

Abstract
The extent to which the BH3-only protein Bid is important for intrinsic (mitochondria-mediated) apoptotic cell death induced by genotoxic stress remains controversial. In the present study, we examine this issue using a panel of gene-manipulated Bax-deficient Jurkat T-lymphocytes. Cells stably depleted of Bid were far less sensitive than control-transfected cells to etoposide-induced apoptosis. In particular, drug-induced Bak activation, cytochrome c release, loss of mitochondrial membrane potential, and caspase activation were all decreased in cells lacking Bid. Reconstitution experiments using recombinant proteins and permeabilized Bid-deficient cells demonstrated that truncated Bid (tBid), but not full-length Bid, potently induced Bak activation and the release of cytochrome c. Further, caspase-8-deficient Jurkat cells efficiently cleaved Bid and were sensitive to drug-induced apoptosis. By comparison, Apaf-1-deficient cells, as well as cells overexpressing full-length X-linked inhibitor of apoptosis protein (XIAP) or the BIR1/BIR2 domains of XIAP, failed to cleave Bid in response to genotoxic stress. These data suggest that tBid plays an important regulatory role in the execution of DNA damage-induced cytochrome c release and apoptosis. However, the fact that cleavage of Bid to tBid is mediated by executioner caspases suggests that a self-amplifying feed forward loop involving caspases, Bid, and mitochondria may help determine irreversible commitment to apoptosis.
Apoptosis is an active form of cell death that plays an essential role during normal embryonic development and in the maintenance of tissue homeostasis in the adult organism (1). Consequently, dysregulation of apoptosis has been implicated as a contributing factor to the onset of different pathological conditions, including cancer. In addition, it is now generally accepted that many genotoxic anticancer drugs are effective against tumor cells for their ability to induce mitochondria-mediated apoptosis (2). Similarly, mutations or the altered expression of pro- and anti-apoptotic proteins can contribute to the development of drug resistance.
Execution of apoptosis is mediated by a family of cysteine-dependent aspartate-specific proteases (caspases). During true mitochondria-mediated apoptosis, members of the Bcl-2 family of proteins are the primary regulators of caspase activation for their role in controlling mitochondrial outer membrane permeabilization (MOMP)2 (3). The process of MOMP results in the release of cytochrome c, second mitochondria-derived activator of caspase (Smac, also known as DIABLO), and Omi (also known as HtrA2) into the cytosol where they converge to promote the activation of caspase-9 within the apoptotic protease-activating factor-1 (Apaf-1) apoptosome complex. The Bcl-2 family contains proteins with opposing functions, and it is generally thought that the induction of MOMP requires the activation of either Bak or Bax triggered by a Bcl-2 homology 3 (BH3)-only protein (4–6). Indeed, evidence in the literature indicates that cells lacking either Bak or Bax exhibit only subtle defects in MOMP, whereas doubly deficient cells are often found to be highly resistant to mitochondria-mediated apoptosis (7, 8).
At present, there are two models for the activation of Bax or Bak by BH3-only proteins. One model argues that BH3-only proteins function as either “sensitizer” (e.g. Bad and Noxa) or “activator” proteins (e.g. truncated Bid (tBid), Bim, and perhaps Puma) (9). In this scenario, a sensitizer protein is needed to displace an activator protein from a prosurvival protein (e.g. Bcl-2, Bcl-xL, or Mcl-1) to activate Bak or Bax. The second model argues that BH3-only proteins bind and inhibit the function of prosurvival Bcl-2 proteins, which normally bind to and inhibit Bak and Bax (10, 11). Of the seven or so known BH3-only proteins (6), Bid is unique in that it requires post-translational modification for activation, most notably involving caspase-8-mediated cleavage to tBid (12–14). Bid normally resides in the cytosol and possibly the nucleus (15). Upon being cleaved, the C-terminal fragment (tBid) is myristoylated at its newly exposed N terminus, translocates to the outer mitochondrial membrane (OMM), and/or activates Bak or Bax protein (16). Recently, it was shown that the N-terminal cleavage fragment of Bid is quickly ubiquitinated for degradation and that this degradation is necessary for the pro-apoptotic function of tBid (17). The same study also concluded that, although full-length Bid is capable of translocating to the OMM, it is not able to induce MOMP on its own (17). A well characterized example of tBid involvement during apoptosis is in the engagement of the mitochondrial apoptotic pathway in so-called type II cells upon activation of the extrinsic pathway (18).
Here, we have investigated whether Bid plays a functional role in the induction of MOMP during apoptosis in response to the genotoxic anticancer drug etoposide. To that end, we used Bax-deficient Jurkat cells that are stably depleted of Bid and evaluated the extent to which these cells underwent drug-induced MOMP. In addition, Jurkat clones in which the intrinsic pathway had been inhibited due to the stable knockdown of Apaf-1 or the overexpression of full-length XIAP or the baculoviral IAP repeats 1 and 2 (BIR1/BIR2) of XIAP were used to gain insight into the molecular requirements necessary for cleavage of Bid to tBid during drug-induced apoptosis. Strikingly, the data showed that etoposide-induced apoptosis was decreased in Bid-deficient Jurkat cells. In particular, cells lacking Bid expression exhibited decreased Bak activation, cytochrome c release, loss of mitochondrial membrane potential (ΔΨ), and caspase activation. Further, incubation of permeabilized Bid-deficient cells with recombinant tBid, but not full-length Bid, induced Bak dimerization and cytochrome c release. Significantly, we also found that cleavage of Bid to tBid occurred strictly downstream of Apaf-1 by a mechanism that required active executioner caspases.
EXPERIMENTAL PROCEDURES
Cell Culture—Wild-type (clones E6.1 and A3) and caspase-8-deficient (clone I 9.2) Jurkat T-lymphocytes were cultured in RPMI 1640 complete medium (Invitrogen) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (HyClone, Logan, UT), 2% (w/v) glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a humidified 5% CO2 incubator. For transfected cells, 1 mg/ml Geneticin (Invitrogen) was substituted for penicillin and streptomycin. Cells were maintained in an exponential growth phase for all experiments. All cells were re-plated in fresh complete nonselective medium prior to apoptosis induction. Apoptosis was induced with etoposide (10 μm, Sigma-Aldrich) or agonistic anti-Fas antibody (100 ng/ml, clone CH-11, MBL International, Woburn, MA). The caspase inhibitor quinoline-Val-Asp-CH2-difluorophenoxy (qVD-OPh, MP Biomedicals, Solon, OH) was used at a final concentration of 20 μm.
