

Abstract
An effective approach of estimating molecular pKa values from simple density functional calculations is proposed in this work. Both the molecular electrostatic potential (MEP) at the nucleus of the acidic atom and the sum of valence natural atomic orbitals are employed for three categories of compounds, amines and anilines, carbonyl acids and alcohols, and sulfonic acids and thiols. A strong correlation between experimental pKa values and each of these two quantities for each of the three categories has been discovered. Moreover, if the MEP is subtracted by the isolated atomic MEP for each category of compounds, we observe a single unique linear relationship between the resultant MEP difference and experimental pKa data of amines, anilines, carbonyl acids, alcohols, sulfonic acids, thiols, and their substituents. These results can generally be utilized to simultaneously estimate pKa values at multiple sites with a single calculation for either relatively small molecules in drug design or amino acids in proteins and macromolecules.
I. Introduction
Knowledge of pKa values, the acid-base dissociation constant, as a measure of the strength of an acid or a base, is essential for the understanding and quantitative treatment of acid-base processes in solution, and is relevant in chemical synthesis, pharmacokinetics, drug design and metabolism, toxicology and environmental protection. There has been an immense interest in the literature to develop new and reliable models to predict and estimate pKa values with approaches using ab initio, density functional theory, molecular modeling and statistical methods.1-12
To compute accurate pKa values according to the thermodynamic cycle (Scheme 1) using ab initio and DFT methods is a challenging task for large systems such as proteins and DNA because the simulations must be carried out in solution. According to the cycle, a number of free energy changes must be simulated:1,13
| (1) |
where R is the Rydberg gas constant and T is the temperature. is the sum of the free energy of deprotonation of the gas-phase species , the free energies of desolvation of the protonated form and solvation of the deprotonated form , and the free energy of solvation for the proton . For large systems, ab initio simulations are still difficult even with the fastest software and hardware.
Scheme 1.

Much recent attention has been devoted to seeking statistical correlations of pKa values with quantum descriptors such as highest occupied molecular orbital (HOMO) energies,14 localized reactive orbital, frontier effective-for-reaction MOs (FERMO),10 electrophilicity or group philicity,15,16 etc. These relationships originated from the idea that proton or electron donor-acceptor reactions are driven by frontier molecular orbitals such as HOMO. However, the relations found were often only applicable within the same family of compounds like phenols, anilines, and azines.
It is our belief that molecular acidity is a property localized to the particular acidic atom and that the impact of the environment is reflected through the changes to that atom. The localized quantities that are relevant to the acidity of the given non-hydrogen acidic atom should be of either electrostatic or quantum nature, or both. In this work, we use two inter-dependent quantum descriptors to effectively and simultaneously estimate molecular pKa values for amines, anilines, carbonyl acids, alcohols, sulfonic acids, thiols, and their substituents. The two quantum descriptors are molecular electrostatic potential (MEP) on the acidic atom, MEP at N, O or S nucleus, and the sum of the valence p natural atomic orbitals, NAO, of the atom. Using MEP, or closely related quantities, to estimate pKa values3,17-20 and other properties 21,22 has a long history in the literature, and frontier orbitals such as FERMO have also been employed in predicting acidity.10 To the best knowledge of the authors, however, this is the first time that quantum descriptors such as MEP at the acidic nucleus and NAO are introduced generally in pKa estimation, and that the interdependence of these two quantities is revealed. In addition, these descriptors are applied to simultaneously estimate pKa values for more than one category of compounds at more than one atom type site.
II. Computational Details
A total of 228 molecular systems (154 primary, secondary and tertiary amines and anilines, 59 carboxylic acids and alcohols, and 15 sulfonic acids and thiols) have been investigated. A full structure optimization was first carried out at the DFT B3LYP/6−311+G(2d,2p) level. When a molecule has more than one stable conformation, all conformers will be examined and the one with the lowest energy will be employed in the subsequent calculations. After structure optimization, single point calculations are performed to obtain the molecular electrostatic potential on each of the nuclei followed by a full NBO 23 analysis. We obtained the initial structure and experimental pKa values are from the literature.24-31 To test the validity and applicability of the relationships presented in the text to other approaches, we also performed the same calculations with the Hartree-Fock method. We examined the results with the inclusion of the solvent effect in terms of the implicit PCM (Polarizable Continuum) Model. All calculations are performed with the Gaussian 03 package 32 with tight self-consistent field convergence and ultra-fine integration grids.
III. Results and Discussion
Figure 1 exhibits linear relationships between experimental pKa values and each of the two quantities for three categories of compounds, amines and anilines (N, blue color), carboxylic acids and alcohols (O, red color), and sulfonic acids and thiols (S, green color) as well as their derivatives. Their respective data of MEP and NAO are shown in Tables 1 to 3. This is seen that reasonable linear relationship is obtained for each category of compounds for each of the two quantum descriptors, giving the correlation coefficient R2 of 0.881, 0.878 and 0.926 for N, O, and S containing compounds, respectively, from the MEP vs. pKa plot, and R2 = 0.905, 0.924, 0.913 for N, O, and S containing compounds, respectively, from the NAO vs. pKa plot. An average correlation coefficient of 0.904 is observed from these correlations.
Figure 1.
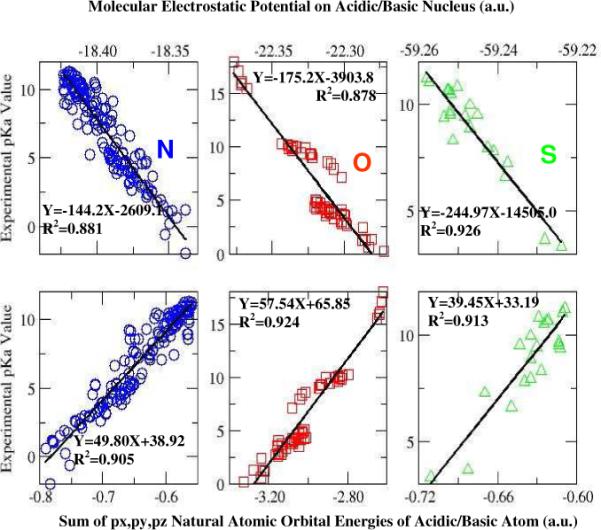
Linear relationships between molecular electrostatic potential on acidic nucleus and experimental pKa values for amines (N), carboxylic acids and alcohols (O), and sulfonic acids and thiols (S) (upper panel); and linear relationships between the sum of three valence NAO 2p/3p orbitals and pKa values (lower panel). See text and supplementary information for calculation details.
