

Abstract
The detection of biomarkers of oxidative stress in brain tissue and cerebrospinal fluid of patients with human immunodeficiency virus, type 1 (HIV)-associated dementia indicates the involvement of stress pathways in the neuropathogenesis of AIDS. Although the biological importance of oxidative stress on events involved in AIDS neuropathogenesis and the HIV-1 proteins responsible for oxidative stress remain to be elucidated, our results point to the activation of hypoxia-inducible factor 1 (HIF-1) upon HIV-1 infection and its elevation in brain cells of AIDS patients with dementia. HIF-1 is a transcription factor that is responsive to oxygen. Under hypoxic conditions, HIF-1α becomes stable and translocates to the nucleus where it dimerizes with aryl hydrocarbon receptor nuclear translocator and modulates gene transcription. Activation of HIF-1 can also be mediated by the HIV-1 accessory protein Vpr. In addition, cellular components, including reactive oxygen species, contribute to the induction of HIF-1α. Our results show that Vpr induces reactive oxygen species by increasing H2O2 production, which can contribute to HIF-1α accumulation. Interestingly, increased levels of HIF-1α stimulated HIV-1 gene transcription through HIF-1 association with HIV-1 long terminal repeat. These observations point to the existence of a positive feedback interplay between HIF-1α and Vpr and that, by inducing oxidative stress via activation of HIF-1, Vpr can induce HIV-1 gene expression and dysregulate multiple host cellular pathways.
Oxidative stress (OS)5 is a phenomenon occurring in cells when production of oxygen radicals exceeds antioxidant capacity (1). An excess of free radicals damages essential macromolecules of the cell, leading to abnormal gene expression, disturbances in receptor activity, cell proliferation, immune perturbation, or cell death. OS is the major player in numerous human diseases such as cancer, ocular degeneration, and neurodegenerative diseases, including AIDS-associated neuropathies (2).
OS is involved in many aspects of HIV disease pathogenesis, including viral replication (3), inflammatory responses, decreased immune cell proliferation, loss of immune function, chronic weight loss, and increased sensitivity to drug toxicity (4). Active replication of HIV in macrophages and microglia represents a reservoir for virus and an important step for HIV neuropathogenesis (5). This process leads to production of inflammatory products and free radical species (6). Because macrophages and microglia function as a long term reservoir for HIV-1, these cells must possess a mechanism to protect themselves against the toxic effects of superoxide anions. In this regard, HIV-1 Tat has been shown to induce neuronal death via TNF-α induction and activation of the OS pathway (7, 8). In another study, injection of HIV-1 Tat in the rat striatum was shown to lead to activation of the OS pathway (9). In addition to Tat, HIV-1 gp120 protein has been shown to cause reactive oxygen species (ROS) production in glial cells leading to neurodegeneration and apoptosis (10). Finally, in a recent study, HIV-1 Vpr protein was shown to be involved in OS pathway (11).
During contact with HIV-infected macrophages or microglia, endothelial
cells might contribute to their own damage as a result of the production of
ROS (12). ROS, including
superoxide (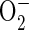 ), hydrogen peroxide
(H2O2), hydroxyl radical (OH), and reactive nitrogen
species such as nitric oxide (NO) and peroxynitrite
(
), hydrogen peroxide
(H2O2), hydroxyl radical (OH), and reactive nitrogen
species such as nitric oxide (NO) and peroxynitrite
(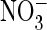 ), are biologically active species
that are increasingly recognized to play major roles in vascular biology
through redox signaling (13).
Cytokines could form a pivotal link in ROS-dependent pathways leading to the
activation of redox-sensitive transcription factors, such as the
hypoxia-inducible factor 1 (HIF-1), whose up-regulation determines the
specificity of cellular responses to oxidative stress
(14).
), are biologically active species
that are increasingly recognized to play major roles in vascular biology
through redox signaling (13).
Cytokines could form a pivotal link in ROS-dependent pathways leading to the
activation of redox-sensitive transcription factors, such as the
hypoxia-inducible factor 1 (HIF-1), whose up-regulation determines the
specificity of cellular responses to oxidative stress
(14).
HIF is an oxygen sensor and master regulator in unicellular and multicellular organisms (15), and is involved in the response to low oxygen levels (hypoxia), inducing changes in gene expression (16). HIF regulates glycolysis, mitochondrial oxygen consumption, erythropoiesis, angiogenesis, and cell survival (17). HIF is a heterodimer of two basic helix-loop-helix/PER-ARNT-SIM proteins containing HIF-1α or HIF-2α and the aryl hydrocarbon nuclear translocator (ARNT or HIF-1β) (18). HIF heterodimers bind to the hypoxia-response element (HRE), a 5′-RCGTG-3′ consensus sequence (19). HIF-1 activation is modulated by TNF-α, ROS, and nitric oxide and/or NO-derived species. ROS contributes to the accumulation and stabilization of HIF-1α (20, 21). Under hypoxic conditions, TNF-α promotes a signaling cascade, which depends on the translocation of NF-κB and leads to accumulation of HIF-1α protein (22). Finally, HIF-1α is phosphorylated (23). In this regard, HIF-1α has been shown to be a substrate for various protein kinase pathways, including the MAPK pathways.
Viral protein R, Vpr is a highly conserved HIV-1 accessory protein, involved in viral replication, transactivation of long terminal repeat (LTR), nuclear localization of the pre-integration complex in nondividing cells, cell cycle arrest, DNA damage, and apoptosis (24–26). Vpr causes apoptosis through suppression of NF-κB activity or through rapid dissipation of the mitochondrial transmembrane potential, through its association with adenine nucleotide translocase leading to the release of cytochrome c, and activation of caspase-3 (27, 28). Finally, Vpr was recently shown to cause neuronal death through convergent pathogenic mechanisms with ensuing in vivo neurodegeneration (29).
We have now identified Vpr as one of the HIV-1 proteins involved in triggering free radical production. This occurs via increased production of ROS leading to HIF-1α accumulation in microglia. Accumulation of HIF-1α then up-regulates the HIV-1 promoter. These data reveal a new function for Vpr regarding its ability to induce OS and shed light on the balance that governs the mechanisms used by HIV-1 to activate the OS pathway and its involvement in neuro-AIDS.
MATERIALS AND METHODS
Plasmids—HIV-1 LTR luciferase reporter plasmid and Vpr expression plasmid were described previously (30). CMV-HIF-1α was a gift from Dr. Steve McKnight (University of Texas Southwestern Medical Center, Dallas). The HIF-1α promoter fused to luciferase reporter plasmid was described previously (31, 32).
Cell Culture, Transfection, and Transduction Assays—The human microglial cell line (33) was maintained in Dulbecco's modified Eagle's medium + 10% FBS. Cells were transfected with 0.5 μg of reporter plasmid (LTR-Luc or HIF-1α-Luc) or co-transfected with 0.5 μg of various expression cDNAs as indicated (34). Separately, the cells were transduced with adeno-Vpr or adeno-null. The amount of DNA used for each transfection was normalized with pcDNA3 vector plasmid. Each transfection was repeated at least three times with different plasmid preparations. Cell extracts were prepared 48 h after transfection, and luciferase assays were performed (Promega, Madison, WI).