Measurement of Caspase Activity—Cells (5 × 105) were pelleted and washed once with ice-cold phosphate-buffered saline (PBS). Cells were resuspended in 25 μl of PBS, added to a microtiter plate, and combined with DEVD-aminomethylcoumarin (Peptide Institute, Osaka, Japan) dissolved in a standard reaction buffer (100 mm Hepes, pH 7.25, 10% sucrose, 10 mm dithiothreitol, 0.1% CHAPS). Cleavage of DEVD-aminomethylcoumarin was monitored by aminomethylcoumarin production in an FLx800 Multi-detection Microplate Reader (BioTek Instruments, Winooski, VT) using 355 nm excitation and 460 nm emission wavelengths.
RNA Interference—The vector-based pSUPER RNAi system (OligoEngine, Seattle, WA) was used to suppress BID gene (RefSeq accession number NM_001196) expression. The gene-specific targeting insert specifies a 19-nucleotide sequence corresponding to nucleotides 35–53 (5′-GGGATGAGTGCATCACAAA-3′) downstream of the transcription start site, which is separated by a 9-nucleotide non-complementary spacer (TTCAAGAGA) from the reverse complement of the same 19-nucleotide sequence. The sequence was ligated into the BglII and XhoI sites of the pSUPER.neo vector, which was subsequently transformed into TOP10 competent cells (Invitrogen) according to the manufacturer's instructions. Several clones were obtained, and the correct insert was verified by sequence analysis.
Transfections—Wild-type Jurkat T-lymphocytes (107) were transfected with 20 μg of plasmid DNA (pSUPER-neo, pSUPER-Apaf-1, pSUPER-Bid, pcDNA3-XIAP, pcDNA3-XIAP-BIR1/BIR2, or pcDNA3-neo) by electroporation using a Bio-Rad Gene Pulser Xcell system (0.4-cm cuvette, 300 V, and 950 microfarads). Cells were allowed to recover in RPMI 1640 complete growth medium for 48 h at 37 °C in a humidified 5% CO2 incubator. Selection of transfected cells was performed in the presence of 1 mg/ml Geneticin for several weeks, at which time serial dilutions were performed to obtain single-cell clones of Bid-silenced cells, Apaf-1-silenced cells, and cells overexpressing full-length XIAP or the BIR1/BIR2 domains of XIAP.
Flow Cytometry for Cell Death and Mitochondrial Membrane Potential (ΔΨ) Measurements—Phosphatidylserine exposure on the outer leaflet of the plasma membrane was detected using the annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit II (BD Pharmingen) according to the manufacturer's instructions. In brief, 106 cells were pelleted following etoposide or anti-Fas treatment and washed in PBS. Next, the cells were resuspended in 100 μl of binding buffer containing annexin V-FITC and propidium iodide. Prior to flow cytometric analysis, 400 μl of binding buffer was added to the cells. For ΔΨ determination, the MitoProbe DiIC1(5) Kit (Invitrogen and Molecular Probes) was used. Briefly, cells (106) were pelleted following drug treatment, washed once in PBS, and resuspended in 1 ml of warm PBS. Next, 5 μl of 10 μm DiIC1(5) were added to the cells, and the mixture was incubated in a humidified 5% CO2 incubator at 37 °C for 15 min. Cells were pelleted, resuspended in 500 μl of PBS, and analyzed by flow cytometry.
Western Blotting—Pelleted cells (5 × 106) were resuspended and lysed in 200 μl of ice-cold lysis buffer (10 mm Tris/HCl, pH 7.4, 10 mm NaCl, 3 mm MgCl2, 1 mm EDTA, 0.1% Nonidet P-40) supplemented with a mixture of protease inhibitors (Complete Mini EDTA-Free, Roche Applied Science). Protein concentrations were determined using the bicinchoninic acid assay (Pierce), and equal amounts were mixed with Laemmli. Western blot analysis was carried out as described previously (19). The antibodies used were rabbit anti-Bak, NT (Millipore), rabbit anti-Bax (Cell Signaling, Danvers, MA), mouse anti-β-actin (clone AC-15, Sigma), rabbit anti-Bid (Cell Signaling), rabbit anti-caspase-3 (clone 8G10, Cell Signaling), rabbit anti-caspase-7 (Cell Signaling), mouse anti-caspase-8 (clone 1C12, Cell Signaling), rabbit anti-caspase-9 (Cell Signaling), mouse anti-cytochrome c (clone 7H8.2C12, BD Pharmingen, San Jose, CA), and rabbit anti-GAPDH (Trevigen, Gaithersburg, MD).
Subcellular Fractionation—Following treatment with etoposide, cells (106) were washed in PBS, resuspended in 50 μl of buffer (140 mm mannitol, 46 mm sucrose, 50 mm KCl, 1 mm KH2PO4, 5 mm MgCl2, 1 mm EGTA, 5 mm Tris, pH 7.4) supplemented with a mixture of protease inhibitors (Complete Mini-EDTA Free) and permeabilized with 3 μg of digitonin (Sigma) on ice for 10 min. Plasma membrane permeabilization was monitored by trypan blue staining, and cell suspensions were centrifuged at 12,000 × g for 10 min at 4 °C. Supernatant and pellet fractions were subjected to Western blot analysis.
Digitonin-permeabilized Cells—Jurkat cells (2.5 × 106) were washed in PBS, resuspended in 125 μl of buffer (140 mm mannitol, 46 mm sucrose, 50 mm KCl, 1 mm KH2PO4, 5 mm MgCl2, 5 mm succinate, 1 mm EGTA, 5 mm Tris, pH 7.4) supplemented with a mixture of protease inhibitors (Complete Mini-EDTA Free). Purified recombinant, truncated or full-length, Bid protein (R & D Systems, Minneapolis, MN) was added at a final concentration ranging from 5 nm to 250 nm. The cells were then permeabilized with 7.5 μg of digitonin (Sigma) and incubated at room temperature (22 °C) for 15 min followed by centrifugation at 12,000 × g for 10 min at 4 °C. Supernatant and pellet fractions were subjected to Western blot analysis.
Determination of Bak Oligomerization and Activation—For detection of Bak oligomerization, cells (106) were harvested, washed, and resuspended in 80 μl of 100 mm EDTA/PBS, incubated with the cross-linking agent bismaleimidohexane (1 mm) for 30 min at room temperature (22 °C), quenched with 100 mm dithiothreitol for 15 min, pelleted, and processed for Western blotting. For detection of activated Bak by flow cytometry, cells (106) were washed in PBS and fixed in 400 μl of 0.25% paraformaldehyde for 5 min, subsequently washed two times with 1% fetal bovine serum in PBS, and incubated in 50 μl of staining buffer (1% fetal bovine serum and 100 μg/ml digitonin in PBS) with a conformation-specific mouse monoclonal antibody against Bak (1:30, AM03, Cal-biochem) for 30 min at room temperature (22 °C). Then, cells were washed and resuspended in 50 μl of staining buffer containing 0.25 μg of Alexa Fluor 488-labeled chicken anti-mouse for 30 min in the dark. Cells were washed again and analyzed by flow cytometry. Analysis and histogram overlays were performed using FlowJo software (Tree Star, Ashland, OR).