Table 1.
Molecular electrostatic potential on the acidic atom nucleus, experimental pKa data and valence NAO energies for amines and anilines (N-containing) calculated at the B3LYP/6−311+G(2d,2p) level of theory. Atomic units.
| Compounds | MEP@N | Exp. pKa | NAO px | NAO py | NAO pz |
|---|---|---|---|---|---|
| Et2NCH | −18.33812 | −2.0 | −0.2591 | −0.2587 | −0.2649 |
| Diethylcyanimide | −18.33841 | 1.2 | −0.2576 | −0.2577 | −0.2647 |
| Acetanilide | −18.34462 | 0.61 | −0.2602 | −0.2556 | −0.2744 |
| NCH2CH2CN3 | −18.34761 | 1.1 | −0.2553 | −0.2538 | −0.2699 |
| HNCH2CN2 | −18.34941 | 0.2 | −0.2449 | −0.2439 | −0.2700 |
| p-Nitrobenzne | −18.35054 | 1.02 | −0.2808 | −0.2539 | −0.2432 |
| EtNCH2CN2 | −18.35225 | −0.6 | −0.2439 | −0.2488 | −0.2598 |
| 3-Methyl-4-nitrobenzene | −18.35365 | 1.5 | −0.2495 | −0.2400 | −0.2782 |
| 4-Chloro-3-nitrobenzene | −18.35567 | 1.9 | −0.2450 | −0.2396 | −0.2758 |
| p-Cyanobenzene | −18.35809 | 1.74 | −0.2734 | −0.2456 | −0.2358 |
| 35-Dimethyl-4-nitrobenzene | −18.36144 | 2.59 | −0.2415 | −0.2323 | −0.2698 |
| m-Nitrobenzene | −18.36192 | 2.5 | −0.2369 | −0.2361 | −0.2698 |
| m-Cyanobenzene | −18.36429 | 2.76 | −0.2345 | −0.2335 | −0.2673 |
| 35-Dibromobenzene | −18.36433 | 2.34 | −0.2303 | −0.2386 | −0.2671 |
| 35-Dichloro-aniline | −18.36477 | 2.37 | −0.2294 | −0.2379 | −0.2666 |
| 3-Methoxy-5-nitrobenzene | −18.36538 | 2.11 | −0.2288 | −0.2367 | −0.2664 |
| 4-Methyl-3-nitrobenzene | −18.36640 | 2.96 | −0.2328 | −0.2294 | −0.2657 |
| 35-Dibromo-4-methoxybenzene | −18.36833 | 2.98 | −0.2258 | −0.2353 | −0.2609 |
| EtNCH2CH2CN2 | −18.36848 | 4.55 | −0.2304 | −0.2426 | −0.2426 |
| 35-Dibromo-4-methylbenzene | −18.36912 | 2.87 | −0.2253 | −0.2327 | −0.2622 |
| Dicyanodiethylamine | −18.36961 | 5.2 | −0.2247 | −0.2261 | −0.2614 |
| HNCH2CH2CN2 | −18.37007 | 5.26 | −0.2360 | −0.2517 | −0.2239 |
| 35-Dibromo-4-hydroxybenzene | −18.37157 | 3.2 | −0.2225 | −0.2292 | −0.2594 |
| PhNMe2 | −18.37325 | 5.1 | −0.2264 | −0.2261 | −0.2335 |
| Dimethylaminoacetonitrile | −18.37434 | 4.2 | −0.2215 | −0.2206 | −0.2435 |
| m-Bromobenzene | −18.37435 | 3.51 | −0.2240 | −0.2243 | −0.2574 |
| 3-Choloro-aniline | −18.37477 | 3.52 | −0.2242 | −0.2229 | −0.2570 |
| m-Chlorobenzene | −18.37477 | 3.34 | −0.2242 | −0.2229 | −0.2570 |
| p-Bromobenzene | −18.37538 | 3.91 | −0.2271 | −0.2185 | −0.2562 |
| m-Fluorobenzene | −18.37572 | 3.59 | −0.2252 | −0.2199 | −0.2560 |
| 3-Fluoro-aniline | −18.37572 | 3.58 | −0.2252 | −0.2199 | −0.2560 |
| 2-Choloro-aniline | −18.37606 | 2.64 | −0.2229 | −0.2240 | −0.2558 |
| 4-Choloro-aniline | −18.37636 | 3.99 | −0.2260 | −0.2175 | −0.2553 |
| p-Chlorobenzene | −18.37636 | 3.98 | −0.2552 | −0.2261 | −0.2175 |
| 2-Fluoro-aniline | −18.37697 | 3.2 | −0.2222 | −0.2186 | −0.2537 |
| PhNEt2 | −18.37729 | 6.6 | −0.2271 | −0.2230 | −0.2306 |
| 3-Chloro-5-methoxybenzene | −18.37780 | 3.1 | −0.2170 | −0.2237 | −0.2538 |
| 3-Bromo-4-methylbenzene | −18.37862 | 3.98 | −0.2199 | −0.2188 | −0.2529 |
| 3-Chloro-4-methylbenzene | −18.37907 | 4.05 | −0.2213 | −0.2162 | −0.2524 |
| CF3CH2N(CH3)2 | −18.38004 | 4.75 | −0.2200 | −0.2169 | −0.2358 |
| 4-Fluoro-aniline | −18.38037 | 4.65 | −0.2214 | −0.2134 | −0.2512 |
| p-Fluorobenzene | −18.38037 | 4.65 | −0.2510 | −0.2216 | −0.2134 |
| Diethylaminoacetonitrile | −18.38089 | 4.5 | −0.2153 | −0.2156 | −0.2405 |
| n-Piperidine-CH2CN | −18.38182 | 4.55 | −0.2370 | −0.2196 | −0.2164 |
| Aminoacetonitrile | −18.38220 | 5.3 | −0.2131 | −0.2222 | −0.2422 |
| H2NCH2CN | −18.38220 | 5.34 | −0.2131 | −0.2222 | −0.2422 |
| 3-Bromo-4-methoxybenzene | −18.38280 | 4.08 | −0.2160 | −0.2129 | −0.2484 |
| Et2NCH2CN | −18.38315 | 4.55 | −0.2150 | −0.2213 | −0.2302 |
