephosphorylation/activation. Irrespective of the mechanism, PDC inhibition in the present study (Fig. 5), illustrated also by the greater rates of muscle glycogenolysis and lactate accumulation (Table 1), was clearly associated with altered muscle energy production and impairment of contractile function, which may explain some of the previously reported adverse functional effects of PPARα agonist administration in patients.
A reduction of CHO oxidation, and by consequence a reduction in muscle tension development, can also occur by reducing pyruvate availability to the PDC reaction. Thus, during prolonged submaximal voluntary contraction, muscle fatigue is closely related to the depletion of glycogen stores (Bergstrom et al. 1967). Furthermore, previous attempts to treat type 2 diabetes by blocking excess hepatic glucose output using non-specific inhibitors of glycogen phosphorylase (GP), the enzyme that controls the rate of glycogen breakdown, ended up also reducing muscle GP activity and muscle tension development (by 35%) during prolonged submaximal contraction (Baker et al. 2006). These data emphasize that irrespective of the site of inhibition, either upstream (pyruvate formation) via GP, or directly at the level of the PDC reaction (pyruvate oxidation), viable activation/flux through the PDC reaction is central to the maintenance of muscle function during prolonged submaximal contraction.
Since intermediary metabolism of both CHO and FFA leads to formation of acetyl groups that are fed into the tricarboxylic acid cycle for energy production, one immediate question is why do FFAs appear to be unable to substitute CHO as a fuel during muscle contraction at the work intensity used in the present study? A possible explanation is that FFA metabolism is unable to sustain a rate of acetyl group supply sufficient to match the energy demand of contraction. Even when plasma FFA concentration was forcibly elevated once muscle CHO availability became reduced by contraction in the rat, it was unable to increase the rate of FFA oxidation to that observed when muscle CHO availability was normal (Turcotte et al. 1994). Taken as a whole, the facts advocate the idea that maintenance of CHO oxidation is critical to mitochondrial energy delivery during contraction and therefore to muscle contractile function. It is worth noting that a reduction in the rate of CHO oxidation following PPAR agonism mediated inhibition of PDC activity is not solely restricted to PPARδ agonists. Thus, a recent study has clearly demonstrated a reduction in the rate of CHO oxidation in hearts from mice overexpressing PPARα (Hopkins et al. 2003).
Administration of the PPARδ agonist GW501516 has been reported to induce a switch in fast twitch muscle towards a slow phenotype in a PPARδ transgenic mouse model (Wang et al. 2004). This would suggest that PPARδ agonism could mimic the positive effects of endurance exercise training, i.e. muscle fibre remodelling towards greater prevalence of type I muscle fibres and increased mitochondrial density, thereby resulting in improved endurance performance. Indeed, Wang et al. (2004) showed improved endurance performance during low intensity running (10 m min−1) in a PPARδ overexpressing mouse, despite intramuscular triglycerides being 2.5-fold lower compared with the wild-type animal. More recent work originating from the same group (Narkar et al. 2008), however, has revealed that direct pharmacological activation of PPARδ receptor is not sufficient to enhance endurance performance in the wild-type mouse, in contrast to the PPARδ overexpressing mouse. Moreover, from an energetic perspective, muscle fuel utilization during exercise in this animal model remains to be demonstrated and, in particular, it remains to be established whether this animal can sustain exercise workloads where CHO becomes the primary substrate utilized.
Generally, administration of PPAR agonists to humans and rodents has been linked to lower blood glucose levels (Berger & Wagner, 2002; Tanaka et al. 2003). With respect to PPARα and γ agonists, the blood glucose lowering effect seems to be attributable to their insulin-sensitizing properties (Tack et al. 1998; Guerre-Millo et al. 2000), which would suggest an increase in both oxidative and non-oxidative routes of glucose disposal. However, based upon the present data, it seems reasonable to suggest that any PPARδ mediated blood glucose lowering effect will be unlikely to be attributable to an increase in oxidative glucose disposal since this route would be inhibited following PPARδ mediated inhibition of PDC activity. Therefore, it seems likely that any blood glucose lowering effect will be exerted through a PPARδ mediated increase in the non-oxidative glucose disposal, i.e. via increased glycogen depositation in liver and/or skeletal muscle, or by diversion of pyruvate to other routes such as fatty acid synthesis. Indeed, recent molecular and functional analyses suggest that PPARδ activation can reduce hepatic glucose output by increasing glycolysis and the pentose phosphate shunt and promoting fatty acid synthesis in the liver (Lee et al. 2006).