RESULTS AND DISCUSSION
Bid Is Cleaved in Response to the DNA-damaging Chemotherapeutic Agent Etoposide—Although it has been reported that overexpression of full-length Bid causes cell death (12) and that full-length Bid can translocate to the OMM in response to an apoptotic stimulus (20–22), it is generally accepted that tBid is far more potent than full-length Bid at activating Bak or Bax to induce MOMP (13, 14, 18). The mechanism responsible for cleavage of Bid to tBid is at least partially understood. Caspase-8-mediated proteolysis of Bid to tBid during receptor-mediated cell killing in type II cells is probably the best characterized example of the involvement of Bid during apoptotic cell death (13, 14). It is also known that cathepsins, calpains, and Granzyme B can cleave Bid, although in most instances the cleavage sites are different from those targeted by caspase-8 (23). It has also been suggested that caspase-3, and possibly caspase-2, can cleave Bid in vitro, although other evidence indicates that Bid is a relatively poor substrate for caspase-2 (24, 25). Irrespective of the mechanism responsible for the cleavage of Bid, it is the C-terminal fragment that promotes apoptosis. Further, caspase-8-mediated cleavage of Bid to tBid exposes a glycine residue at the newly formed N terminus that, in turn, undergoes myristoylation to facilitate translocation of tBid to mitochondria where it promotes MOMP induction (16).
Although tBid is a potent inducer of Bak/Bax-controlled MOMP during receptor-mediated cell killing, whether tBid is an important upstream regulator of MOMP during true intrinsic apoptotic cell death is controversial (15, 26–28). Thus, we set out to determine the extent to which Bid or tBid is functionally important for MOMP induction and apoptosis in response to DNA damage. As illustrated in Fig. 1A, Western blot analysis of Jurkat whole-cell lysates obtained at 6 h following incubation with etoposide (10 μm) revealed that Bid had been cleaved to tBid. Drug-induced cleavage of Bid to tBid was accompanied by an increase in the percentage of cells undergoing apoptosis (28%) as determined by annexin V-FITC and propidium iodide co-staining (Fig. 1B). Further, in agreement with our previous findings, incubation of cells with 10 μm etoposide for 6 h resulted in the proteolytic cleavage of caspase-9, -3, and -7, and a concomitant increase in caspase (DEVDase) activity (Fig. 1, C and D).
FIGURE 1.

Etoposide-induced Bid cleavage and apoptosis in wild-type Jurkat T cells. A, wild-type cells (106/ml) were cultured with DMSO or 10 μm etoposide for 6 h, harvested, and lysed for Western blotting. B–D, duplicate aliquots of cells in A were harvested and processed for cell death determination by flow cytometric analysis of annexin V-FITC and propidium iodide staining (B), Western blotting of caspases (C), or caspase (DEVDase) activity measurements (D). In B, quadrants are defined as live (lower left), early apoptotic (lower right), late apoptotic (upper right), and necrotic (upper left). Numbers refer to the percentage of cells in each quadrant. Casp, caspase; RFU, relative fluorescence units.
Stable Knockdown of Bid Desensitizes Jurkat Cells to Genotoxic Drug-induced Apoptosis—Having shown that tBid is generated in wild-type Jurkat cells undergoing DNA damage-induced apoptosis, we next used a vector-based system that directs the synthesis of short hairpin RNA (shRNA) to produce stable silencing of Bid. We hypothesized that such cells would shed light on whether Bid was important for genotoxic stress-induced apoptosis or if cleavage of Bid to tBid (Fig. 1A) was largely a bystander event in this setting with little biological significance.
Two single-cell Bid-deficient Jurkat clones (#7 and #13) were used for these studies, and the extent to which Bid was suppressed was confirmed by Western blot analysis (Fig. 2A). As mentioned previously, the requirement for Bid cleavage to tBid upon activation of the extrinsic pathway in type II cells has been well characterized and is widely accepted (13, 14, 29). Because Jurkat cells are considered to be of type II origin, we first tested the sensitivity of these cells to apoptosis induced by agonistic anti-Fas antibody (100 ng/ml). As anticipated, Bid-deficient cells were highly resistant to anti-Fas-induced apoptosis with clone #13 exhibiting the greatest degree of resistance as assessed by annexin V-FITC and propidium iodide co-staining (Fig. 2B) and proteolytic processing of caspases (Fig. 2C, lanes 6 and 9 versus 3). Next, having demonstrated that Bid was strictly required for receptor-mediated apoptosis in Jurkat cells, we incubated the cells for 6 h in the presence of etoposide (10 μm) and evaluated the different clones for their sensitivity to apoptosis induction. The results indicated that both Bid-deficient clones were less sensitive than control-transfected cells to etoposide-induced apoptosis with the degree of resistance paralleling the extent to which Bid expression had been suppressed (Fig. 2B). In agreement with these findings, Western blot analysis of cell lysates obtained at 6 h post-etoposide treatment showed that proteolytic processing of caspase-9, -3, and -7 was decreased in Bid-deficient cells (Fig. 2C, lanes 5 and 8 versus 2). Overall, these data suggest that Bid plays an important function during genotoxic drug-induced apoptosis in Jurkat cells.
FIGURE 2.

Bid-deficient Jurkat cells are less susceptible to etoposide-induced apoptosis. A, vector control or two single-cell Bid-deficient Jurkat clones (#7 and #13) were harvested and lysed for Western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. B, cells (106/ml) were cultured in the presence or absence of DMSO, 10 μm etoposide, or 100 ng/ml agonistic anti-Fas antibody for 6 h, harvested, and processed for cell death determination by flow cytometric analysis of annexin V-FITC and propidium iodide staining. Quadrants are defined as live (lower left), early apoptotic (lower right), late apoptotic (upper right), and necrotic (upper left). Numbers refer to the percentage of cells in each quadrant. C, duplicate aliquots of cells in B were harvested and lysed for Western blotting. shRNA, short hairpin RNA; Casp, caspase.