| Beta-dimethylaminopropionitrile | −18.38334 | 7.0 | −0.2132 | −0.2147 | −0.2379 |
| m-Hydroxybenzene | −18.38508 | 4.17 | −0.2158 | −0.2104 | −0.2468 |
| Aniline | −18.38581 | 4.58 | −0.2460 | −0.2171 | −0.2078 |
| 35-Dimethoxybenzene | −18.38610 | 3.82 | −0.2072 | −0.2164 | −0.2456 |
| CF3CH2NHCH3 | −18.38635 | 6.05 | −0.2172 | −0.2356 | −0.2054 |
| n-Piperidine-CCH3CN | −18.38648 | 9.22 | −0.2131 | −0.2111 | −0.2345 |
| CF3CH2NH2 | −18.38776 | 5.7 | −0.2105 | −0.2225 | −0.2334 |
| 3-Methoxyl-aniline | −18.38807 | 4.2 | −0.2120 | −0.2080 | −0.2438 |
| m-Methoxybenzene | −18.38807 | 4.2 | −0.2120 | −0.2080 | −0.2438 |
| m-Methylbenzene | −18.38808 | 4.69 | −0.2118 | −0.2078 | −0.2442 |
| Et2NC(CH3)2CN | −18.38892 | 9.13 | −0.2132 | −0.2152 | −0.2278 |
| 4-Methyl-aninile | −18.38929 | 5.08 | −0.2127 | −0.2041 | −0.2426 |
| p-Methylbenzene | −18.38929 | 5.12 | −0.2127 | −0.2041 | −0.2426 |
| n-Methyleamphetamine-(CH2)2CN | −18.38970 | 6.95 | −0.2235 | −0.2194 | −0.2149 |
| p-Hydroxybenzene | −18.38980 | 5.5 | −0.2113 | −0.2036 | −0.2416 |
| Et2N(CH2)2CN | −18.38981 | 7.65 | −0.2236 | −0.2098 | −0.2185 |
| 35-dimethylbenzene | −18.39012 | 4.91 | −0.2032 | −0.2132 | −0.2408 |
| m-Aminobenzene | −18.39058 | 4.88 | −0.2401 | −0.2062 | −0.2099 |
| 34-Dimethylbenzene | −18.39133 | 5.17 | −0.2101 | −0.2025 | −0.2404 |
| Beta-diethylaminopropionitrile | −18.39143 | 7.6 | −0.2083 | −0.2145 | −0.2247 |
| 2-Amino-2-cyanopropane | −18.39167 | 5.3 | −0.2083 | −0.2261 | −0.2214 |
| 4-Methoxyl-aniline | −18.39174 | 5.36 | −0.2105 | −0.2037 | −0.2363 |
| p-Methoxybenzene | −18.39174 | 5.29 | −0.2093 | −0.2017 | −0.2397 |
| Beta-aminopropionitrile | −18.39304 | 7.7 | −0.2045 | −0.2237 | −0.2235 |
| Phenyl_OHOHOHH | −18.39345 | 8.58 | −0.2052 | −0.2034 | −0.2490 |
| n-Amphetamine-(CH2)2CN | −18.39407 | 7.23 | −0.2031 | −0.2009 | −0.2390 |
| Epinephrine | −18.39415 | 8.55 | −0.2037 | −0.2082 | −0.2311 |
| 3-Amino-4-hydroxybenzene | −18.39512 | 5.7 | −0.2066 | −0.1988 | −0.2352 |
| p-Aminobenzene | −18.39595 | 6.08 | −0.2049 | −0.1972 | −0.2352 |
| Triethanolamine | −18.39601 | 7.77 | −0.2013 | −0.2013 | −0.2265 |
| Arterenol | −18.39616 | 8.55 | −0.2080 | −0.2286 | −0.2127 |
| Et2N(CH2)3CN | −18.39621 | 9.29 | −0.2048 | −0.2040 | −0.2221 |
| 2-Methyleanilne-Et2 | −18.39666 | 7.18 | −0.2021 | −0.2068 | −0.2168 |
| n-Methylmorpholine | −18.39918 | 7.41 | −0.2213 | −0.2059 | −0.1981 |
| n-Allylmorpholine | −18.39975 | 7.05 | −0.2057 | −0.1994 | −0.2200 |
| nn-Dimethyl-2−2-aminoethoxyethanol | −18.40040 | 9.1 | −0.1961 | −0.1968 | −0.2199 |
| Et2N(CH2)4CN | −18.40046 | 10.08 | −0.2171 | −0.2020 | −0.1992 |
| n-Benzoylpiperazine | −18.40053 | 7.78 | −0.1995 | −0.1954 | −0.2259 |
| Beta-difluoroethylamine | −18.40078 | 7.52 | −0.1947 | −0.2375 | −0.1934 |
| Triallylamine | −18.40090 | 8.31 | −0.2141 | −0.2036 | −0.2028 |
| Dimethylethanolamine | −18.40128 | 10.3 | −0.1984 | −0.1966 | −0.2157 |
| n-Ethylmorpholine | −18.40141 | 7.7 | −0.1999 | −0.1975 | −0.2229 |
| Benzyldimethylamine | −18.40151 | 8.93 | −0.1980 | −0.2175 | −0.1954 |
| Et2N(CH2)5CN | −18.40182 | 10.46 | −0.2068 | −0.2020 | −0.2067 |
| Allyldimethylamine | −18.40272 | 8.72 | −0.1945 | −0.1943 | −0.2155 |
| Diallylmethylamine | −18.40366 | 8.79 | −0.1940 | −0.1989 | −0.2138 |
| n-Carbethoxypiperazine | −18.40371 | 8.28 | −0.1972 | −0.1896 | −0.2246 |
| (CH3)3N | −18.40449 | 9.76 | −0.1920 | −0.1920 | −0.2165 |
| Morpholine | −18.40649 | 8.36 | −0.2266 | −0.1900 | −0.1864 |
| Alpha-benzylpyrroline | −18.40658 | 7.08 | −0.2027 | −0.2096 | −0.1946 |
| n-Allylpiperidine | −18.40716 | 9.69 | −0.1915 | −0.1915 | −0.2156 |
| Triethylenediamine | −18.40816 | 8.8 | −0.2232 | −0.1849 | −0.1849 |