Recombinant Adenoviruses—Vpr cDNA (288 bp) was excised from pcDNA3-Vpr and cloned into the EcoRI and NheI sites of the adenovirus-shuttle plasmid pDC515 under the control of the murine cytomegalovirus promoter (purchased from Microbix Inc., Ontario, Canada). Adeno-Vpr recombinant shuttle containing Vpr sequence (pDC515-Vpr) was transfected into HEK-293 cells with pBHGfrt (del) E1, 3FLP, and a plasmid that provides adenovirus type 5 genome deleted in E1 and E3 genes. Plaques of recombinant adenovirus arising as a result of frt/FLP recombination were isolated, grown, and purified by cesium chloride density equilibrium banding as described previously (35). Empty shuttle plasmid, pDC515, was used to construct control adenoviral vector (Adeno-null, a virus without a transgene). Adeno-Vpr or adeno-Null was used at an m.o.i. of five plaque-forming units per cell.
Hydrogen Peroxide Production Assay—Microglia were grown in Dulbecco's modified Eagle's medium with 10% FBS and seeded in 6-well plates. Cells were transduced with adeno-null or adeno-Vpr virus at an m.o.i. of 5 for 48 h. Uninfected cells were also collected as a control. Cells were lysed by the freeze and thaw method three times. Protein was measured using Bio-Rad protein assay reagent. Equal amounts of protein were used in H2O2 assays with the QuantiChrom™ peroxide assay kit (DIOX-250) from BioAssay Systems. The kit is designed to measure peroxide concentration in biological samples without any pretreatment. The method utilizes the chromogenic Fe3+ xylenol orange reaction, in which a purple complex is formed when Fe2+ provided in the reagent is oxidized to Fe3+ by peroxides present in the sample. The intensity of the color, measured at 540–610 nm, is an accurate measure of peroxide level in the sample. A standard curve was generated as recommended by the manufacturer using known amounts of H2O2 (conversions: 1 μm H2O2 equals 34 ng/ml).
Western Blot—Microglia were infected with adeno-null, adeno-Vpr, or adeno-Tat virus at an m.o.i. of 5 at 37 °C. Twenty four hours post-infection, 30 μg of cell extracts were subjected to Western blot analysis using anti-HIF-1α, -Vpr, or anti-Grb2, antibodies using 1/1000 dilution. For HIF-1α detection, two different anti-HIF-1α antibodies were used. The HIF-1α antibodies were purchased either from BD Biosciences or from Abcam (Cambridge, MA). Interestingly and according to the manufacturer's catalogue, antibody purchased from BD Biosciences detects two bands of HIF-1α, whereas Abcam antibody detects only one band of HIF-1α. Note that all Western blot assays were performed at least three times.
RNA Interference—Transient knockdown of HIF-1α was performed with an HIF-1α-specific siRNA (Dharmacon Research Inc., Lafayette, CO). Twenty four hours after cell plating, microglial cells were rinsed once with Opti-MEM. siRNA was added at a final concentration of 50 nm. Western blot was performed with protein extracts from untransfected cells or cells transfected with HIF-1α-specific siRNA using anti-HIF-1α antibody. For a loading control, anti-Grb2 antibody was used. Control nontargeting siRNA was also used (Dharmacon).
Tissue Acquisition—Archival autopsy samples from the frontal lobe of HIV-encephalitis (HIVE) cases and matching normal brain samples have been collected from the Manhattan Brain Bank at the Mount Sinai School of Medicine, New York, under Institutional Review Board approval by Temple University Office for Human Subjects Protection.
Immunohistochemistry—The tissue, which was formalin-fixed and paraffin-embedded, was sectioned at 5 μm thickness and placed on electromagnetically charged glass slides. Sections were processed for immunohistochemistry as described previously (36). A rabbit polyclonal anti-HIF-1α antibody was utilized as primary antibody. After rinsing with PBS, sections were incubated for 1 h at room temperature with biotinylated anti-rabbit secondary antibody, followed by 1 h of incubation with avidin-biotin-peroxidase complexes (ABC Elite Kit, Vector Laboratories, Burlingame, CA) and developed with diaminobenzidine, counterstained with hematoxylin, and mounted for histological evaluation.
ROS Measurement—The trafficking of
2,3,4,5,6-pentafluorodihydrotetramethylfosamine (redox sensor red CC-1;
Invitrogen) was used to detect reactive oxygen intermediates
(37). Redox Sensor Red CC-1 is
oxidized in the presence of 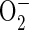 and
H2O2. Briefly, microglial cells mock-, ad-null-, or
adeno-Vpr-infected were loaded at 37 °C for 20 min with 1 μm
of Redox Sensor Red CC-1 and a mitochondria-specific dye, MitoTracker Green FM
(50 nm; Molecular Probes). Culture slides were washed with PBS and
visualized with a Nikon fluorescence microscope (Nikon Eclipse E800) equipped
with a triple-filter cube and charge-coupled device camera (Nikon DXM1200).
Note that all ROS measurement experiments were performed at least three
times.
and
H2O2. Briefly, microglial cells mock-, ad-null-, or
adeno-Vpr-infected were loaded at 37 °C for 20 min with 1 μm
of Redox Sensor Red CC-1 and a mitochondria-specific dye, MitoTracker Green FM
(50 nm; Molecular Probes). Culture slides were washed with PBS and
visualized with a Nikon fluorescence microscope (Nikon Eclipse E800) equipped
with a triple-filter cube and charge-coupled device camera (Nikon DXM1200).
Note that all ROS measurement experiments were performed at least three
times.
Chromatin Immunoprecipitation (ChIP) Assay—HeLa 3T1 (HL3T1) cells were grown overnight in 100-mm dishes to 60–70% confluency; cells were then transfected with 50 nm of siRNA-HIF-1α and/or transduced with Ad-Vpr or Ad-Null at an m.o.i. of 5 using Lipofectamine transfection reagent (Roche Applied Sciences). Plates were returned to the incubator for 40–48 h. Cells were cross-linked with formaldehyde and harvested, and ChIP was performed. For these studies, only 5 × 106 cells were used per immunoprecipitation reaction because the plasmid is present at a high copy number. The remainder of the procedure followed standard protocols for ChIP analysis, as recommended by the manufacturer (Upstate Biotechnology, Inc.). The resulting DNA was analyzed by PCR using the following HIV-LTR primers: coding (-120/+66) 5′-aactggtaccatcgagcttgct-3′ and noncoding (+66/-120) 5′-ttgaggatccagcagtgggttc-3′. Antibody used in the ChIP procedure was against HIF-1α (purchased from BD Biosciences) as well as rabbit anti-mouse IgG. All ChIP assays were performed twice.
RNA Extraction and Northern Blot Analysis—RNA was purified using the RNeasy kit from Ambion (Austin, TX) and used for Northern blot analysis as described previously (38). For generation of probes for Northern blot analysis, cDNA of human full-length HIF-1α (∼2.48 kb) or 5.8 S rRNA cDNAs was labeled using a random primed labeling reaction with Klenow enzyme and [α-32P]dCTP. The Northern blot experiment was performed three times.
HIV-1 Infection—The human U-937 monocytic cell line was maintained in RPMI + 10% FBS, 100 units/ml penicillin, 50 μg/ml streptomycin-G. Cells in log phase were infected with pNL4-3, pNL4-3 ΔVpr, or JR-FL strains of HIV-1 as follows. Fifty nanograms of p24-containing virus stock were added per 1 × 106 cells. Cells were incubated with virus stock in a small volume of serum-free medium for 2 h at 37 °C. The cells were then washed twice with PBS, and fresh medium containing 2% of FBS was added (500,000 cells/ml). All infection experiments were performed three times.
Statistical Analyses—Densitometric analyses were performed using EZQuant-Gel software (EZQuant Biology, Israel). The obtained data are expressed as the mean ± S.E. Statistical analysis of parameters was performed using the one-way ANOVA test, followed by the unpaired Student's t test, with p value less than 0.05 considered statistically significant.