DNA Damage-induced Mitochondrial Events Are Attenuated in Bid-deficient Jurkat Cells—Studies from numerous laboratories have shown that MOMP is tightly regulated by pro- and anti-apoptotic proteins of the Bcl-2 family (6). In particular, MOMP is widely accepted to require the activation of a multidomain protein, notably Bak or Bax. The presence and activation of either Bak or Bax is thought to be sufficient to induce MOMP. In fact, because the deletion of either gene alone produces only subtle defects in apoptosis, whereas doubly-deficient cells exhibit severe apoptosis deficiencies (7), Bak and Bax are generally thought to be redundant in function. The activation of Bak and Bax coincides with their homo-oligomerization and in most instances is thought to depend on the presence and prior activation of a BH3-only Bcl-2 family member. However, the mechanism by which BH3-only proteins activate Bak or Bax is controversial (30). As mentioned previously, one model suggests that BH3-only proteins activate Bak and Bax directly, whereas the second model indicates BH3-only proteins activate Bak and Bax indirectly by neutralizing the anti-apoptotic function of pro-survival Bcl-2 family proteins.
Because we had observed that etoposide-induced apoptosis was decreased in Bid-deficient Jurkat cells, we next evaluated mitochondrial events that characteristically define MOMP, such as the dimerization and activation of Bak or Bax, the release of intermembrane space proteins, and the loss of ΔΨ. In a recent paper, we showed that Jurkat (E6.1) cells express Bak, but not Bax, protein (31). Those findings were confirmed in Fig. 3A, and we also determined that silencing Bid had no effect on the expression level of Bak protein (lane 4 versus 3 and 2). Therefore, we next examined the extent to which Bak had undergone oligomerization in response to etoposide, using the cross-linking agent bismaleimidohexane. As illustrated in Fig. 3B, cross-linked complexes consistent with Bak dimers were detected in control-transfected cells following treatment with 10 μm etoposide, whereas the extent of drug-induced dimerization was markedly decreased in the Bid-deficient cells as illustrated in lane 4 of Fig. 3B. These findings were in line with results obtained using an active conformation-specific monoclonal Bak antibody and flow cytometric analysis where activation causes a shift to the right of the resulting histogram (Fig. 3C).
FIGURE 3.

Inhibition of etoposide-induced mitochondrial apoptotic changes in cells lacking Bid. A, whole-cell lysates of Jurkat (E6.1) and MCF-7 (positive control for Bax expression) cells were subjected to SDS-PAGE and Western blotted. B and C, cells (106/ml) were cultured with DMSO or 10 μm etoposide for 6 h and processed for determination of Bak oligomerization by Western blotting (B) or Bak activation by flow cytometric analysis (C). Numbers in C refer to the percentage increase in Bak-associated fluorescence between DMSO- and etoposide-treated samples. D, duplicate aliquots of cells in B and C were harvested and processed for subcellular fractionation. Supernatant (s) and pellet (p) fractions were analyzed by Western blotting. E, duplicate aliquots of cells in B and C were harvested and processed for mitochondrial membrane potential (ΔΨ) determination by flow cytometry. Reduced DiIC1(5) fluorescence is indicative of a loss of ΔΨ, and numbers refer to the percentage of cells that underwent a dissipation of ΔΨ. shRNA, short hairpin RNA; BMH, bismaleimidohexane; Cyt c, cytochrome c.
Next, consistent with the inhibition of Bak activation in cells lacking Bid, the release of cytochrome c in response to etoposide was also markedly inhibited (Fig. 3D). Finally, as illustrated in Fig. 3E, the loss of ΔΨ in response to etoposide was also decreased in the Bid-deficient cell line (Fig. 3E). Combined, these data strongly suggest that Bid functions to promote Bak-controlled MOMP during DNA damage-induced apoptosis in Jurkat cells.
Cleaved Bid (tBid) Mediates MOMP and Is Generated Independently of Caspase-8 in Response to DNA Damage—A previous study reported that the expression of sublethal levels of wild-type or uncleavable Bid in bid-/- mouse embryonic fibroblasts resulted in a similar sensitization of cells to DNA damage-induced apoptosis that was accompanied by an enhanced localization of Bid to mitochondria and release of cytochrome c (20). In this regard, because we had demonstrated that cells lacking Bid were resistant to drug-induced apoptosis, we next sought to determine if the phenotype we observed was due to the absence of full-length Bid or tBid. To address this issue, we made several attempts to rescue the phenotype of our Bid-deficient cells by reintroducing wild-type human Bid and mutants of Bid that were uncleavable or unable to be myristoylated. Although it had been reported previously that overexpression of Bid is lethal to Jurkat cells (12), we reasoned that reintroduction of wild-type or mutant Bid into cells that were deficient in this protein would yield viable cell clones. Surprisingly, however, all attempts to express wild-type or mutant Bid into Bid-silenced cells were unsuccessful, because we found that cells simply could not be transfected with any form of Bid and remain viable. Because our efforts to rescue the phenotype in whole cells failed, we next performed experiments in which we tried to reconstitute the phenotype using permeabilized cells. Specifically, recombinant full-length and caspase-8-cleaved Bid were used to treat permeabilized Bid-deficient Jurkat cells to evaluate more precisely whether Bid, tBid, or both could promote the engagement of the mitochondrial pathway. As shown in Fig. 4A, when Bid-deficient Jurkat cells were permeabilized with digitonin and incubated with 5, 10, 25, 50, 100, or 250 nm recombinant tBid for 15 min at 22 °C, cytochrome c was released into the cytosol. By comparison, full-length Bid failed to stimulate the release of any cytochrome c, even at the highest concentration of 250 nm. Consistent with the cytochrome c release data, only tBid (25 nm) induced the oligomerization of Bak when added to digitonin-permeabilized Bid-deficient cells (Fig. 4B). These findings indicate that tBid, and not full-length Bid, is most probably responsible for drug-induced activation of Bak and the induction of MOMP in response to genotoxic stress.
FIGURE 4.

tBid, but not Bid, mediates MOMP and is generated independently of caspase-8 in response to etoposide. A, Bid-deficient (clone #13) Jurkat cells (2.5 × 106) were permeabilized with 7.5 μg of digitonin and incubated with recombinant tBid (top panels) or Bid (bottom panels) protein (5–250 nm) at 22 °C for 15 min. Subsequently, supernatant (s) and pellet (p) fractions were obtained, subjected to SDS-PAGE, and Western blotted. B, digitonin-permeabilized Bid-deficient cells as in A were incubated in the presence or absence of either 25 nm tBid or Bid and processed for determination of Bak oligomerization by Western blotting. C, wild-type cells (106/ml) were preincubated in the presence or absence of 20 μm qVD-OPh for 1 h and subsequently incubated in the presence or absence of DMSO or 10 μm etoposide for an additional 6 h at which time cells were harvested and processed for Western blot analysis. D, whole-cell lysates of wild-type (A3) and caspase-8-deficient (I9.2) Jurkat cells were subjected to SDS-PAGE and Western blotted. E and F, cells (106/ml) were incubated in the absence or presence of DMSO, 10 μm etoposide, or 100 ng/ml agonistic anti-Fas antibody for 6 h, harvested, and lysed for Western blotting (E) or processed for cell death determination by flow cytometric analysis of annexin V-FITC and propidium iodide staining (F). Quadrants in F are defined as live (lower left), early apoptotic (lower right), late apoptotic (upper right), and necrotic (upper left). Numbers refer to the percentage of cells in each quadrant. shRNA, short hairpin RNA; Cyt c, cytochrome c; Conc, concentration; BMH, bismaleimidohexane; Casp, caspase.