| Benzyldiethylamine | −18.40854 | 9.48 | −0.1925 | −0.1982 | −0.2000 |
| Ethanolamine | −18.40857 | 9.5 | −0.2035 | −0.1917 | −0.2085 |
| Diallylamine | −18.40900 | 9.29 | −0.1851 | −0.2090 | −0.1964 |
| n-Methylpiperidine | −18.40921 | 10.08 | −0.1921 | −0.1888 | −0.2137 |
| Dimethyl-n-propylamine | −18.40975 | 9.99 | −0.2059 | −0.1935 | −0.1897 |
| Dimethylethylamine | −18.40982 | 9.99 | −0.2122 | −0.1881 | −0.1896 |
| Dimethyl-n-butylamine | −18.40987 | 10.02 | −0.2058 | −0.1931 | −0.1894 |
| Benzylmethylamine | −18.40990 | 9.58 | −0.1984 | −0.1980 | −0.1922 |
| n-Methylpyrrolidine | −18.41092 | 10.46 | −0.1933 | −0.1872 | −0.2151 |
| Allylmethylamine | −18.41106 | 10.11 | −0.1840 | −0.1799 | −0.2186 |
| 1n-Propylpiperidine | −18.41122 | 10.48 | −0.1882 | −0.1880 | −0.2124 |
| n-Methyltrimethyleneimine | −18.41175 | 10.4 | −0.2146 | −0.1886 | −0.1822 |
| nn-Dimethylcyclohexylamine | −18.41175 | 10.0 | −0.1880 | −0.2009 | −0.1949 |
| 2−2-Aminoethoxyethanol | −18.41184 | 9.5 | −0.2142 | −0.1957 | −0.1804 |
| Methyldiethylamine | −18.41187 | 10.29 | −0.1904 | −0.1890 | −0.2055 |
| 12-Dimethylpyrrolidine | −18.41198 | 10.26 | −0.1905 | −0.1899 | −0.2141 |
| Benzylethylamine | −18.41266 | 9.68 | −0.1935 | −0.1817 | −0.2097 |
| (CH3)2NH | −18.41295 | 10.64 | −0.1972 | −0.1984 | −0.1805 |
| Benzylamine | −18.41331 | 9.34 | −0.1858 | −0.2148 | −0.1899 |
| Alpha-ethylpyrroline | −18.41335 | 7.43 | −0.2103 | −0.1905 | −0.1864 |
| (C2H5)3N | −18.41377 | 10.65 | −0.1880 | −0.1898 | −0.2031 |
| n-Ethylpiperidine | −18.41388 | 10.4 | −0.2055 | −0.1933 | −0.1871 |
| (C3H7)3N | −18.41393 | 10.65 | −0.1859 | −0.1870 | −0.2043 |
| (C4H9)3N | −18.41423 | 10.89 | −0.1860 | −0.1877 | −0.2011 |
| Allylamine | −18.41470 | 9.49 | −0.1821 | −0.2101 | −0.1925 |
| Quinuclidine | −18.41526 | 11.0 | −0.1784 | −0.1793 | −0.2155 |
| Phenyl_HHHH | −18.41548 | 9.78 | −0.1815 | −0.1869 | −0.2123 |
| Beta-Phenylethylamine | −18.41599 | 9.83 | −0.1835 | −0.2026 | −0.1958 |
| Methoxypropylamine | −18.41712 | 10.1 | −0.1786 | −0.1910 | −0.2078 |
| Phenyl_ohohohch3 | −18.41714 | 8.55 | −0.1840 | −0.1757 | −0.2045 |
| Ethylenediamine | −18.41724 | 9.98 | −0.2037 | −0.1961 | −0.1754 |
| Piperidine | −18.41752 | 11.22 | −0.1781 | −0.1762 | −0.2156 |
| Gama-Phenylpropylamine | −18.41761 | 10.2 | −0.1779 | −0.2052 | −0.1929 |
| Diisobutylamine | −18.41775 | 10.5 | −0.2177 | −0.1766 | −0.1763 |
| i_(C3H7)3N | −18.41776 | 11.05 | −0.1827 | −0.1822 | −0.2029 |
| CH3NH2 | −18.41803 | 10.62 | −0.2121 | −0.1870 | −0.1735 |
| (C2H5)2NH | −18.41862 | 10.98 | −0.1815 | −0.1868 | −0.1949 |
| NH3 | −18.41865 | 9.21 | −0.1827 | −0.1827 | −0.2301 |
| (C3H7)2NH | −18.41886 | 11.0 | −0.1898 | −0.1843 | −0.1897 |
| Pyrrolidine | −18.41906 | 11.27 | −0.1752 | −0.1790 | −0.2161 |
| (C4H9)2NH | −18.41920 | 11.25 | −0.1925 | −0.1789 | −0.1884 |
| Trimethyleneimine | −18.41950 | 11.29 | −0.2115 | −0.1780 | −0.1745 |
| 1-Ethylr-2-methylpyrrolidine | −18.41964 | 10.64 | −0.1926 | −0.1873 | −0.2037 |
| C2H5NH2 | −18.42005 | 10.63 | −0.2052 | −0.1899 | −0.1730 |
| C3H7NH2 | −18.42022 | 10.53 | −0.2001 | −0.1937 | −0.1728 |
| C4H9NH2 | −18.42049 | 10.59 | −0.2073 | −0.1857 | −0.1724 |
| Phenyl_HHOHH | −18.42093 | 8.9 | −0.1749 | −0.1782 | −0.2127 |
| i_(C3H7)2NH | −18.42188 | 11.0 | −0.1761 | −0.1941 | −0.1906 |
| i_C3H7NH2 | −18.42234 | 10.63 | −0.1932 | −0.1948 | −0.1775 |
| Phenyl_HOHOHH | −18.42246 | 8.93 | −0.1776 | −0.1933 | −0.1943 |
| Cyclohexylaime | −18.42277 | 9.82 | −0.2120 | −0.1806 | −0.1712 |
| Cyclohexylamine | −18.42277 | 10.49 | −0.2120 | −0.1806 | −0.1712 |
| Di-sec-butylamine | −18.42332 | 11.01 | −0.1732 | −0.2087 | −0.1774 |
| Cycloheptylamine | −18.42339 | 9.99 | −0.1740 | −0.1709 | −0.2184 |
Table 3.
