

Abstract
NADPH-cytochrome P450 oxidoreductase (CYPOR) catalyzes the transfer of electrons to all known microsomal cytochromes P450. A CYPOR variant, with a 4-amino acid deletion in the hinge connecting the FMN domain to the rest of the protein, has been crystallized in three remarkably extended conformations. The variant donates an electron to cytochrome P450 at the same rate as the wild-type, when provided with sufficient electrons. Nevertheless, it is defective in its ability to transfer electrons intramolecularly from FAD to FMN. The three extended CYPOR structures demonstrate that, by pivoting on the C terminus of the hinge, the FMN domain of the enzyme undergoes a structural rearrangement that separates it from FAD and exposes the FMN, allowing it to interact with its redox partners. A similar movement most likely occurs in the wild-type enzyme in the course of transferring electrons from FAD to its physiological partner, cytochrome P450. A model of the complex between an open conformation of CYPOR and cytochrome P450 is presented that satisfies mutagenesis constraints. Neither lengthening the linker nor mutating its sequence influenced the activity of CYPOR. It is likely that the analogous linker in other members of the diflavin family functions in a similar manner.
NADPH-cytochrome P450 oxidoreductase (CYPOR)4 is a ∼78-kDa, multidomain, microsomal diflavin protein that shuttles electrons from NADPH → FAD → FMN to members of the ubiquitous cytochrome P450 superfamily (1, 2). In humans, the cytochromes P450 (cyt P450) are one of the most important families of proteins involved in the biosynthesis and degradation of a vast number of endogenous compounds and the detoxification and biodegradation of most foreign compounds. CYPOR also donates electrons to heme oxygenase (3), cytochrome b5 (4), and cytochrome c (5).
The FAD receives a hydride anion from the obligate two electron donor NADPH and passes the electrons one at a time to FMN. The FMN then donates electrons to the redox partners of CYPOR, again one electron at a time. Cyt P450 accepts electrons at two different steps in its complex reaction cycle. Ferric cyt P450 is reduced to the ferrous protein, and oxyferrous cyt P450 receives the second of the two electrons to form the peroxo (Fe+3OO)2- cyt P450 intermediate (6). In vivo, CYPOR cycles between the one- and three-electron reduced forms (7, 8). Although the one-electron reduced form is an air-stable, neutral blue semiquinone (FMNox/sq, -110 mV), it is the FMN hydroquinone (FMNsq/hq, -270 mV), not the semiquinone, that donates an electron to its redox partners (8–11). CYPOR is the prototype of the mammalian diflavin-containing enzyme family, which includes nitric-oxide synthase (12), methionine synthase reductase (13, 14), and a novel reductase expressed in the cytoplasm of certain cancer cells (15). CYPOR is also a target for anticancer therapy, because it reductively activates anticancer prodrugs (16).
CYPOR consists of an N-terminal single α-helical transmembrane anchor (∼6 kDa) responsible for its localization to the endoplasmic reticulum and the soluble cytosolic portion (∼66 kDa) capable of reducing cytochrome c. Crystal structures of the soluble form of the wild-type and several mutant CYPORs are available (17, 18). The first ∼170 amino acids of the soluble domain are highly homologous to flavodoxin and bind FMN (FMN domain), whereas the C-terminal portion of the soluble protein consists of a FAD- and NADPH-binding domain with sequence and structural similarity to ferredoxin-NADP+ oxidoreductase (FAD domain). A connecting domain, possessing a unique sequence and structure, joins the FMN and FAD domains and is partly responsible for the relative orientation of the FMN and FAD domains. In the crystal structure, a convex anionic surface surrounds FMN. In the wild-type crystal structure, the two flavin isoalloxazine rings are in van der Waals contact, poised for efficient interflavin electron transfer (17). Based on the juxtaposition of the two flavins, an extrinsic electron transfer rate of ∼1010 s-1 is predicted (19). However, the experimentally observed electron transfer rate between the two flavins is 30–55 s-1 (20, 21). This modest rate and slowing of electron transfer in a viscous solvent (75% glycerol) suggest that interflavin electron transfer is likely conformationally gated. Moreover, the “closed” crystal structure, in which the flavins are in contact, is difficult to reconcile with mutagenesis studies that indicate the acidic amino acid residues on the surface near FMN are involved in interacting with cyt P450 (22). The first structural insight into how cyt P450 might interact with the FMN domain of CYPOR was provided by the crystal structure of a complex between the heme and FMN-containing domains of cyt P450 BM3 (23). In this complex, the methyl groups of FMN are oriented toward the heme on the proximal surface of cyt P450 BM3. Considered together, these three observations, the slow interflavin electron transfer, the mutagenesis data, and the structure of the complex between the heme and FMN domains of cyt P450 BM3, suggest that CYPOR will undergo a large conformational rearrangement in the course of shuttling electrons from NADPH to cyt P450. In addition, crystal structures of various CYPOR variants indicate that the FMN domain is highly mobile with respect to the rest of the molecule (18).