As mentioned previously, it is widely accepted that caspase-8-mediated cleavage of Bid is required for execution of extrinsic apoptotic cell death in type II cells. It has also been suggested that other caspases may have the ability to cleave Bid, such as caspase-2 (24, 25) and caspase-3 (32, 33). Additional lines of evidence have shown that non-caspase proteases, such as calpains (34, 35) and cathepsins (36, 37), are also capable of cleaving full-length Bid. Furthermore, a different study reported that tBid is important during DNA damage-induced apoptosis and that the cleavage of Bid to tBid in this context was not caspase-dependent. Instead, cleavage was suggested to occur by an unidentified aspartate protease (38). The same study concluded that the importance of tBid for genotoxic stress-induced apoptosis was that it functioned to promote MOMP upstream of caspase activation.
To determine if the cleavage of Bid to tBid was a caspase-mediated event in our experimental system, we incubated wild-type Jurkat cells with the caspase inhibitor qVD-OPh (20 μm) for 1 h prior to the addition of etoposide (10 μm). As illustrated in Fig. 4C, pretreatment of cells with qVD-OPh completely prevented drug-induced generation of tBid. Next, because of early reports indicating that caspase-8 is important during DNA damage-induced apoptosis (39–41) and because caspase-8 is the best characterized protease responsible for the cleavage of Bid, we used caspase-8-deficient Jurkat cells (Fig. 4D) to determine whether caspase-8 is necessary for mitochondria-mediated apoptosis and/or Bid cleavage. As expected, these cells were highly resistant to receptor-mediated apoptosis induced by anti-Fas (Fig. 4F). By comparison, caspase-8-deficient cells underwent etoposide-induced apoptosis to the same extent as A3 control cells (Fig. 4F), suggesting that caspase-8 is dispensable for etoposide-induced apoptosis. Furthermore, cleavage of Bid to tBid was prevented in caspase-8 null cells incubated in the presence of anti-Fas, whereas Bid was cleaved to a similar extent in caspase-8-deficient and A3 control cells treated with etoposide (Fig. 4E). Combined, these data suggest that caspase-8 is dispensable for etoposide-induced cleavage of Bid to tBid.
Executioner Caspases Mediate Bid Cleavage During Etoposide-induced Apoptosis—To extend these findings and to determine whether drug-induced Bid cleavage was mediated by a caspase other than caspase-8, we first used Apaf-1-deficient Jurkat cells described previously that are totally resistant to etoposide-induced apoptosis (42). As illustrated in Fig. 5A, cleavage of Bid to tBid did not occur in Apaf-1-deficient cells incubated in the presence of 10 μm etoposide for 6 h. Because Bid cleavage appeared to require apoptosome-dependent caspase activation, we were next interested to determine the extent to which this was mediated by an initiator or executioner caspase protease. To distinguish between these two possibilities, we used Jurkat cells described recently that overexpress either full-length XIAP or the BIR1/BIR2 domains of XIAP (31). Full-length XIAP inhibits apoptosis by preventing caspase-9, -3, and -7 activity, whereas the BIR1/BIR2 domains of XIAP can inhibit caspase-3 and -7, but not caspase-9 (43, 44). During either mitochondria-mediated apoptosis or receptor-mediated apoptosis in type II cells, the prosurvival activity of XIAP is neutralized by Smac and Omi, which are released into the cytosol from the intermembrane space of mitochondria as a consequence of MOMP. In agreement with our previous findings (31), Jurkat cells that overexpressed either full-length XIAP or the BIR1/BIR2 domains of XIAP were impaired in their ability to activate caspases and undergo apoptosis, exhibiting 7 and 12% apoptotic cells, respectively, in response to etoposide (data not shown). Western blot analysis of duplicate cell aliquots revealed that control-transfected, but not XIAP- or BIR1/BIR2-transfected, cells had cleaved Bid in response to etoposide treatment (Fig. 5B). Importantly, the absence of etoposide-induced Bid cleavage in cells overexpressing either XIAP or the BIR1/BIR2 domains of XIAP was accompanied by a marked decrease in Bak activation in these cells incubated under the same conditions (Fig. 5C). Taken together, these data suggest that etoposide-induced cleavage of Bid to tBid and tBid-mediated activation of Bak occur by a mechanism that requires the prior activation of executioner caspases.
FIGURE 5.

Cleavage of Bid in response to etoposide is mediated by executioner caspases. A, wild-type, control-transfected, and Apaf-1-deficient Jurkat cells (106/ml) were incubated in the presence or absence of DMSO or 10 μm etoposide for 6 h, harvested, and lysed for Western blotting. B, control-transfected, XIAP-overexpressing, and Myc-BIR1/BIR2-expressing Jurkat cells (106/ml) were incubated in the presence or absence of DMSO or 10 μm etoposide for 6 h, harvested, and lysed for Western blotting. C, duplicate aliquots of cells in B were processed for determination of Bak activation by flow cytometric analysis. Numbers in C refer to the percentage increase in Bak-associated fluorescence between DMSO- and etoposide-treated samples. shRNA, short hairpin RNA.
Concluding Remarks—A key step during true intrinsic apoptosis is the induction of MOMP mediated by an active multidomain pro-apoptotic protein Bak or Bax (30). In fact, it is generally accepted that the presence and activation of either Bak or Bax are strictly required for MOMP. BH3-only proteins are most often responsible for the activation of Bak and Bax, which involves their homo-oligomerization and insertion into the OMM to form pores through which intermembrane space proteins, such as cytochrome c and Smac, are released into the cytosol to activate caspases. As mentioned previously, the best characterized example of a BH3-only protein being responsible for the activation of a multidomain protein is during receptor-mediated apoptosis in type II cells where tBid activates Bak/Bax to induce MOMP. A similar emergent role for a BH3-only protein in the activation of Bak or Bax during true intrinsic apoptotic cell death is currently lacking.