Molecular electrostatic potential on the acidic atom nucleus, experimental pKa data and valence NAO energies for sulfonic acids and thiols (S-containing) calculated at the B3LYP/6−311+G(2d,2p) level of theory. Atomic units.
| Compounds | MEP@S | Exp.pKa | NAO px | NAO py | NAO pz |
|---|---|---|---|---|---|
| Methyl_thioglycolate | −59.24106 | 7.8 | −0.1978 | −0.2189 | −0.2228 |
| Ethyl_mercaptan | −59.25179 | 10.5 | −0.1780 | −0.2011 | −0.2457 |
| o-aminothiophenol | −59.23813 | 6.59 | −0.1853 | −0.2519 | −0.2122 |
| HOCH2CH(OH)CH2-thiol | −59.25194 | 9.51 | −0.1779 | −0.1971 | −0.2385 |
| CH2=CHCH2-thiol | −59.24700 | 9.96 | −0.1837 | −0.2240 | −0.2273 |
| n-C4H9-thiol | −59.25272 | 10.66 | −0.1765 | −0.2004 | −0.2445 |
| t-C5H11-thiol | −59.25812 | 11.21 | −0.2142 | −0.1733 | −0.2211 |
| C2H5OCOCH2-thiol | −59.24254 | 7.95 | −0.1964 | −0.2176 | −0.2211 |
| C2H5OCH2CH2-thiol | −59.25366 | 9.38 | −0.1833 | −0.2073 | −0.2218 |
| HOCH2CH(OH)CH2-thiol | −59.25194 | 9.66 | −0.1779 | −0.1971 | −0.2385 |
| n-C3H7-thiol | −59.25237 | 10.65 | −0.1771 | −0.2006 | −0.2449 |
| Thioglycolic_acid | −59.22815 | 3.67 | −0.2144 | −0.2535 | −0.2149 |
| Mercaptoethanol | −59.24517 | 9.5 | −0.1849 | −0.2072 | −0.2513 |
| Cysteamine | −59.25026 | 10.81 | −0.1798 | −0.2027 | −0.2466 |
| Thioacetic_acid | −59.22381 | 3.33 | −0.2240 | −0.2678 | −0.2193 |
Moreover, if one given number, the MEP evaluated for the isolated neutral acidic atom, is subtracted from the MEP value on the acidic nucleus for each of the three categories of compounds and then all MEP differences of the three categories are plotted together against the experimental pKa data, one single linear relationship, as shown Fig. 2, is obtained with the correlation coefficient R2=0.896. The aforementioned reference MEP value (isolated atoms of N, O, and S) employed in this work is −18.28 a.u. for amine and aniline compounds, −22.20 a.u. for carboxylic acids and alcohols, and −59.12 a.u. for sulfonic acids and thiols.
Figure 2.
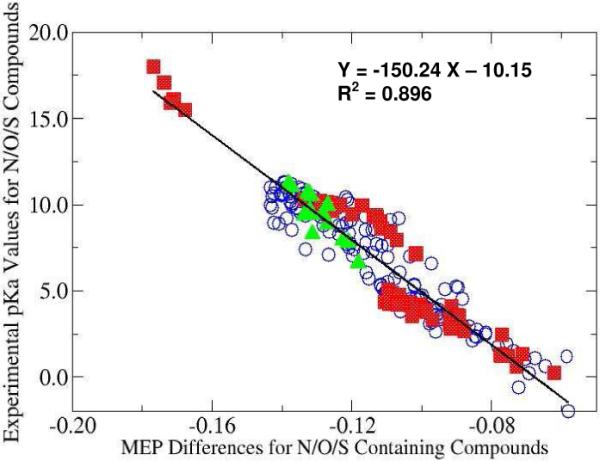
Linear relationship between the MEP difference and experimental pKa values for all 228 data points. The MEP reference value for N, O, and S compounds is −18.28, −22.20, and −59.12 a.u., respectively. Symbols: N: open blue cycle; O: filled red square; S: filled green triangle
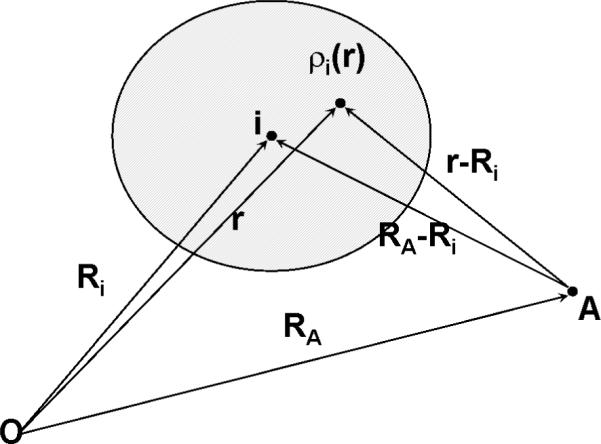
The universality of the above linear relationship between the MEP difference [MEP (in molecule) – MEP (neutral isolated atom)] and pKa values for different kinds of compounds can be understood in this manner. The molecular electrostatic potential on a nuclear RA can be expressed as follows:
| (2) |
This quantity is system dependent because it is a function of {Zi}. However, if one uses the sum of atomic electron densities as the zeroth-order approximation for the total molecular electron density, plus a local environment dependent correction,
| (3) |
and inserts it into the MEP formula, the first term of the MEP can be arranged to cancel approximately, leaving the correction term dependent only on the local environment of the nucleus. To demonstrate, let us rewrite Eq. (3) as
| (4) |
With Eq. (4), we have
| (5) |
To obtain the second term at the right-hand side of Eq. (5), we employed the approximation that Ri and RA are separated (i.e., Atoms A and i are not overlapped) so when calculating MEP at RA from contributions of atoms Ri, we assume r ≈Ri or |r - RA| ≈ |Ri – RA|. That is,
| (6) |
The physical meaning of the above approximation is that the electrostatic potential at points A outside a spherical charge distribution ρi(r) is equal to the electrostatic potential generated by the point charge Zi from the center of the spherical atom i (Scheme 2). To get the last equality of Eq. (6), we used
| (7) |
The last term of Eq. (6) absorbed approximations from Eq. (7). Since
| (8) |
with Eqs. (2), (5), and (8), there arrives
| (9) |