A prospective look towards the efficacy of use of PPARδ agonists as antidiabetic drugs
While the general observation that PPARδ agonism can increase fat oxidation in resting muscle has potential positive implications to type II diabetic patients, this also has negative implications with respect to patient mobility and quality of life, not least because the present data clearly show that PPARδ agonism can impair muscle function by inhibiting CHO oxidation during exercise at an intensity where CHO is the mainstay of energy delivery. The prevalence of type II diabetes increases with age and therefore it is likely that PPAR agonists will be prescribed most commonly to middle aged and elderly patients. Given muscle oxygen delivery and the ability of muscle to utilize oxygen during exercise markedly decreases with age (Conley et al. 2000), this clearly suggests activities associated with daily living will become more dependent on muscle CHO utilization in the elderly since less oxygen is required for CHO oxidation than for fat oxidation. This therefore raises an important point concerning the therapeutic potential of PPARδ agonist drugs, namely the necessity for further investigation into the effect of PPARδ agonist administration on muscle fuel metabolism and function at different exercise intensities. Clearly, based upon the present data the therapeutic potential of PPARδ agonism may not be as positive as proposed by some (Wang et al. 2004; Narkar et al. 2008).
PPARδ agonists have not yet been licensed for clinical use in the UK, and to our knowledge anywhere in the world, and therefore we do not have information regarding the doses that would be administered clinically without having side-effects. However, PPARα agonists (e.g. Bezafibrate) are routinely administered up to a dose of 400 mg day−1, i.e. 5 mg day−1 (kg bm)−1 for an 80 kg person. The highest dose of PPARδ agonist used in the present experiment (100 mg day−1 (kg bm)−1) was clearly above this dose. However, in addition to pharmacogenetic and environmental factors (e.g. sex, age, diet, training status) affecting drug metabolism, a number of genetic factors controlling skeletal muscle energy metabolism are likely to influence individual susceptibility to an agonist-induced change in muscle metabolism. Given that preclinical studies are generally carried out in young, healthy genetically ‘normal’ volunteers/animals it is not uncommon that higher drug doses are required in order to obtain similar changes to those seen in patient groups. Nevertheless, it would be useful to evaluate whether the potential beneficial role of PPARδ agonism as an antidiabetic approach can be preserved when the drug dose is lowered to minimize its negative impact on muscle contractile function. Alternative strategies to counteract the detrimental effect of PPARδ agonism on muscle function may include coadministration with pharmacological activators of PDC, such as dichloroacetate.
In conclusion, the present data clearly demonstrate that PPARδ agonism, induced by GW610742X administration, inhibits CHO oxidation in rat muscle during contraction by blunting PDC activation, and consequently results in the activation of oxygen independent energy production and reduced muscle tension development. This perturbation of metabolism may at least partly underlie reports of muscle fatigue during PPARα agonist therapy in insulin resistant and type II diabetic patients (Davidson et al. 2007).
References
- Abbot EL, McCormack JG, Reynet C, Hassall DG, Buchan KW, Yeaman SJ. Diverging regulation of pyruvate dehydrogenase kinase isoform gene expression in cultured human muscle cells. FEBS J. 2005;272:3004–3014. doi: 10.1111/j.1742-4658.2005.04713.x. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Greenhaff PL, MacInnes A, Timmons JA. The experimental type 2 diabetes therapy glycogen phosphorylase inhibition can impair aerobic muscle function during prolonged contraction. Diabetes. 2006;55:1855–1861. doi: 10.2337/db05-1687. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Timmons JA, Greenhaff PL. Glycogen phosphorylase inhibition in type 2 diabetes therapy: a systematic evaluation of metabolic and functional effects in rat skeletal muscle. Diabetes. 2005;54:2453–2459. doi: 10.2337/diabetes.54.8.2453. [DOI] [PubMed] [Google Scholar]