Some evidence in the literature indicates that the activation of Bak or Bax following DNA damage can be mediated by p53-dependent up-regulation of the BH3-only protein Puma (45, 46). However, Jurkat cells, which are highly susceptible to DNA damage-induced apoptosis, possess mutant p53 (47). In addition, cycling T lymphoma cells and mitogenically activated T lymphocytes from p53-/- mice were shown to be sensitive to genotoxic stress-induced apoptosis (48). In this regard, p53-induced up-regulation of Puma is unlikely to be the sole mediator of Bak/Bax-dependent apoptosis in response to DNA damage. In fact, accumulating evidence in the literature suggests that the activation of Bak or Bax during true intrinsic apoptosis requires the presence of either Bid or Bim, irrespective of p53 status (49).
Recent studies investigating a potential role for Bid during DNA damage-induced apoptosis have produced conflicting results (15, 26, 27). On one hand, embryonic fibroblasts and myeloid progenitor cells from Bid-deficient mice were shown to be less susceptible to etoposide-induced apoptosis (15, 27), whereas another study reported that Bid plays no role in DNA damage-induced apoptosis (26). The fact that each of these studies used cells that were cultivated from bid-/- mice on a C57BL/6 background makes it difficult to reconcile the different findings in the absence of additional experimentation.
As mentioned previously, a separate study using Jurkat T cells suggested that tBid was required for apoptosis signaling during DNA damage-induced apoptosis (38). The authors also concluded that Bid cleavage occurred independently of caspase activity. Rather, it was speculated that an unidentified aspartate protease was responsible for the cleavage of Bid to tBid, which, in turn, was critical for MOMP and the activation of all caspases.
Our findings using a panel of gene-manipulated Jurkat cells also support a role for cleavage of Bid to tBid in eliciting cytochrome c release and apoptosis in response to etoposide. However, unlike Werner et al. (38), our data suggest that most, if not all, Bid cleavage occurs downstream of MOMP by a mechanism that depends on active executioner caspases (Fig. 6). In this regard, our current data support and extend a model of the intrinsic pathway in which downstream caspases and mitochondria forge a circuit of positive amplification to ensure irreversible commitment to apoptosis. Indeed, it is tempting to speculate that some cell types may be spared death if feed forward amplification of early mitochondrial apoptotic events is inhibited. We envision a scenario where genotoxic stress triggers initial MOMP and the release of intermembrane space proteins by activating Bak or Bax by an as yet poorly understood mechanism that may or may not involve a BH3-only protein (Fig. 6). The initial induction of MOMP would not necessarily mark the “point of no return” for an injured cell unless, or until, the release of intermembrane space proteins, including cytochrome c, exceeded a threshold necessary to trigger apoptosome formation resulting in caspase-9 activation and the subsequent activation of caspase-3/7. In turn, active caspase-3 (or -7) would mediate feed forward amplification of initial MOMP by cleaving Bid to tBid to increase the activation of Bak or Bax.
FIGURE 6.
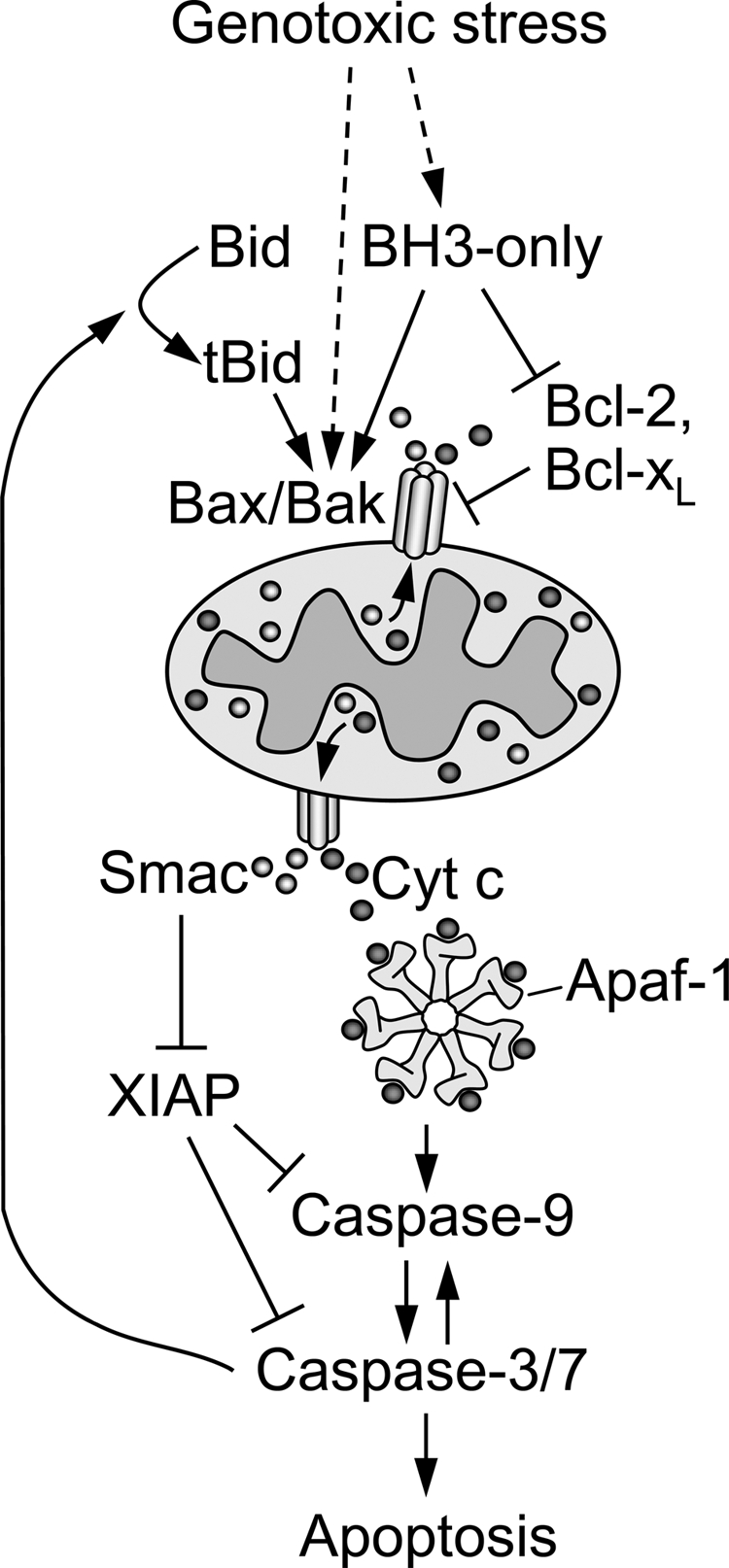
Hypothetical scheme of etoposide-induced apoptosis. Genotoxic stress in response to etoposide induces initial MOMP by generating a still poorly defined death signal that activates Bax/Bak. As a consequence of early MOMP, intermembrane space proteins, including cytochrome c and Smac, are released into the cytosol. The initial release of these proteins into the cytosol per se is not an irreversible commitment point to apoptotic cell death. Instead, the point of commitment occurs when the accumulation of Smac is sufficient to inhibit XIAP-mediated inhibition of caspase-9, -3, and -7 and/or the accumulation of cytochrome c surpasses a threshold needed for the conversion of sufficient numbers of monomeric Apaf-1 molecules to heptameric apoptosome signaling platforms necessary for initial caspase-9 activation. The ensuing caspase-9 activity would need to be sufficiently strong to activate enough executioner caspase-3 or -7 molecules to kill a cell outright and/or to elicit feed forward amplification of MOMP by cleaving Bid to tBid. BH3, Bcl-2 homology 3; Cyt c, cytochrome c.