From the model density, Eq. (3), we know that the correction terms, gi(|r-Ri|,NAOv,i), depends on differences in electron density between the atoms; these will be functions of the NAOs of the valence shells of the atoms. Since these local differences will be positive or negative, the r.h.s of Eq. (9) will thus be relatively small (see Fig. 2), due to the significant cancellations in integration over the corrections. As seen in Fig. 2, the r.h.s of Eq. (9) is indeed small for the large number of molecules studied; it is remarkable that these small numbers vary systematically with the pKa values.
Scheme 2.
A strong correlation between the MEP on the acidic nucleus and the sum of the atom's valence natural atomic orbitals is observed. As an illustrative example, Figure 3 exhibits the relationship for amines and anilines. A similar correlation is seen for O and S containing compounds as well (not shown). Notice that the valence natural atomic orbitals employed in this study are 2p orbitals for nitrogen and oxygen and 3p orbitals for sulfur. We considered to add 2s/3s atomic orbitals in the summation but no significantly different results were obtained. The strong correlation between the MEP on a nucleus and the valence NAO indicates that the correction term in Eq. (3), fi(|r-Ri|), is dominated by the contribution from the valence part of NAOs of the atom.
Figure 3.

Strong linear relationship between MEP on N and the sum of nitrogen 2Px/2Py/2Pz NAO for N-containing compounds (amines and anilines) at the level of B3LYP/6−311+G(2d,2p). Atomic units.
The MEP data are from DFT gas phase calculations at the B3LYP/6−311+G(2d,2p) level. Taking the solvent effect into account does not destroy the correlation between MEP on the nucleus and experimental pKa data. An example is illustrated in Fig. 4 for the N-containing compounds, where one can see that the correlation coefficient is similar to that of the gas phase results. Also, we performed MEP calculations at other levels of theory, such as Hartree-Fock theory (Fig. 5) or with different density functionals; no significantly difference in the correlation was seen. In addition, for amines we also considered the protonated, conjugate species, but no statistically significant correlation between MEP at N and pKa data is observed (results not shown).
Figure 4.

The impact of the solvent effect on the correlation between MEP on N and experimental pKa data for N-containing compounds (amines and anilines). The implicit PCM (Polarizable Continuum Model) and 6−311+G(2d,2p) basis set were used.
Figure 5.

The strong linear relationship between MEP on N nucleus and experimental pKa data for amines and anilines using the Hartree-Fock method and 6−311+G(2d,2p) basis set.
One possible application of these results is to estimate pKa values with a single DFT calculation for amino acids and peptides where different pKa values at different atom sites are possible. As an illustrative example, we estimated pKa values of cysteinylcysteine, which has four acidic sites, O, S1, S2, and N. Using the relationships in Fig. 1, we obtained the pKa values to be 3.5 (O), 6.9 (S1), 8.2 (S2) and 9.8, respectively, whereas experimental data give 2.7, 7.3, 9.4, and 10.9, respectively. Similar results are obtained when the relationship in Fig. 2 is employed. In both cases, reasonable pKa values are obtained and the order of acidity of the four atoms is correctly predicted.
IV. Conclusions
An effective approach of estimating molecular pKa values from simple gas-phase density functional calculations is proposed in this work, using either the molecular electrostatic potential on the nucleus of the acidic atom or the sum of valence natural atomic orbitals. A strong correlation between experimental pKa values and each of these two quantities has been discovered. Moreover, if the MEP is subtracted by a given reference value for each category of compounds, we observe a single unique linear relationship between the MEP difference and experimental pKa data of amines, anilines, carbonyl acids, alcohols, sulfonic acids, thiols, and their substituents. With a single DFT calculation these results can conveniently be utilized to simultaneously estimate pKa values at multiple sites of small molecules in drug design and of amino acids in proteins and macromolecules.
Table 2.