RESULTS
Induction of HIF-1α Protein in the Brain of HIV-1-infected Patients—First, we examined the level of HIF-1α in brain tissue from HIVE patients. Fig. 1A shows white matter of an HIVE case, where numerous reactive astrocytes with enlarged nuclei and abundant eosinophilic cytoplasm are present (arrows). HIF-1α was found in low levels in the cytoplasm of astrocytes in normal brain, compared with the robust immunolabeling that was observed in the cytoplasm of reactive astrocytes and in macrophages and microglial nodules in HIVE white matter, indicating increased levels of HIF-1α in HIVE.
FIGURE 1.

Induction of HIF-1α in AIDS patients with HIVE. A, white matter of a normal brain showing small normal astrocytes. In contrast, the white matter of an HIVE case shows numerous reactive astrocytes with enlarged nuclei and abundant stellar cytoplasm. HIF-1α was weakly detected in the cytoplasm of normal astrocytes. In a case of HIVE, there is robust immunoreactivity for HIF-1α in the cytoplasm of enlarged reactive astrocytes as well as in macrophages (perivascular cuffs) (×400) and in microglial nodules. B and D, representative Western blots. HIV-1 infection enhances HIF-1α levels. Cell lysates were prepared from uninfected (lane 1) and HIV-infected (lane 2) microglial cultures, and 30 μg of cell lysate were analyzed for HIF-1α using anti-HIF-1α, -Vpr, and -Grb2 antibodies (B). Induction of HIF-1α in HIV-infected but not in HIVΔVpr-infected cells. Cell lysates were prepared from microglial cells, uninfected (lane 1), HIF-transfected (lane 2), HIV-infected (lane 3) or HIVΔVpr-infected cells (lane 4). Thirty micrograms of cell lysate were used (D). C and E, histograms represent the variations in HIF-1α levels obtained by Western assays and measured relatively to Grb2 using densitometric analysis (EZQuant-Gel software) (mean ± S.E.; n = 3; one-way analysis of variance and ANOVA test; **, compared with the Mock control group, p < 0.05). The control was set arbitrarily at 100%.
Induction of HIF-1α in HIV-1-infected Microglia—Next, we examined the levels of HIF-1α in HIV-infected microglia. Primary cultures of human microglial cells (1 × 106) were infected with 50 ng (p24) of the JR-FL strain of HIV-1 in 200 μl of serum-free medium for 2 h at 37 °C. Cells were washed twice with PBS and maintained in fresh medium containing 2% FBS (500,000 cells/ml). After 5 days, cells were harvested, and total protein extracts were prepared and analyzed by Western blot. As shown in Fig. 1B, HIF-1α was induced in HIV-1-infected cells compared with the mock-infected cells. Infection of the cells with HIV-1 was verified, in parallel, by Western blot using anti-Vpr antibody. The levels of the housekeeping protein Grb-2 served as internal controls for equal protein loading. Densitometry was used to quantitate the fold changes in HIF-1α protein prepared from uninfected or infected cells as observed in Fig. 1, B and C.
Expression of HIF-1α in the Absence of Vpr in HIV-1-infected Cells—To investigate the importance of Vpr in HIF-1α accumulation during the course of HIV-1 infection, human microglial cells were infected with HIV-1pNL4–3 or HIV-1pNL4–3ΔVpr (HIVΔVpr, where Vpr is deleted) or transfected with pcDNA3-HIF-1α. After 48 h, protein extracts were prepared and analyzed (30 μg of lysate) by Western blot. HIF-1α was induced in HIV-infected or HIF-transfected cells (Fig. 1D, HIF panel, compare lanes 1–3). HIF-1α was weakly induced in cells infected with Vpr-deleted HIV-1 (HIVΔVpr) (Fig. 1D, HIF panel, lane 4), indicating a small role for the other viral proteins, such as Tat, in HIF-1α induction. The efficiency of infection was monitored by Western blot using anti-Vpr antibody. As shown in Fig. 1D, Vpr was detected only in extracts from HIV-1-infected cells and not from mock-infected or HIVΔVpr cells (Vpr panel, lane 3). Grb2 levels are shown as a loading control. Note that HIV-1pNL4–3 is a T-cell tropic virus and cannot infect primary microglia. However, our experiment was performed in a microglial cell line, and the ability of HIV-1pNL4–3 to replicate in this particular microglial cell line was confirmed by PCR using several LTR primers (data not shown). Differences in the levels of expression of HIF-1α protein were further evaluated by densitometric analysis using Scion Image software (Scion Image Corp.). As shown in Fig. 1, HIF-1α protein levels increased by almost 3-fold (Fig. 1C) and 2-fold (Fig. 1E) in HIV-infected cells, respectively. Almost no change was observed in the HIF-1α protein levels in cells infected with HIV-1 that lacks Vpr (Fig. 1E, lane 4).
Induction of HIF-1α by Adenovirus-Vpr—Next, we examined whether induction of HIF-1α by Vpr is dose-dependent. Microglial cells were infected with different Ad-Vpr concentrations (1, 2, 5, 10, 15, and 20 m.o.i.) for 72 h after which the cells were washed and cellular proteins were collected and analyzed by Western blot. As shown in Fig. 2A, HIF-1α was induced by Ad-Vpr at an m.o.i. as low as 1 (lane 2). Almost no difference was observed in HIF-1α induction in cells infected with Ad-Vpr at m.o.i. of 5, 10, 15, and 20 (Fig. 2A, compare lane 4 with lanes 5–7). Differences in the levels of expression of HIF-1α protein were further evaluated by densitometric analysis (Fig. 2B).
FIGURE 2.

Induction of HIF-1α by Vpr is dose- and time-dependent. A, representative Western blot. Cell lysates were prepared from microglial cells, uninfected or infected with Ad-Vpr and Ad-null or HIV-infected. Thirty micrograms of cell lysate prepared from microglial cells were analyzed for HIF-1α and Vpr. Induction of HIF-1α was measured relative to Grb2, and the values obtained with EZQuant-Gel software are presented in B. The control was set arbitrarily at 100% (mean ± S.E.; n = 5). For statistical analysis, the results were analyzed using the Student's t test statistical significance level, p < 0.05 is indicated by **; **, compared with the Mock control group (one-way analysis of variance and ANOVA test). C, microglial cells were mock-infected or infected with two concentrations of Ad-Vpr as well as with Ad-null or HIV-1 as indicated. Cell lysates were prepared and analyzed by Western blot as described in A. The arrowheads indicate the position of each protein. Anti-Grb2 antibody was used to show equal protein loading.
Because Ad-Vpr at an m.o.i. of 5 was enough to induce HIF-1α, it was necessary to examine whether this concentration is physiologically significant. To this end, microglial cells were infected with Ad-Vpr at an m.o.i. of 1 and 5, and Ad-Null at an m.o.i. of 5, whereas U937 cells were infected with JR-FL strain of HIV-1. Cellular proteins were prepared from Ad-null- or Ad-Vpr-infected cells 48 post-infection and were collected 8 days post-infection from U937 cells infected with HIV-1. Thirty micrograms of extracts were subjected to Western blot analysis using anti-Vpr antibody. As shown in Fig. 2C, the amount of Vpr detected in extracts collected from HIV-1-infected cells was considerably higher than the amount of Vpr detected in cells infected with Ad-Vpr (compare lane 5 to lanes 2 and 3). Vpr was not detected in mock- or Ad-null-infected cells (Fig. 2C, lanes 1 and 4). Based on these results, we used Ad-Vpr at an m.o.i. of 5 in all experiments.