Acknowledgments
We are grateful to Beth Walsh for technical help and Dr. Joyce Slusser for invaluable assistance with flow cytometry. We thank Dr. Emad Alnemri for the pcDNA3-XIAP and pcDNA3-myc-BIR1/BIR2 mammalian expression vectors.
This work was supported, in whole or in part, by National Institutes of Health Grants K22 ES011647 (to J. D. R.), P20 RR016475 from the IDeA Networks of Biomedical Research Excellence Program of the National Center for Research Resources, and T32 ES007079. The Flow Cytometry Core is supported in part by NIH Grant P20 RR016443 from the NCRR.
Footnotes
The abbreviations used are: MOMP, mitochondrial outer membrane permeabilization; Smac, second mitochondria-derived activator of caspase; Apaf-1, apoptotic protease activating factor-1; BH3, Bcl-2 homology 3; tBid, truncated Bid; OMM, outer mitochondrial membrane; ΔΨ, mitochondrial membrane potential; XIAP, X-linked inhibitor of apoptosis protein; BIR, baculoviral inhibitor of apoptosis protein repeat; PBS, phosphate-buffered saline; FITC, fluorescein isothiocyanate; qVD-OPh, quinoline-Val-Asp-CH2-difluorophenoxy; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid; shRNA, short hairpin RNA.
References
- 1.Danial, N. N., and Korsmeyer, S. J. (2004) Cell 116, 205-219 [DOI] [PubMed] [Google Scholar]
- 2.Lowe, S. W., and Lin, A. W. (2000) Carcinogenesis 21, 485-495 [DOI] [PubMed] [Google Scholar]
- 3.Adams, J. M., and Cory, S. (1998) Science 281, 1322-1326 [DOI] [PubMed] [Google Scholar]
- 4.Chipuk, J. E., Bouchier-Hayes, L., and Green, D. R. (2006) Cell Death Differ. 13, 1396-1402 [DOI] [PubMed] [Google Scholar]
- 5.Ow, Y. L., Green, D. R., Hao, Z., and Mak, T. W. (2008) Nat. Rev. Mol. Cell Biol. 9, 532-542 [DOI] [PubMed] [Google Scholar]
- 6.Youle, R. J., and Strasser, A. (2008) Nat. Rev. Mol. Cell Biol. 9, 47-59 [DOI] [PubMed] [Google Scholar]
- 7.Lindsten, T., Ross, A. J., King, A., Zong, W. X., Rathmell, J. C., Shiels, H. A., Ulrich, E., Waymire, K. G., Mahar, P., Frauwirth, K., Chen, Y., Wei, M., Eng, V. M., Adelman, D. M., Simon, M. C., Ma, A., Golden, J. A., Evan, G., Korsmeyer, S. J., MacGregor, G. R., and Thompson, C. B. (2000) Mol. Cell 6, 1389-1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei, M. C., Zong, W. X., Cheng, E. H., Lindsten, T., Panoutsakopoulou, V., Ross, A. J., Roth, K. A., MacGregor, G. R., Thompson, C. B., and Korsmeyer, S. J. (2001) Science 292, 727-730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim, H., Rafiuddin-Shah, M., Tu, H. C., Jeffers, J. R., Zambetti, G. P., Hsieh, J. J., and Cheng, E. H. (2006) Nat. Cell Biol. 8, 1348-1358 [DOI] [PubMed] [Google Scholar]
- 10.Chen, L., Willis, S. N., Wei, A., Smith, B. J., Fletcher, J. I., Hinds, M. G., Colman, P. M., Day, C. L., Adams, J. M., and Huang, D. C. (2005) Mol. Cell 17, 393-403 [DOI] [PubMed] [Google Scholar]
- 11.Willis, S. N., Fletcher, J. I., Kaufmann, T., van Delft, M. F., Chen, L., Czabotar, P. E., Ierino, H., Lee, E. F., Fairlie, W. D., Bouillet, P., Strasser, A., Kluck, R. M., Adams, J. M., and Huang, D. C. (2007) Science 315, 856-859 [DOI] [PubMed] [Google Scholar]
- 12.Wang, K., Yin, X. M., Chao, D. T., Milliman, C. L., and Korsmeyer, S. J. (1996) Genes Dev. 10, 2859-2869 [DOI] [PubMed] [Google Scholar]
- 13.Li, H., Zhu, H., Xu, C. J., and Yuan, J. (1998) Cell 94, 491-501 [DOI] [PubMed] [Google Scholar]
- 14.Luo, X., Budihardjo, I., Zou, H., Slaughter, C., and Wang, X. (1998) Cell 94, 481-490 [DOI] [PubMed] [Google Scholar]
- 15.Zinkel, S. S., Hurov, K. E., Ong, C., Abtahi, F. M., Gross, A., and Korsmeyer, S. J. (2005) Cell 122, 579-591 [DOI] [PubMed] [Google Scholar]
- 16.Zha, J., Weiler, S., Oh, K. J., Wei, M. C., and Korsmeyer, S. J. (2000) Science 290, 1761-1765 [DOI] [PubMed] [Google Scholar]
- 17.Tait, S. W., de Vries, E., Maas, C., Keller, A. M., D'Santos, C. S., and Borst, J. (2007) J. Cell Biol. 179, 1453-1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yin, X. M., Wang, K., Gross, A., Zhao, Y., Zinkel, S., Klocke, B., Roth, K. A., and Korsmeyer, S. J. (1999) Nature 400, 886-891 [DOI] [PubMed] [Google Scholar]
- 19.Robertson, J. D., Enoksson, M., Suomela, M., Zhivotovsky, B., and Orrenius, S. (2002) J. Biol. Chem. 277, 29803-29809 [DOI] [PubMed] [Google Scholar]
- 20.Sarig, R., Zaltsman, Y., Marcellus, R. C., Flavell, R., Mak, T. W., and Gross, A. (2003) J. Biol. Chem. 278, 10707-10715 [DOI] [PubMed] [Google Scholar]