Molecular electrostatic potential on the acidic atom nucleus, experimental pKa data and valence NAO energies for carbonyl acids and alcohols (O-containing) calculated at the B3LYP/6−311+G(2d,2p) level of theory. Atomic units.
| Compounds | MEP@O | Exp.pKa | NAO px | NAO py | NAO pz |
|---|---|---|---|---|---|
| 2,2-dimethyl-propionic_acid | −22.31973 | 5.05 | −0.3292 | −0.3599 | −0.3459 |
| propionic_acid | −22.31848 | 4.87 | −0.3342 | −0.3574 | −0.3475 |
| butyric_acid | −22.31914 | 4.82 | −0.3308 | −0.3594 | −0.3468 |
| acetic_acid | −22.31648 | 4.76 | −0.3327 | −0.3633 | −0.3495 |
| p-methyl-benzoic_acid | −22.32054 | 4.37 | −0.3292 | −0.3575 | −0.3449 |
| vinyl-acetic_acid | −22.31360 | 4.35 | −0.3384 | −0.3628 | −0.3520 |
| phenyl-acetic_acid | −22.31572 | 4.31 | −0.3433 | −0.3513 | −0.3529 |
| m-methyl-benzoic_acid | −22.31932 | 4.27 | −0.3302 | −0.3588 | −0.3459 |
| succinic_acid | −22.31061 | 4.21 | −0.3391 | −0.3674 | −0.3545 |
| benzoic_acid | −22.31684 | 4.19 | −0.3594 | −0.3347 | −0.3484 |
| p-fluoro-benzoic_acid | −22.31179 | 4.14 | −0.3383 | −0.3661 | −0.3534 |
| 3-chloro-propionic_acid | −22.30156 | 4.1 | −0.3543 | −0.3709 | −0.3653 |
| p-chloro-benzoic_acid | −22.31041 | 3.98 | −0.3396 | −0.3673 | −0.3546 |
| p-bromo-benzoic_acid | −22.31011 | 3.97 | −0.3400 | −0.3676 | −0.3549 |
| m-fluoro-benzoic_acid | −22.30922 | 3.87 | −0.3400 | −0.3690 | −0.3555 |
| m-chloro-benzoic_acid | −22.30871 | 3.83 | −0.3405 | −0.3696 | −0.3560 |
| glycolic_acid | −22.31175 | 3.83 | −0.3401 | −0.3651 | −0.3545 |
| m-bromo-benzoic_acid | −22.30859 | 3.81 | −0.3454 | −0.3650 | −0.3562 |
| formic_acid | −22.30199 | 3.75 | −0.3755 | −0.3448 | −0.3622 |
| m-cyano-benzoic_acid | −22.29973 | 3.6 | −0.3518 | −0.3764 | −0.3647 |
| p-cyano-benzoic_acid | −22.29935 | 3.55 | −0.3776 | −0.3512 | −0.3651 |
| methoxy-acetic_acid | −22.31280 | 3.54 | −0.3392 | −0.3575 | −0.3493 |
| 3-butynoic_acid | −22.30687 | 3.32 | −0.3485 | −0.3665 | −0.3586 |
| fumaric_acid | −22.30203 | 3.05 | −0.3461 | −0.3749 | −0.3614 |
| bromo-acetic_acid | −22.30017 | 2.86 | −0.3585 | −0.3595 | −0.3721 |
| chloro-acetic_acid | −22.29858 | 2.81 | −0.3549 | −0.3761 | −0.3666 |
| 2-chloro-propionic_acid | −22.30170 | 2.8 | −0.3512 | −0.3574 | −0.3769 |
| fluoro-acetic_acid | −22.29786 | 2.66 | −0.3578 | −0.3697 | −0.3640 |
| cyano-acetic_acid | −22.28694 | 2.44 | −0.3750 | −0.3751 | −0.3781 |
| nitro-acetic_acid | −22.28111 | 1.32 | −0.3826 | −0.3807 | −0.3833 |
| dichloro-acetic_acid | −22.28739 | 1.3 | −0.3664 | −0.3735 | −0.3844 |
| oxalic_acid | −22.28724 | 1.25 | −0.3617 | −0.3853 | −0.3765 |
| difluoro-acetic_acid | −22.28535 | 1.24 | −0.3745 | −0.3736 | −0.3732 |
| trichloro-acetic_acid | −22.28290 | 0.63 | −0.3907 | −0.3656 | −0.3759 |
| trifluoro-acetic_acid | −22.27212 | 0.23 | −0.3770 | −0.4020 | −0.3881 |
| t-butanol | −22.37672 | 18.0 | −0.2766 | −0.2942 | −0.2904 |
| isopropanol | −22.37374 | 17.1 | −0.2817 | −0.2867 | −0.2970 |
| n-propanol | −22.37101 | 16.1 | −0.2748 | −0.2956 | −0.2993 |
| ethanol | −22.37177 | 15.9 | −0.2740 | −0.2962 | −0.2994 |
| methanol | −22.36778 | 15.5 | −0.2875 | −0.2867 | −0.3014 |
| p-amino-phenol | −22.34360 | 10.3 | −0.3147 | −0.3091 | −0.3188 |
| p-methoxy-phenol | −22.33902 | 10.21 | −0.3236 | −0.3096 | −0.3231 |
| p-methyl-phenol | −22.33704 | 10.14 | −0.3223 | −0.3155 | −0.3248 |
| m-methyl-phenol | −22.33582 | 10.08 | −0.3358 | −0.3047 | −0.3259 |
| phenol | −22.33335 | 9.98 | −0.3259 | −0.3197 | −0.3283 |
| p-hydroxy-phenol | −22.33679 | 9.96 | −0.3218 | −0.3157 | −0.3253 |
| p-fluoro-phenol | −22.32713 | 9.95 | −0.3317 | −0.3257 | −0.3344 |
| m-amino-phenol | −22.33835 | 9.87 | −0.3332 | −0.3022 | −0.3233 |
| m-methoxy-phenol | −22.33569 | 9.65 | −0.3359 | −0.3049 | −0.3257 |
| m-hydroxy-phenol | −22.33002 | 9.44 | −0.3410 | −0.3106 | −0.3312 |
| p-chloro-phenol | −22.32374 | 9.38 | −0.3354 | −0.3289 | −0.3374 |
| p-bromo-phenol | −22.32300 | 9.36 | −0.3363 | −0.3296 | −0.3381 |
| m-fluoro-phenol | −22.32288 | 9.28 | −0.3477 | −0.3185 | −0.3380 |
| m-bromo-phenol | −22.32181 | 9.03 | −0.3488 | −0.3198 | −0.3391 |
| m-chloro-phenol | −22.32212 | 9.02 | −0.3490 | −0.3187 | −0.3387 |
| m-cyano-phenol | −22.31146 | 8.61 | −0.3586 | −0.3301 | −0.3490 |
| m-nitro-phenol | −22.30897 | 8.4 | −0.3617 | −0.3323 | −0.3514 |
| p-cyano-phenol | −22.30726 | 7.95 | −0.3451 | −0.3525 | −0.3526 |
| p-nitro-phenol | −22.30165 | 7.15 | −0.3506 | −0.3583 | −0.3576 |
Acknowledgment
This work was supported in part by the National Institute of Health (HL-06350), NSF (FRG DMR-0804549) and the Intramural Research Program of NIH, NIEHS. We acknowledge the use of the computational resources provided by Research Computing Center at University of North Carolina at Chapel Hill and the Biomedical Unit of the Pittsburgh Supercomputer Center.