Expression of HIF-1α in the Presence of Tat and Vpr—In light of the previous results showing the ability of Tat and Vpr to induce oxidative stress (6, 11, 39, 40), we examined the effect of Tat (101 amino acids) and Vpr on HIF-1α expression in microglial cells. Human microglia were transduced with Ad-Tat, Ad-Vpr, or Ad-Null at an m.o.i. of 5. After 48 h, cells were harvested and analyzed by Western blot using anti-HIF-1α antibody (Abcam). Mock-infected cells or cells transfected with pcDNA3 were used as a negative control, whereas pcDNA3-HIF1α-transfected cells were used as a positive control (Fig. 3A, lanes 3 and 6, respectively). HIF-1α was weakly detected in control cells (Fig. 3A, lanes 1–3). Infection of cells with Ad-Tat had no significant effect on HIF-1α in microglial cells (Fig. 3A, lane 4), whereas the expression of Vpr significantly increased HIF-1α production (lane 5). Anti-Tat and -Vpr antibodies were used as controls to show expression of both proteins. Anti-Grb2 was used as a control for equal protein loading. Differences in the levels of expression of HIF-1α protein were further evaluated by densitometric analysis (Fig. 3B).
FIGURE 3.

The HIV-1 protein, Vpr, enhances HIF-1α levels. A, representative Western blot. Cell lysates were prepared from uninfected (lane 1) or Ad-Null- (lane 2), Ad-Tat- (lane 4), or Ad-Vpr (lane 5)-infected microglial cultures, as well as from pcDNA3 (lane 3) or pcDNA3-HIF1α (lane 6)-transfected cells and analyzed. Anti-Tat and Vpr antibodies were used as a control to show expression of both proteins. Anti-Grb2 antibody was used to show equal protein loading. B, histograms represent the values of HIF-1α bands obtained by Western assays as measured relatively to Grb2 using densitometric analysis using EZQuant-Gel software (mean ± S.E.; n = 3). For statistical analysis, the results were analyzed using the Student's t test statistical significance level, p < 0.05 is indicated by **; **, compared with the Mock control group (one-way analysis of variance and ANOVA test).
Vpr Induces HIF-1α at the Level of Transcription—Induction of HIF-1α by Vpr gave us a rationale to investigate the ability of Vpr to affect expression of HIF-1α at the RNA level. To this end, microglial cells were infected with Ad-null or Ad-Vpr for 48 h at an m.o.i. of 5 after which total RNA was prepared and analyzed by Northern blot using HIF-1α cDNA as a probe. As shown in Fig. 4A, HIF-1α RNA was induced in Vpr-infected cells but not in mock- or Ad-null-infected cells (compare lane 3 with lanes 1 and 2). Glyceraldehyde-3-phosphate dehydrogenase was used as the RNA loading control. Fig. 4B displays the fold changes in mRNA prepared from uninfected or infected cells as indicated.
FIGURE 4.

Transcriptional regulation of HIF-1α by Vpr. A and B, representative Northern blot. Microglia were infected with Ad-null or Ad-Vpr at m.o.i. of 5 or transfected with 0.5 μg of HIF-1α-luc alone or in combination with 5 m.o.i. of Ad-Vpr or 1 μg of HIF-1α expression plasmid, 50 nm siRNA-Sp1, or p65 as described. Cells infected with Ad-null or Ad-Vpr were analyzed using 20 μg of total RNA. Induction of HIF-1α was measured relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) with EZQuant-Gel software, and the values are presented in B (mean ± S.E.; n = 3; one-way analysis of variance and ANOVA test; **, compared with the Mock control group, p < 0.05). C, transfected cells were subjected to luciferase assay 48 h after being transfected with the HIF-1α promoter construct. Data represent the mean value of five separate transfection experiments performed with several DNA preparations. For statistical analysis, the results were analyzed using the Student's t test statistical significance level, p < 0.05 is indicated by **; **, compared with the Mock control group (one-way analysis of variance and ANOVA test). D and F, representative Western blots. Cells were transfected with 40 nm siRNA-Sp1, siRNA-p65, or nontargeting siRNA3 (ns-siRNA) as indicated. Thirty micrograms of extract were assayed with anti-Sp1, -p65, or anti-Grb2 antibodies. Arrows indicate the positions of each protein. E and G, histograms represent the induction of Sp1 and p65, respectively, relatively measured to Grb2 using densitometric analysis (EZQuant-Gel software) (mean ± S.E.; n = 3; one-way analysis of variance and ANOVA test; **, compared with the Mock control group, p < 0.05). Note that in E, the 105- and 95-kDa isoforms (bands) of Sp1 are shown in black and gray, respectively.
Next, we examined the ability of Vpr to induce the activity of the HIF-1α promoter. As shown in Fig. 4C, infection of microglial cells with Ad-Vpr at an m.o.i. of 5 stimulated the HIF-1α promoter (lane 2). In light of earlier studies suggesting the cooperativity of Vpr with Sp1 and NF-κB (26, 27), and the presence of both Sp1- and NF-κB-binding sites within the HIF-1α promoter, we investigated the importance of Sp1 and the p65 subunit of NF-κB in Vpr activation of the HIF-1α promoter. We used siRNA (50 nm) to suppress expression of Sp1 and NF-κB-p65 in the transfected cells and found that the presence of Sp1 and, to a lesser degree, p65 is essential for Vpr activation of the HIF-1α promoter (Fig. 4C, compare lane 2 with lanes 4 and 6). As a control, nontargeting siRNA (ns-siRNA) (50 nm) was used. Addition of ns-siRNA did not affect the ability of Vpr to activate the HIF-1α promoter (Fig. 4C, compare lanes 2 and 8). Finally, we determined whether addition of HIF-1α affects the ability of Vpr to regulate the HIF-1α promoter. Interestingly, the activation of the HIF-1α promoter by HIF-1α (Fig. 4C, lane 9) was increased by the presence of Vpr (lane 10). These results point to a functional association between Vpr and HIF-1α and that Vpr activates HIF-1α at transcriptional and translational levels. Fig. 4, D and F, illustrates results from Western blots showing suppression of Sp1 and p65 in cells treated with siRNA targeting Sp1 and p65, respectively. Nontargeting siRNA was also used as a control. Differences in the levels of expression of Sp1 and p65 proteins were further evaluated by densitometric analysis (Fig. 4, E and G).
Vpr Induces Mitochondrial ROS Production in Microglia—As the generation of excess mitochondrial ROS induces a number of redox reactions that can induce HIF-1α expression, in the next series of studies, we investigated the effect of Vpr on mitochondrial ROS (41). Microglia were infected with Ad-Null and Ad-Vpr virus at an m.o.i. of 5 for 48 h as described under “Materials and Methods” and then treated with the redox-sensitive probe Redox Sensor Red CC-1 and the mitochondria-specific dye MitoTracker Green FM. As shown in Fig. 5A, only Vpr-infected cells exhibit bright yellow/orange fluorescence in mitochondria because of the co-localization of oxidized Red CC-1 and MitoTracker Green, whereas oxidation of Red CC-1 in cytoplasm shows red/orange fluorescence, indicative of increased ROS production in the mitochondria and cytosolic compartments (Fig. 5A, compare Vpr panels to mock or ad-null panels). These results point to the ability of Vpr to induce mitochondrial ROS in microglia. Fig. 5B shows quantification of the ROS levels. In comparison with mock- or Ad-null-infected cells, ROS production was ∼15-fold higher in the cells infected with Ad-Vpr. Infection of the cells with Ad-null had no effect on ROS production.