- Berger J, Wagner JA. Physiological and therapeutic roles of peroxisome proliferator-activated receptors. Diabetes Technol Ther. 2002;4:163–174. doi: 10.1089/15209150260007381. [DOI] [PubMed] [Google Scholar]
- Bergstrom J, Hermansen L, Hultman E, Saltin B. Diet, muscle glycogen and physical performance. Acta Physiol Scand. 1967;71:140–150. doi: 10.1111/j.1748-1716.1967.tb03720.x. [DOI] [PubMed] [Google Scholar]
- Carvalho AA, Lima UW, Valiente RA. Statin and fibrate associated myopathy: study of eight patients. Arq Neuropsiquiatr. 2004;62:257–261. doi: 10.1590/s0004-282x2004000200013. [DOI] [PubMed] [Google Scholar]
- Cederblad G, Carlin JI, Constantin-Teodosiu D, P Harper, Hultman E. Radioisotopic assays of CoASH and carnitine and their acetylated forms in human skeletal muscle. Anal Biochem. 1990;185:274–278. doi: 10.1016/0003-2697(90)90292-h. [DOI] [PubMed] [Google Scholar]
- Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000;526:203–210. doi: 10.1111/j.1469-7793.2000.t01-1-00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin D, Constantin-Teodosiu D, Layfield R, Tsintzas K, Bennett AJ, Greenhaff PL. PPARdelta agonism induces a change in fuel metabolism and activation of an atrophy programme, but does not impair mitochondrial function in rat skeletal muscle. J Physiol. 2007;583:381–390. doi: 10.1113/jphysiol.2007.135459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Carlin JI, Cederblad G, Harris RC, Hultman E. Acetyl group accumulation and pyruvate dehydrogenase activity in human muscle during incremental exercise. Acta Physiol Scand. 1991a;143:367–372. doi: 10.1111/j.1748-1716.1991.tb09247.x. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Cederblad G, Hultman E. A sensitive radioisotopic assay of pyruvate dehydrogenase complex in human muscle tissue. Anal Biochem. 1991b;198:347–351. doi: 10.1016/0003-2697(91)90437-x. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Cederblad G, Hultman E. PDC activity and acetyl group accumulation in skeletal muscle during isometric contraction. J Appl Physiol. 1993;74:1712–1718. doi: 10.1152/jappl.1993.74.4.1712. [DOI] [PubMed] [Google Scholar]
- Davidson MH, Armani A, McKenney JM, Jacobson TA. Safety considerations with fibrate therapy. Am J Cardiol. 2007;99:3C–18C. doi: 10.1016/j.amjcard.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Franc S, Dejager S, Bruckert E, Chauvenet M, Giral P, Turpin G. A comprehensive description of muscle symptoms associated with lipid-lowering drugs. Cardiovasc Drugs Ther. 2003;17:459–465. doi: 10.1023/b:card.0000015861.26111.ab. [DOI] [PubMed] [Google Scholar]
- Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J. 2003;375:365–371. doi: 10.1042/BJ20030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter PM, Olijhoek JK, van der Graaf Y, Algra A, Rabelink TJ, Visseren FL. Prevalence of the metabolic syndrome in patients with coronary heart disease, cerebrovascular disease, peripheral arterial disease or abdominal aortic aneurysm. Atherosclerosis. 2004;173:363–369. doi: 10.1016/j.atherosclerosis.2003.12.033. [DOI] [PubMed] [Google Scholar]
- Graham TL, Mookherjee C, Suckling KE, Palmer CN, Patel L. The PPARδ agonist GW0742X reduces atherosclerosis in LDLR (–/–) mice. Atherosclerosis. 2005;181:29–37. doi: 10.1016/j.atherosclerosis.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Guerre-Millo M, Gervois P, Raspe E, Madsen L, Poulain P, Derudas B, Herbert JM, Winegar DA, Willson TM, Fruchart JC, Berge RK, Staels B. Peroxisome proliferator-activated receptor a activators improve insulin sensitivity and reduce adiposity. J Biol Chem. 2000;275:16638–16642. doi: 10.1074/jbc.275.22.16638. [DOI] [PubMed] [Google Scholar]
- Harris RC, Hultman E, Nordesjo LO. Glycogen, glycolytic intermediates and high-energy phosphates determined in biopsy samples of musculus quadriceps femoris of man at rest. Methods and variance of values. Scand J Clin Lab Invest. 1974;33:109–120. [PubMed] [Google Scholar]