- 21.Tafani, M., Karpinich, N. O., Hurster, K. A., Pastorino, J. G., Schneider, T., Russo, M. A., and Farber, J. L. (2002) J. Biol. Chem. 277, 10073-10082 [DOI] [PubMed] [Google Scholar]
- 22.Valentijn, A. J., and Gilmore, A. P. (2004) J. Biol. Chem. 279, 32848-32857 [DOI] [PubMed] [Google Scholar]
- 23.Yin, X. M. (2006) Gene (Amst.) 369, 7-19 [DOI] [PubMed] [Google Scholar]
- 24.Bonzon, C., Bouchier-Hayes, L., Pagliari, L. J., Green, D. R., and Newmeyer, D. D. (2006) Mol. Biol. Cell 17, 2150-2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo, Y., Srinivasula, S. M., Druilhe, A., Fernandes-Alnemri, T., and Alnemri, E. S. (2002) J. Biol. Chem. 277, 13430-13437 [DOI] [PubMed] [Google Scholar]
- 26.Kaufmann, T., Tai, L., Ekert, P. G., Huang, D. C., Norris, F., Lindemann, R. K., Johnstone, R. W., Dixit, V. M., and Strasser, A. (2007) Cell 129, 423-433 [DOI] [PubMed] [Google Scholar]
- 27.Kamer, I., Sarig, R., Zaltsman, Y., Niv, H., Oberkovitz, G., Regev, L., Haimovich, G., Lerenthal, Y., Marcellus, R. C., and Gross, A. (2005) Cell 122, 593-603 [DOI] [PubMed] [Google Scholar]
- 28.Zinkel, S. S., Hurov, K. E., and Gross, A. (2007) Cell 130, 9-10; author reply 10–11 [DOI] [PubMed] [Google Scholar]
- 29.Scaffidi, C., Fulda, S., Srinivasan, A., Friesen, C., Li, F., Tomaselli, K. J., Debatin, K. M., Krammer, P. H., and Peter, M. E. (1998) EMBO J. 17, 1675-1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chipuk, J. E., and Green, D. R. (2008) Trends Cell Biol. 18, 157-164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shawgo, M. E., Shelton, S. N., and Robertson, J. D. (2008) J. Biol. Chem. 283, 35532-35538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bossy-Wetzel, E., and Green, D. R. (1999) J. Biol. Chem. 274, 17484-17490 [DOI] [PubMed] [Google Scholar]
- 33.Slee, E. A., Keogh, S. A., and Martin, S. J. (2000) Cell Death Differ. 7, 556-565 [DOI] [PubMed] [Google Scholar]
- 34.Chen, M., He, H., Zhan, S., Krajewski, S., Reed, J. C., and Gottlieb, R. A. (2001) J. Biol. Chem. 276, 30724-30728 [DOI] [PubMed] [Google Scholar]
- 35.Mandic, A., Viktorsson, K., Strandberg, L., Heiden, T., Hansson, J., Linder, S., and Shoshan, M. C. (2002) Mol. Cell. Biol. 22, 3003-3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stoka, V., Turk, B., Schendel, S. L., Kim, T. H., Cirman, T., Snipas, S. J., Ellerby, L. M., Bredesen, D., Freeze, H., Abrahamson, M., Bromme, D., Krajewski, S., Reed, J. C., Yin, X. M., Turk, V., and Salvesen, G. S. (2001) J. Biol. Chem. 276, 3149-3157 [DOI] [PubMed] [Google Scholar]
- 37.Droga-Mazovec, G., Bojic, L., Petelin, A., Ivanova, S., Romih, R., Repnik, U., Salvesen, G. S., Stoka, V., Turk, V., and Turk, B. (2008) J. Biol. Chem. 283, 19140-19150 [DOI] [PubMed] [Google Scholar]
- 38.Werner, A. B., Tait, S. W., de Vries, E., Eldering, E., and Borst, J. (2004) J. Biol. Chem. 279, 28771-28780 [DOI] [PubMed] [Google Scholar]
- 39.Friesen, C., Herr, I., Krammer, P. H., and Debatin, K. M. (1996) Nat. Med. 2, 574-577 [DOI] [PubMed] [Google Scholar]
- 40.Kasibhatla, S., Brunner, T., Genestier, L., Echeverri, F., Mahboubi, A., and Green, D. R. (1998) Mol. Cell 1, 543-551 [DOI] [PubMed] [Google Scholar]
- 41.Petak, I., Tillman, D. M., Harwood, F. G., Mihalik, R., and Houghton, J. A. (2000) Cancer Res. 60, 2643-2650 [PubMed] [Google Scholar]
- 42.Franklin, E. E., and Robertson, J. D. (2007) Biochem. J. 405, 115-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott, F. L., Denault, J. B., Riedl, S. J., Shin, H., Renatus, M., and Salvesen, G. S. (2005) EMBO J. 24, 645-655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shiozaki, E. N., Chai, J., Rigotti, D. J., Riedl, S. J., Li, P., Srinivasula, S. M., Alnemri, E. S., Fairman, R., and Shi, Y. (2003) Mol. Cell 11, 519-527 [DOI] [PubMed] [Google Scholar]
- 45.Jeffers, J. R., Parganas, E., Lee, Y., Yang, C., Wang, J., Brennan, J., MacLean, K. H., Han, J., Chittenden, T., Ihle, J. N., McKinnon, P. J., Cleveland, J. L., and Zambetti, G. P. (2003) Cancer Cell 4, 321-328 [DOI] [PubMed] [Google Scholar]
- 46.Villunger, A., Michalak, E. M., Coultas, L., Mullauer, F., Bock, G., Ausserlechner, M. J., Adams, J. M., and Strasser, A. (2003) Science 302, 1036-1038 [DOI] [PubMed] [Google Scholar]
- 47.Cheng, J., and Haas, M. (1990) Mol. Cell. Biol. 10, 5502-5509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strasser, A., Harris, A. W., Jacks, T., and Cory, S. (1994) Cell 79, 329-339 [DOI] [PubMed] [Google Scholar]
- 49.Letai, A. G. (2008) Nat. Rev. Cancer 8, 121-132 [DOI] [PubMed] [Google Scholar]