FIGURE 5.

Induction of ROS production by Vpr at 48 h. A, microglia were infected with Ad-null, Ad-Vpr, or mock-infected. Cells were loaded with the redox-sensitive dye Redox Sensor Red CC-1 (red panels) and the mitochondrial-specific dye MitoTracker Green FM (green panels). Induction of ROS production and its localization in the mitochondria (yellow/orange and arrows) or in the cytoplasm (red/orange and arrows) is also shown. B, total ROS fluorescence was averaged from individual cells and expressed as a percentage of mitofluorescence. Measurements were made from 100 cells per field. The columns (means ± S.E.) represent the average of five fields. Statistically significant differences were seen between experimental values and controls (p < 0.05; **, compared with the Mock control group, Student's t test). The experiment was carried out three times, with similar results.
Vpr Induces H2O2 Production in Microglia—As a component of ROS, H2O2 has been shown to cause activation of the HIV-1 LTR in patients with latent HIV-1 (13, 42). Furthermore, exogenous H2O2 was shown to prevent HIF-1α accumulation in the liver cell line Hep3B (43). Therefore, we investigated the effect of Vpr on endogenous H2O2 secretion and whether this secretion will affect endogenous levels of HIF-1α. To this end, microglial cells were infected in duplicate with Ad-Null and Ad-Vpr virus at an m.o.i. of 5 and at various times post-infection (3–48 h). The level of H2O2 was determined from one set of cells using the QuantiChrom™ peroxide assay kit (DIOX-250, BioAssay Systems), which is designed to measure H2O2. The method utilizes the chromogenic Fe3+ xylenol orange reaction, in which a purple complex formed when Fe2+ provided in the reagent is oxidized to Fe3+ by peroxides present in the sample. The intensity of the color (540–610 nm) measures H2O2. A standard curve was generated according to the manufacturer's protocol using known amounts of H2O2. As shown in Fig. 6A, in the presence of Vpr, the level of H2O2 production was increased at 12 h post-infection, reached a maximum level at 24 h, and then decreased at later time points (36 and 48 h). Note that we observed an unexplained “boost” in H2O2 secretion/production at 48 h (data not shown). Infection of the cells with Ad-Null did not lead to any increase in the production of H2O2 (Fig. 6A).
FIGURE 6.

Induction of H2O2 in Vpr-infected cells in the absence of HIF-1α. Representative Western blots are shown. A and C, assay for H2O2 showing production of H2O2 in Vpr-infected microglia (A) or in HIV-1-infected cells (C). Expression of Vpr, HIF-1α, and Grb2 was analyzed as indicated. Induction of HIF-1α was measured relative to Grb2, and the values are presented in B. (Conversions: 1 μm H2O2 equals 34 ng/ml.) The experiments were carried out three times, with similar results. D, data shown here are from a single representative experiment that was replicated three times and statistically significant. For statistical analysis, the results of the H2O2 secretion were analyzed using the Student's t test statistical significance level, p < 0.05 is indicated by ** in D only; **, compared with the Mock control group (one-way analysis of variance and ANOVA test). Even though not labeled, note that the results presented in B were also statistically significant.
The other set of cells was used for Western analysis. Briefly, the cell lysates were prepared from these cells and subjected to Western blot analysis using anti-Vpr, anti-HIF-1α, and anti-Grb2 antibodies. As shown in Fig. 6A, Vpr was detected as early as 6 h post-infection (Vpr panel). It peaks at 24 h, relatively decreases as of 36 h, and remains constant at 48 h. Endogenous levels of HIF-1α were induced as early as 12 h post-infection (Fig. 6A, HIF-1α panel) and remained invariable up until 48 h. Differences in the levels of expression of HIF-1α protein were further evaluated by densitometric analysis (Fig. 6B). These data led us to conclude that secretion of H2O2 did promote HIF-1α accumulation in microglial cells and not as previously reported in Hep3B (43).
To investigate the ability of HIV-1 infection to induce H2O2 secretion, microglial cells (1 × 106) were infected with the JR-FL strain of HIV-1 (50 ng of p24-containing virus stock). Our results showed an increase in the level of H2O2 upon HIV-1 infection at 24 h post-infection (Fig. 6C), suggesting that expression of Vpr induces H2O2 production during the course of HIV-1 infection of microglial cells.
Regulation of the HIV-1 Promoter by HIF-1α and/or Vpr in Microglia—To examine the impact of HIF-1α induction on HIV-1, microglial cells were transfected in triplicate with HIV-1-Luc alone or with 1.0 μg of CMV-HIF-1α and Ad-Vpr. After 48 h, the cells were processed for luciferase assay, whereas one set was analyzed by Western blot. As shown in Fig. 7A, transfection of HIF-1α (lane 5) or infection of the cells with Ad-Vpr (lane 2) activated the HIV-1 LTR promoter when compared with the LTR alone (lane 1). Similar to the results obtained with the HIF-1α promoter, activation of the LTR by HIF-1α was further increased in cells that were expressing Vpr (Fig. 7A, lane 6), suggesting cooperativity between HIF-1α and Vpr upon LTR transcription. To further demonstrate the specificity of this functional interaction, and to examine the involvement of HIF-1α in Vpr-mediated activation of the HIV-1 promoter, the LTR was transfected with 50 nm siRNA-HIF-1α alone or in the presence of Ad-Vpr. Interestingly, inhibition of HIF-1α led to silencing of the LTR in the presence and absence of Vpr (Fig. 7A, compare lanes 1 and 2 with lanes 3 and 4). To further confirm the dependence of Vpr on the presence of HIF-1α to activate the HIV-1 promoter, the cells were transfected with non-targeting siRNA in the presence or absence of Vpr. As shown in Fig. 7A, activation of the LTR by Vpr was not affected by the presence of ns-siRNA (compare lanes 1 and 2 with lanes 7 and 8). In parallel, protein extracts were prepared from one set of the cells and analyzed by Western blot using anti-HIF-1α, anti-Vpr, and anti-Grb2 antibodies. As expected, infection of the cells with Vpr led to an increase in the levels of HIF-1α (Fig. 7A, lane 2). Vpr failed to induce endogenous levels of HIF-1α in the presence of siRNA-HIF-1α (Fig. 7A, lane 4) but not in the presence of ns-siRNA (lane 8). Vpr was only detected in lanes where it was introduced (Fig. 7A, Vpr panel). Anti-Grb2 was used to control equal protein loading. Differences in the levels of expression of HIF-1α protein were further evaluated by densitometric analysis (Fig. 7B).
FIGURE 7.

Transcription regulation of the HIV-1 promoter by Vpr and HIF-1α. A, microglia were transfected in triplicate with 0.5 μg of LTR-luc in the presence or absence of 1 μg of CMV-HIF-1α and/or infected with Ad-Vpr at an m.o.i. of 5 as indicated or Ad-Null at an m.o.i. of 5 (data not shown). siRNA-HIF-1α and ns-siRNA were also transfected alone or in the presence of Ad-Vpr (lanes 5–8). Data represent the mean value of three separate transfection experiments. Luciferase activity was determined after 48 h. Expression of Vpr, HIF-1α, and Grb2 was analyzed by Western blot as indicated using one set of transfected/infected cells (mean ± S.E.; n = 5; one-way analysis of variance and ANOVA test). B, data shown here are from a single representative experiment that was replicated five times and statistically significant. For the statistical analysis, the results were analyzed using the Student's t test statistical significance level, p < 0.05 is indicated by **; **, compared with the Mock control group (one-way analysis of variance and ANOVA test). C, interaction of HIF-1α with HIV-LTR DNA by ChIP in Vpr-infected microglia in the presence and absence of siRNA-HIF-1α or ns-siRNA. Anti-HIF-1α (lanes 3), pre-immune serum (lanes 2), and no antibody (lanes 1) were used.