Consideration of how the reductase would undergo a reorientation to interact with its redox partners led us to hypothesize the existence of a structural element in the reductase that would regulate the conformational changes and the relative dynamic motion of the domains. Our attention focused on the hinge region between the FMN and the connecting domain, because it is often disordered and highly flexible in the crystal structure (supplemental Fig. S1). The length and sequence of the hinge have been altered by site-directed mutagenesis, and the effects of the mutations on the catalytic properties of each mutant have been determined. The results demonstrate that lengthening the linker or altering its sequence do not modify the properties of CYPOR. In contrast, deletion of four amino acids markedly disrupts electron transfer from FAD to FMN, whereas the ability of the FMN domain to donate electrons to cyt P450 remains intact. The hinge deletion variant has been crystallized in three “open” conformations capable of interacting with cyt P450.
EXPERIMENTAL PROCEDURES
Determination of CYPOR Activity Using Cytochrome c and Ferricyanide—The ability of CYPOR hinge mutants to reduce cytochrome c and ferricyanide was measured in 270 mm potassium phosphate buffer, pH 7.7, at 30 °C as described previously (24). Details of the assay are provided in the supplemental materials. To ensure that the loss of cytochrome c activity observed with the two (ΔT236, G237) (ΔTG) and four (ΔT236, G237, E238, E239) (ΔTGEE) amino acid deletion mutants was not due to a dissociation of flavins from the CYPOR, the reduction of cytochrome c assays was also measured in the presence of 240 nm free FMN and FAD. The added flavins did not significantly increase the activity of the CYPOR. The concentration of free FMN and FAD was determined using their extinction coefficients (εFMN445 nm = 12.5 mm-1cm-1; εFAD450 nm = 11.3 mm-1 cm-1) (11). The kcat and Km for cytochrome c and NADPH were determined by following the kinetics of cytochrome c reduction at varying concentrations of cytochrome c (65, 28, 18.5, 9.2, and 1.9 μm) and NADPH (60, 30, 15, 9, 3, and 1.2 μm). The kinetic parameters were obtained by fitting the initial rate versus concentration of the substrate using the Michaelis-Menten equation.
Reduction of ferricyanide by CYPOR was measured by following the bleaching of the absorbance at 420 nm. Typically, 10 nm CYPOR was preincubated with 500 μm oxidized ferricyanide for 5 min at 30 °C. The reaction was started by the addition of NADPH to a final concentration of 100 μm. The concentration of the reduced ferricyanide was calculated using an extinction coefficient at 420 nm of 1.02 mm-1 cm-1 (11).
Measurement of Benzphetamine Metabolism by Cyt P450 2B4—Cyt P450 2B4 was expressed and purified as described previously (25) (details in supplemental materials). The metabolism of benzphetamine at 30 °C under steady-state conditions was determined by measuring formaldehyde formation using Nash's reagent as previously described (26).
Kinetics of the Reduction of CYPOR by NADPH—The experiments were performed at 25 °C under anaerobic conditions using a High-Tech SF61DX2 stopped-flow spectrophotometer equipped with a temperature-controlled circulating water bath, housed in an anaerobic Belle Technology glove box (Hi-Tech, Salisbury, England) as previously described (8). Additional experimental details are included in the supplemental materials.
Determination of the Rate of Reduction of Ferric Cyt P450 2B4 by CYPOR in the Presence and Absence of Benzphetamine—The rate of reduction of ferric cyt P450 2B4 in the presence of CO by wild-type, ΔTG, and ΔTGEE CYPOR was determined as previously described (27). Details are in the supplemental materials.