- Hennig G, Loffler G, Wieland OH. Active and inactive forms of pyruvatedehydrogenase in skeletal muscle as related to the metabolic and functional state of the muscle cell. FEBS Lett. 1975;59:142–145. doi: 10.1016/0014-5793(75)80361-9. [DOI] [PubMed] [Google Scholar]
- Hopkins TA, Sugden MC, Holness MJ, Kozak R, Dyck JR, Lopaschuk GD. Control of cardiac pyruvate dehydrogenase activity in peroxisome proliferator-activated receptor-a transgenic mice. Am J Physiol Heart Circ Physiol. 2003;285:H270–H276. doi: 10.1152/ajpheart.00852.2002. [DOI] [PubMed] [Google Scholar]
- Howlett RA, Parolin ML, Dyck DJ, Hultman E, Jones NL, Heigenhauser GJ, Spriet LL. Regulation of skeletal muscle glycogen phosphorylase and PDH at varying exercise power outputs. Am J Physiol Regul Integr Comp Physiol. 1998;275:R418–R425. doi: 10.1152/ajpregu.1998.275.2.R418. [DOI] [PubMed] [Google Scholar]
- Illingworth JA, Mullings R. Pyruvate dehydrogenase activation after an increase in cardiac output. Biochem Soc Trans. 1976;4:291–292. doi: 10.1042/bst0040291. [DOI] [PubMed] [Google Scholar]
- Kim YI, Lee FN, Choi WS, Lee S, Youn JH. Insulin regulation of skeletal muscle PDK4 mRNA expression is impaired in acute insulin-resistant states. Diabetes. 2006;55:2311–2317. doi: 10.2337/db05-1606. [DOI] [PubMed] [Google Scholar]
- Lee CH, Olson P, Hevener A, Mehl I, Chong LW, Olefsky JM, Gonzalez FJ, Ham J, Kang H, Peters JM, Evans RM. PPARd regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci U S A. 2006;103:3444–3449. doi: 10.1073/pnas.0511253103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FN, Zhang L, Zheng D, Choi WS, Youn JH. Insulin suppresses PDK-4 expression in skeletal muscle independently of plasma FFA. Am J Physiol Endocrinol Metab. 2004;287:E69–E74. doi: 10.1152/ajpendo.00461.2003. [DOI] [PubMed] [Google Scholar]
- Motojima K. A metabolic switching hypothesis for the first step in the hypolipidemic effects of fibrates. Biol Pharm Bull. 2002;25:1509–1511. doi: 10.1248/bpb.25.1509. [DOI] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, Kang H, Shaw RJ, Evans RM. AMPK and PPARd agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters SJ, Harris RA, Wu P, Pehleman TL, Heigenhauser GJ, Spriet LL. Human skeletal muscle PDH kinase activity and isoform expression during a 3-day high-fat/low-carbohydrate diet. Am J Physiol Endocrinol Metab. 2001;281:E1151–E1158. doi: 10.1152/ajpendo.2001.281.6.E1151. [DOI] [PubMed] [Google Scholar]
- Sznaidman ML, Haffner CD, Maloney PR, Fivush A, Chao E, Goreham D, Sierra ML, LeGrumelec C, Xu HE, Montana VG, Lambert MH, Willson TM, Oliver WR, Jr, Sternbach DD. Novel selective small molecule agonists for peroxisome proliferator-activated receptor delta (PPARδ) – synthesis and biological activity. Bioorg Med Chem Lett. 2003;13:1517–1521. doi: 10.1016/s0960-894x(03)00207-5. [DOI] [PubMed] [Google Scholar]
- Tack CJ, Ong MK, Lutterman JA, Smits P. Insulininduced vasodilatation and endothelial function in obesity/insulin resistance. Effects of troglitazone. Diabetologia. 1998;41:569–576. doi: 10.1007/s001250050948. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, Watanabe M, Magoori K, Ioka RX, Tachibana K, Watanabe Y, Uchiyama Y, Sumi K, Iguchi H, Ito S, Doi T, Hamakubo T, Naito M, Auwerx J, Yanagisawa M, Kodama T, Sakai J. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci U S A. 2003;100:15924–15929. doi: 10.1073/pnas.0306981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsintzas K, Jewell K, Kamran M, Laithwaite D, Boonsong T, Littlewood J, Macdonald I, Bennett A. Differential regulation of metabolic genes in skeletal muscle during starvation and refeeding in humans. J Physiol. 2006;575:291–303. doi: 10.1113/jphysiol.2006.109892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turcotte LP, Hespel PJ, Graham TE, Richter EA. Impaired plasma FFA oxidation imposed by extreme CHO deficiency in contracting rat skeletal muscle. J Appl Physiol. 1994;77:517–525. doi: 10.1152/jappl.1994.77.2.517. [DOI] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARδ. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Peters JM, Harris RA. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor α. Biochem Biophys Res Commun. 2001;287:391–396. doi: 10.1006/bbrc.2001.5608. [DOI] [PubMed] [Google Scholar]