The functional interplay between HIF-1α and Vpr prompted us to examine whether there is a physical interaction between the two proteins. Microglia were infected with Ad-Vpr or Ad-Null at an m.o.i. of 5 and lysates harvested after 48 h. Three hundred micrograms of extracts were then subjected to immunoprecipitation/Western blot analysis using anti-Vpr or anti-HIF-1α antibodies. These experiments failed to show an interaction between these two proteins (data not shown).
Identification of HRE within the HIV-LTR—Because HIF-1 activates the HIV-1 LTR, we next investigated that mechanism. First, we examined if HIF-1 can activate the LTR directly since the LTR contains a predicted HRE located between -71 and -67 (RCGTG, where R could be an A or G) (16). Therefore, we performed ChIP to examine if HIF-1 can bind to this site. As shown in Fig. 7C, microglial cells were transfected with 0.5 μg of the HIV-1 promoter alone or in the presence of 50 nm of siRNA-HIF-1α, ns-siRNA, and/or infected with Ad-Vpr (lanes 3) or Ad-Null (data not shown) at an m.o.i. of 5. After 48 h, cells were cross-linked with formaldehyde and harvested, and ChIP was performed. The remainder of the procedure followed standard protocols for ChIP analysis as described previously (34). The resulting DNA was analyzed by PCR with HIV-LTR primers. Anti-HIF-1α antibody (Fig. 7C, lanes 3) and rabbit anti-mouse IgG (lanes 2) were used in the procedure. Endogenous HIF-1 binds to HIV-LTR DNA. Western blot showed expression of Vpr and HIF-1α (data not shown). Interestingly, addition of Vpr increased the association of HIF-1 with the DNA (Fig. 7C, Vpr panel, lane 3). As expected, addition of siRNA-HIF-1α but not ns-siRNA abolished HIF-1 interaction with the LTR DNA in the absence or presence of Vpr.
DISCUSSION
Recent advances in the field of oxidative stress have highlighted the importance of reduction-oxidation (redox)-sensitive transcription factors in cellular regulation. Redox-sensitive transcription factors are often associated with the development and progression of many human disease states. For example, OS occurs in brain tissues of patients undergoing HIV infection and is implicated in cell death of both astroglia and neurons and hence plays a role in the pathogenesis of neuro-AIDS. For this reason, OS is a potential target for therapeutic interventions for possible clinical applications. HIF-1 is a crucial transcription factor that is a master regulatory element in sensing hypoxic conditions and integrating an adaptive response via gene expression of redox-sensitive enzymes and cofactors. The signal transduction components that link the availability of oxygen to the activation of HIF-1α are poorly defined. Thus, a better understanding of the processes that trigger and modulate free radical formation in the brain tissue of AIDS patients requires answers to the following questions. What are the mechanisms that trigger free radical overproduction in neuro-AIDS? What is the impact of HIV-1 infection on HIF-1α accumulation? Which viral protein(s) induces OS and accumulation of HIF-1α in the central nervous system and what are the mechanisms used? Does induction of HIF-1α by HIV-1 protein(s) happen at the transcriptional or post-transcriptional level? What is the impact of OS induction and HIF-1α accumulation on HIV-1 gene expression and replication?
To address these questions, we examined the involvement of viral (Vpr) and cellular (HIF-1α) regulatory proteins in oxidative stress-induced neuropathogenesis of HIV-1 infection. Our data indicate that Vpr induced HIF-1α accumulation and ROS secretion in microglia. In turn, HIF-1 binds and up-regulates the HIV-1 promoter.
Microglial cells were chosen because hypoxia has been shown to induce microglia activation, and animal studies have also shown that neuronal cell death is correlated with microglial activation in other situations, e.g. following cerebral ischemia (18). Thus, it is likely that toxic inflammatory mediators produced by activated microglia, e.g. HIV-1- or Ad-Vpr-infection, under hypoxic conditions may exacerbate neuronal injury following cerebral ischemia via induction of HIF-1α. Although the role of HIF-1 in microglia activation under hypoxia remains to be elucidated, it has been shown that exposure of primary rat microglial cultures, as well as an established microglial cell line, to hypoxia induced accumulation of HIF-1α. In turn, HIF-1 induces the expression of inducible NO synthase and ROS secretion, indicating that hypoxia could lead to the inflammatory activation of microglia (18). Thus, during cerebral ischemia, hypoxia may not only directly damage neurons but may also promote neuronal injury indirectly via microglial activation.
Earlier work has shown that Vpr causes cell death through its association with the adenine nucleotide translocase protein, which promotes the permeability of the mitochondrial membrane, leading to the release of cytochrome c and activation of the caspase-3 pathway (44). These events are involved in inducing OS, suggesting the possibility of involvement of Vpr in the induction of OS. Recent studies demonstrated that the dose-dependent H2O2 stress response promotes increased survival for Schizosaccharomyces pombe cells expressing HIV-1 Vpr (11). These results are in conflict with our data. However, several studies demonstrated that ROS might play different roles (pro- or antiapoptotic) according to the cell types.
Our data presented in Fig. 5 indicate that Vpr may induce NADPH oxidase ROS production, but this possibility remains to be elucidated. It has been demonstrated that, in addition to its involvement in phagocytosis, NADPH oxidase-derived ROS is also implicated in facilitating cell-to-cell communication (45). Therefore, activation of NADPH oxidase pathway could point to a novel mechanism used by Vpr to facilitate virus trafficking. Thus, it is possible that Vpr helps in creating a suitable environment for HIV-1 to “travel” between the cells (data not shown). However, our preliminary results demonstrated that Vpr failed to induce production of internal calcium from microglia (data not shown) leading to the activation of the inositol 1,4,5-trisphosphate pathway and preventing cell connection. Both pathways are under investigation.
In addition to the oxidative stress pathway, the TNF-α and p38 MAPK pathways have also been shown to induce HIF-1α (16, 23). We further confirmed this relation by examining the ability of Vpr to induce these pathways leading to HIF-1α up-regulation. Interestingly, Vpr was capable of activating TNF-α at the transcriptional and translational levels leading to HIF-1α induction. Furthermore, we found that Vpr promotes phosphorylation of the p38 MAPK protein leading to accumulation of HIF-1α (data not shown). Up-regulation of HIF-1α by Vpr through the p38 MAPK pathway was further confirmed using inhibitor of p38 MAPK (data not shown).
Activation and accumulation of HIF-1α by a viral protein are not without a precedent. In this regard, it has been shown that HIF-1α can be induced and accumulated in cells infected with HTLV-1 and HHV8 (46, 47). Furthermore, HIF-1α was also shown to be suppressed in cells infected with a temperature-sensitive Moloney murine leukemia virus mutant (48). In addition to its diverse regulation by different viruses, HIF-1α has also been shown to be regulated positively as well as negatively in different cell lines. In this regard, it has been shown that H2O2 blocks accumulation of HIF-1α in the liver cell line, Hep3B (43). These latest results differ from our data presented in Fig. 6. However, this difference could be due to variation between the cell types used as well as to the presence of HIV-1 Vpr protein, which may have the ability to overcome the negative effect of H2O2 (data not shown).