References
- 1.Jorgensen WL, Briggs JM, Gao JJ. Am. Chem. Soc. 1987;109:6857. [Google Scholar]
- 2.Potter MJ, Gilson MK, McCammon JA. J. Am. Chem. Soc. 1994;116:10298. [Google Scholar]
- 3.Rajasekaran E, Jayaram B, Honig BJ. Am. Chem. Soc. 1994;116:8238. [Google Scholar]
- 4.Alagona G, Ghio C, Kollman PA. J. Am. Chem. Soc. 1995;117:9855. [Google Scholar]
- 5.Jorgensen WL, Briggs JM. J. Am. Chem. Soc. 1989;111:4190. [Google Scholar]
- 6.Lim C, Bashford D, Karplus MJ. Phys. Chem. 1991;95:5610. [Google Scholar]
- 7.Namazian M, Heidary H. Theochem-J. Mol. Struct. 2003;620:257. [Google Scholar]
- 8.Nielsen JE, Mccammon JA. Protein Sci. 2003;12:1894. doi: 10.1110/ps.03114903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fitch CA, Garcia-Moreno B. Biophys. J. 2004;86:86A. [Google Scholar]
- 10.da Silva G, Kennedy EM, Dlugogorski BZ. J Phys Chem A. 2006;110:11371. doi: 10.1021/jp0639243. [DOI] [PubMed] [Google Scholar]
- 11.Ohno K, Sakurai MJ. Comput. Chem. 2006;27:906. doi: 10.1002/jcc.20372. [DOI] [PubMed] [Google Scholar]
- 12.MacDermaid CM, Kaminski GA. J. Phys. Chem. B. 2007;111:9036. doi: 10.1021/jp071284d. [DOI] [PubMed] [Google Scholar]
- 13.Pliego JR. Chem. Phys. Lett. 2003;367:145. [Google Scholar]
- 14.De Proft F, Amira S, Choho K, Geerlings PJ. Phys. Chem. B. 1995;98:5227. [Google Scholar]; Machado HJS, Hinchliffe A. Theochem-J. Mol. Struct. 1995;339:255. [Google Scholar]
- 15.Deka RC, Roy RK, Hirao K. Chem. Phys. Lett. 2004;389:186. [Google Scholar]
- 16.Gupta K, Roy DR, Subramanian V, Chattaraj PK. Theochem-J. Mol. Struct. 2007;812:13. [Google Scholar]
- 17.Nagy P, Novak K, Szasz G. Theochem-J. Mol. Struct. 1989;60:257. [Google Scholar]
- 18.Brinck T, Murray JS, Politzer P, Carter RE. J. Org. Chem. 1991;56:2934. [Google Scholar]
- 19.Gross KC, Seybold PG, Peralta-Inga Z, Murray JS, Politzer PJ. Org. Chem. 2001;66:6919. doi: 10.1021/jo010234g. [DOI] [PubMed] [Google Scholar]
- 20.Ma Y, Gross KC, Hollingsworth CA, Seybold PG, Murray JS. J. Mol. Model. 2004;10:235. [Google Scholar]
- 21.Politzer P. Theor. Chem Acc. 2004;111:395. [Google Scholar]
- 22.Politzer P, Ma YG, Jalbout AF, Murray JS. Mol. Phys. 2005;103:15. [Google Scholar]
- 23.Glendening ED, Badenhoop J,K, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F. NBO 5.0. Theoretical Chemistry Institute, University of Wisconsin; Madison: 2001. [Google Scholar]
- 24.Haiting Lu Xi Chen, Chang-Guo Zhan. J. Phys. Chem. B. 2007;111:10599. doi: 10.1021/jp072917r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liptak Matthew D., Shields George C. J. Am. Chem. Soc. 2001;123:7314. doi: 10.1021/ja010534f. [DOI] [PubMed] [Google Scholar]
- 26.Liptak Matthew D., Gross Kevin C., Seybold Paul G., Feldgus Steven, Shields George C. J. Am. Chem. Soc. 2002;124:6421. doi: 10.1021/ja012474j. [DOI] [PubMed] [Google Scholar]
- 27.Schruumann G, Cossi M, Barone V, Tomasi J. J. Phys. Chem. A. 1998;102:6706. [Google Scholar]
- 28.Gross Kevin C., Seybold Paul G. J. Org. Chem. 2001;66:6919. doi: 10.1021/jo010234g. [DOI] [PubMed] [Google Scholar]
- 29.da Silva Rodrigo R., Ramalho Teodorico C., Santos Joana M., Figueroa-Villar J. Daniel. J. Phys. Chem. A. 2006;110:1031. doi: 10.1021/jp054434y. [DOI] [PubMed] [Google Scholar]
- 30.Chaudry UA, Popelier PLA. J. Org. Chem. 2004;69:233. doi: 10.1021/jo0347415. [DOI] [PubMed] [Google Scholar]
- 31.Lide DR. Handbook of Chemistry and Physics. 88th ed. CRC Press; Boca Raton, New York: 2007. [Google Scholar]
- 32.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr., Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Gaussian, Inc.; Pittsburgh, PA: 2003. revision E.01. [Google Scholar]