On the other hand, however, we found that Vpr is involved in ROS secretion and HIF-1α accumulation, and the involvement of other viral proteins such as gp120 cannot be ruled out. In this regard, gp120 has been shown to induce ROS secretion in monocytic cells that can produce neuronal damage (49). ROS produced in glial cells by gp120 exposure has been shown to cause neurodegeneration by inducing the synthesis of interleukin-1β (50). In addition to Vpr and gp120, HIV-1 Tat has been shown to induce ROS secretion by inhibiting expression of manganese-dependent superoxide dismutase in HeLa and T-cells via a pathway that involves NF-κB (51). Tat also induces apoptosis of hippocampal neurons by a mechanism that involves oxidative stress (52). Oxidative damage has also been demonstrated to be induced by the injection of HIV-1 Tat protein in the rat striatum (9). Finally, Tat protein was found to induce oxidative and inflammatory pathways in brain endothelium (53). Hence, these observations, in addition to our data, suggest a role for gp120 and Tat proteins, in addition to Vpr, on HIF-1α accumulation and ROS secretion.
Furthermore, activation of the HIF-1α promoter by Vpr through Sp1 is not without precedent. Several groups, including ours, demonstrated the ability of Vpr to activate the HIV-1 LTR and the p21WAF1 promoters through its association with Sp1 transcription factor (Refs. 26, 54 and references within). In this regard, because no physical association between Vpr and HIF-1α was observed, it will be interesting to examine whether Sp1 mediates the interaction of Vpr with HIF-1α in these cells. Furthermore, Vpr has previously been shown to cause apoptosis through suppression of NF-κB (27). Thus, association of HIF-1α with NF-κB within the HIV-1 LTR might facilitate the ability of Vpr to activate the HIV-1 promoter and to induce apoptosis. To examine this last possibility, we performed studies using microglial cells where we examined the status of cleaved caspase-3 in the presence and absence of Vpr and/or HIF-1α. Interestingly, overexpression of HIF-1α prevented Vpr from cleaving caspase-3 (data not shown). Therefore, we concluded that accumulation of HIF-1α in microglial cells might explain the lack of apoptosis observed in these cells in the presence of HIV-1.
These observations led us to suggest that Vpr plays a key role in inducing OS in HIV-1-infected patients. In addition, accumulation of HIF-1α in microglia following Vpr addition might have a positive impact on cell survival and might explain the inability of Vpr to cause microglial cell death. In future studies, we plan to identify the mechanisms used by Vpr to induce OS leading to HIF-1α accumulation and to examine the impact of this induction/accumulation on Vpr functions and on HIV-1 gene expression and replication. Furthermore, because Vpr can be released and taken up by neurons, and because neurons undergo degeneration, interplay between HIF-1α and Vpr should also be examined in neurons. Completion of these studies will help in developing successful therapeutic approaches against HIV infection and against neurodegeneration often observed in AIDS patients.
Acknowledgments
We thank past and present members of the Department of Neuroscience and Center for Neurovirology for their support and sharing of reagents and ideas. We also thank Dr. Martyn White for comments on the manuscript.
This work was supported, in whole or in part, by a National Institutes of Health grant (to B. E. S. and S. A.).
Footnotes
The abbreviations used are: OS, oxidative stress; ROS, reactive oxygen species; HIF-1, hypoxia-inducible factor 1; TNF-α, tumor necrosis α; LTR, long terminal repeat; siRNA, short interfering RNA; m.o.i., multiplicity of infection; ChIP, chromatin immunoprecipitation; MAPK, mitogen-activated protein kinase; FBS, fetal bovine serum; ANOVA, analysis of variance; PBS, phosphate-buffered saline; HRE, hypoxia-response element; HIVE, HIV-encephalitis; ns-siRNA, nontargeting siRNA.
References
- 1.Blomgren, K., Leist, M., and Groc, L. (2007) Apoptosis 12, 993-1010 [DOI] [PubMed] [Google Scholar]
- 2.Sacktor, N., Haughey, N., Cutler, R., Tamara, A., Turchan, J., Pardo, C., Vargas, D., and Nath, A. (2004) J. Neuroimmunol. 157, 176-184 [DOI] [PubMed] [Google Scholar]
- 3.Hervouet, E., Simonnet, H., and Godinot, C. (2007) Biochimie (Paris) 89, 1080-1088 [DOI] [PubMed] [Google Scholar]
- 4.Kimura, H., Sawada, T., Oshima, S., Kozawa, K., Ishioka, T., and Kato, M. (2005) Curr. Drug Targets Inflamm. Allergy 4, 489-495 [DOI] [PubMed] [Google Scholar]
- 5.Mollace, V., Nottet, H. S., Clayette, P., Turco, M. C., Muscoli, C., Salvemini, D., and Perno, C. F. (2001) Trends Neurosci. 24, 411-416 [DOI] [PubMed] [Google Scholar]
- 6.Price, T. O., Uras, F., Banks, W. A., and Ercal, N. (2006) Exp. Neurol. 201, 193-202 [DOI] [PubMed] [Google Scholar]
- 7.New, D. R., Maggirwar, S. B., Epstein, L. G., Dewhurst, S., and Gelbard, H. A. (1998) J. Biol. Chem. 273, 17852-17858 [DOI] [PubMed] [Google Scholar]
- 8.Shi, B., Raina, J., Lorenzo, A., Busciglio, J., and Gabuzda, D. (1998) J. Neurovirol. 4, 281-290 [DOI] [PubMed] [Google Scholar]
- 9.Aksenov, M. Y., Hasselrot, U., Bansal, A. K., Wu, G., Nath, A., Anderson, C., Mactutus, C. F., and Booze, R. M. (2001) Neurosci. Lett. 305, 5-8 [DOI] [PubMed] [Google Scholar]
- 10.Israel, N., and Gougerot-Pocidalo, M. A. (1997) Cell. Mol. Life Sci. 53, 864-870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antal, J., and Pesti, M. (2006) Folia Microbiol. (Praha.) 51, 406-412 [DOI] [PubMed] [Google Scholar]
- 12.Flores, S. C., Marecki, J. C., Harper, K. P., Bose, S. K., Nelson, S. K., and McCord, J. M. (1993) Proc. Natl. Acad. Sci. U. S. A. 90, 7632-7636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu, W. S. (2006) Cancer Metastasis Rev. 25, 695-705 [DOI] [PubMed] [Google Scholar]
- 14.Chin, B. Y., Jiang, G., Wegiel, B., Wang, H. J., Macdonald, T., Zhang, X. C., Gallo, D., Cszimadia, E., Bach, F. H., Lee, P. J., and Otterbein, L. E. (2007) Proc. Natl. Acad. Sci. U. S. A. 104, 5109-5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordan, J. D., and Simon, M. C. (2007) Opin. Genet. Dev. 17, 71-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semenza, G. L. (2006) Expert Opin. Ther. Targets 10, 267-280 [DOI] [PubMed] [Google Scholar]
- 17.Vaux, E. C., Metzen, E., Yeates, K. M., and Ratcliffe, P. J. (2001) Blood 98, 296-302 [DOI] [PubMed] [Google Scholar]
- 18.Makino, Y., Cao, R., Svensson, K., Bertilsson, G., Asman, M., Tanaka, H., Cao, Y., Berkenstam, A., and Poellinger, L. (2001) Nature 414, 550-554 [DOI] [PubMed] [Google Scholar]
- 19.Schweppe, R. E., Cheung, T. H., and Ahn, N. G. (2006) J. Biol. Chem. 281, 20993-21003 [DOI] [PubMed] [Google Scholar]
- 20.Simon, M. C. (2006) Adv. Exp. Med. Biol. 588, 165-170 [DOI] [PubMed] [Google Scholar]
- 21.Haddad, J. J. (2004) Int. Immunopharmacol. 4, 1249-1285 [DOI] [PubMed] [Google Scholar]
- 22.Zhou, J., Schmid. T., and Brune, B. (2003) Mol. Biol. Cell 14, 2216-2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ke, Q., and Costa, M. (2006) Mol. Pharmacol. 70, 1469-1480 [DOI] [PubMed] [Google Scholar]
- 24.Amini, S., Khalili, K., and Sawaya, B. E. (2004) DNA Cell Biol. 23, 249-260 [DOI] [PubMed] [Google Scholar]
- 25.Nitahara-Kasahara, Y., Kamata, M., Yamamoto, T., Zhang, X., Miyamoto, Y., Muneta, K., Iijima, S., Yoneda, Y., Tsunetsugu-Yokota, Y., and Aida, Y. (2007) J. Virol. 81, 5284-5293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sawaya, B. E., Khalili, K., Mercer, W. E., Denisova, L., and Amini, S. (1998) J. Biol. Chem. 273, 20052-20057 [DOI] [PubMed] [Google Scholar]
- 27.Ayyavoo, V., Mahboubi, A., Mahalingam, S., Ramalingam, R., Kudchodkar, S., Williams, W. V., Green, D. R., and Weiner, D. B. (1997) Nat. Med. 3, 1117-1123 [DOI] [PubMed] [Google Scholar]
- 28.Jacotot, E., Ravagnan, L., Loeffler, M., Ferri, K. F., Vieira, H. L., Zamzami, N., Costantini, P., Druillennec, S., Hoebeke, J., Briand, J. P., Irinopoulou, T., Daugas, E., Susin, S. A., Cointe, D., Xie, Z. H., Reed, J. C., Roques, B. P., and Kroemer, G. (2000) J. Exp. Med. 191, 33-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones, G. J., Barsby, N. L., Cohen, E. A., Holden, J., Harris, K., Dickie, P., Jhamandas, J., and Power, C. (2007) J. Neurosci. 27, 3703-3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sawaya, B. E., Khalili, K., Gordon, J., Taube, R., and Amini, S. (2000) J. Biol. Chem. 275, 35209-35214 [DOI] [PubMed] [Google Scholar]
- 31.Bonello, S., Zähringer, C., BelAiba, R. S., Djordjevic, T., Hess, J., Michiels, C., Kietzmann, T., and Görlach, A. (2007) Arterioscler. Thromb. Vasc. Biol. 27, 755-761 [DOI] [PubMed] [Google Scholar]
- 32.Minet, E., Ernest, I., Michel, G., Roland, I., Remacle, J., Raes, M., and Michiels, C. (1999) Biochem. Biophys. Res. Commun. 261, 534-540 [DOI] [PubMed] [Google Scholar]
- 33.Janabi, N., Peudenier, S., Héron, B., Ng, K. H., and Tardieu, M. (1995) Neurosci. Lett. 195, 105-108 [DOI] [PubMed] [Google Scholar]
- 34.Mukerjee, R., Sawaya, B. E., Khalili, K., and Amini, S. (2007) J. Cell. Biochem. 100, 1210-1216 [DOI] [PubMed] [Google Scholar]
- 35.Eldeen, M. B., Deshmane, S. L., Simbiri, K., Khalili, K., Amini, S., and Sawaya, B. E. (2006) J. Neuroimmunol. 176, 174-180 [DOI] [PubMed] [Google Scholar]
- 36.Saunders, M., Eldeen, M. B., Valle, L. D., Reiss, K., Peruzzi, F., Mameli, G., Gelman, B. B., Khalili, K., Amini, S., and Sawaya, B. E. (2005) Apoptosis 10, 1419-1431 [DOI] [PubMed] [Google Scholar]
- 37.Chintapalli, J., Yang, S., Opawumi, D., Goyal, S. R., Shamsuddin, N., Malhotra, A., Reiss, K., and Meggs, L. G. (2007) Am. J. Physiol. 292, F523-F530 [DOI] [PubMed] [Google Scholar]
- 38.Sweet, T., Khalili, K., Sawaya, B. E., and Amini, S. (2003) J. Cell. Biochem. 90, 884-891 [DOI] [PubMed] [Google Scholar]
- 39.Valcour, V., and Shiramizu, B. (2004) Mitochondrion (Kidlington) 4, 119-129 [DOI] [PubMed] [Google Scholar]
- 40.Wallace, D. R., Dodson, S., Nath, A., and Booze, R. M. (2006) Synapse 59, 51-60 [DOI] [PubMed] [Google Scholar]
- 41.Le Bras, M., Clement, M. V., Pervaiz, S., and Brenner, C. (2005) Histol. Histopathol. 20, 205-219 [DOI] [PubMed] [Google Scholar]
- 42.Kurata, S. (1996) J. Biol. Chem. 271, 21798-21802 [DOI] [PubMed] [Google Scholar]
- 43.Huang, L. E., Arany, Z., Livingston, D. M., and Bunn, H. F. (1996) J. Biol. Chem. 271, 32253-32259 [DOI] [PubMed] [Google Scholar]
- 44.Jacotot, E., Ferri, K. F., El Hamel, C., Brenner, C., Druillennec, S., Hoebeke, J., Rustin, P., Metivier, D., Lenoir, C., Geuskens, M., Vieira, H. L., Loeffler, M., Belzacq, A. S., Briand, J. P., Zamzami, N., Edelman, L., Xie, Z. H., Reed, J. C., Roques, B. P., and Kroemer, G. (2001) J. Exp. Med. 193, 509-519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun, G. Y., Horrocks, L. A., and Farooqui, A. A. (2007) J. Neurochem. 103, 1-16 [DOI] [PubMed] [Google Scholar]
- 46.Tomita, M., Semenza, G. L., Michiels, C., Matsuda, T., Uchihara, J. N., Okudaira, T., Tanaka, Y., Taira, N., Ohshiro, K., and Mori, N. (2007) Biochem. J. 406, 317-323 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Cai, Q., Murakami, M., Si, H., and Robertson, E. S. (2007) J. Virol. 81, 10413-10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lungu, G. F., Stoica, G., and Wong, P. K. (2008) J. Neurovirol. 14, 239-251 [DOI] [PubMed] [Google Scholar]
- 49.Foga, I. O., Nath, A., Hasinoff, B. B., and Geiger, J. D. (1997) J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 16, 223-229 [DOI] [PubMed] [Google Scholar]
- 50.Viviani, B., Corsini, E., Binaglia, M., Galli, C. L., and Marinovich, M. (2001) Neuroscience 107, 51-58 [DOI] [PubMed] [Google Scholar]
- 51.Westendorp, M. O., Shatrov, V. A., Schulze-Osthoff, K., Frank, R., Kraft, M., Los, M., Krammer, P. H., Droge, W., and Lehmann, V. (1995) EMBO J. 14, 546-554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kruman, I. I., Nath, A., and Mattson, M. P. (1998) Exp. Neurol. 154, 276-288 [DOI] [PubMed] [Google Scholar]
- 53.Toborek, M., Lee, Y. W., Pu, H., Malecki, A., Flora, G., Garrido, R., Hennig, B., Bauer, H. C., and Nath, A. (2003) J. Neurochem. 84, 169-179 [DOI] [PubMed] [Google Scholar]
- 54.Amini, S., Saunders, M., Kelley, K., Khalili, K., and Sawaya, B. E. (2004) J. Biol. Chem. 279, 46046-46056 [DOI] [PubMed] [Google Scholar]