Determination of the Kinetics of Product Formation by Oxyferrous Cyt P450 2B4 in the Presence of Two-electron Reduced Wild-type and Variant CYPOR—The rate of oxidation of cyclohexane and benzphetamine by cyt P450 2B4 in the presence of the two-electron reduced CYPOR was performed at 30 and 15 °C, respectively, using a QFM-400 chemical quench flow apparatus (BioLogic, France) as described (28). Briefly, a protein complex between oxidized reductase and ferric cyt P450 was preformed in the presence of dilauroyl l-3-phosphatidyl choline at a molar ratio of 1:1:60 (CYPOR:cyt P450:dilauroyl l-3-phosphatidyl choline). The substrate (cyclohexane or benzphetamine) was added to the protein mixture at a final concentration of 1 mm. Then the preformed cyt P450·CYPOR complex was stoichiometrically reduced with dithionite under anaerobic conditions to the ferrous state for cyt P450 and to the two-electron reduced state for CYPOR. The solution containing the reduced, preformed cyt P450·CYPOR complex was loaded into syringe 1 of the chemical quenched-flow apparatus and rapidly mixed with aerobic buffer ([O2] ≈ 0.32 mm) containing 1 mm of substrate from syringe 2. The reaction was quenched with 1 m NaOH at different times. The samples were collected, and the product in each sample was quantified as described (28). Cyclohexanol was measured by a gas-chromatography mass spectrometry assay (Agilent Technologies HP6890/MSO5893). Norbenzphetamine was quantified using a high-pressure liquid chromatography-mass spectrometer assay (ThermoFinnigan TSQ).
Crystallization and Data Collection—The Δ56 form of the ΔTGEE mutant (hereafter referred to as the soluble form), in which the first 56 residues of the N terminus, including the membrane anchor of ΔTGEE, were deleted, was constructed and expressed in a manner similar to that of the full-length ΔTGEE protein (details in supplemental materials). The purification procedure (details in supplemental materials) was also similar except that 0.1% Triton X-100 rather than 0.3% Triton X-100 was used, and an affinity column (2′,5′-ADP-Sepharose) was included at the final step. Prior to crystallization setup, 2× and 20× molar excesses of FMN and NADP+, respectively, were added to the protein sample followed by two cycles of concentration/dilution to remove excess cofactors. The protein solution was concentrated to 15 mg/ml in 50 mm HEPES, pH 7.5. Crystals were grown using the hanging drop method by mixing 1.8 μl of the protein solution and 2 μl of reservoir solution (100 mm HEPES, pH 7.5, 150 mm NH4SCN, and 15% polyethylene glycol 5000 monomethylester). Small thin plate-shaped crystals appeared within a week. Crystals were soaked in a cryoprotectant solution (reservoir buffer plus 10% glycerol) for ∼5 s and then flash frozen in liquid nitrogen prior to data collection. A 3.4-Å data set was collected at the SBC 19ID beamline, Advanced Photon Source. Data processing was done with the program HKL2000 (29). The crystals belong to the space group P21 with unit cell dimensions of a = 109.1Å, b = 93.1Å, and c = 125.7 Å and β = 100.0° and contained three molecules of the ΔTGEE mutant per asymmetric unit.
Structure Determination—Molecular replacement methods were employed for phase determination. Not surprisingly, no reasonable solutions were obtained using either the program, MOLREP in CCP4 (CCP4, Programs for protein crystallography (30)) or Phaser (31), when the wild-type rat CYPOR structure (PDB ID, 1AMO) was used as the search model. However, when separate domains of CYPOR, i.e. the FAD domain and the FMN domain, were used as search models, Phaser successfully located three FAD domains and two FMN domains sequentially with the rotation function Z score/translation function Z scores/log likelihood gain of 18.5/15.4/367, 15.7/37.6/1294, 12.7/42.9/2392, 7.8/40.4/3211, and 2.6/15.1/4688, respectively, with an R value of 40.9%. Because all the cofactors were excluded from the search models, the strong electron densities for FAD and FMN at expected positions confirmed the correctness of the solution. The third FMN domain could not be located at this stage, although there was a large enough empty space for an FMN domain in the crystal packing. When four cofactors (three FAD and one FMN) were added to the model, then the linker region between the two flavin domains of the second molecule was clearly visible in the |Fo| - |Fc| difference Fourier map (Fig. 2). In addition, the adenosine pyrophosphate portion of NADP+ in all three FAD domains could be identified. Further refinements were carried out using the program CNS (32) and manual model adjustments with COOT (33), yielding R-factors of 33.3% and 25.1% for R-free and R-crystal, respectively. During the refinement, tight non-crystallographic symmetry restraints for the three FAD domains and two FMN domains were used until the last cycle of refinement, which gave final R-factors of 27.3% and 21.4% for R-free and R-crystal, respectively. At this stage, all three FAD domains are well defined. However, only one FMN domain is clearly defined and the B-factors of the β-sheets and loop areas of the second FMN domain were extremely high (∼200 Å2). Therefore, all but 78 residues, mostly corresponding to α helices, were excluded from the subsequent refinement. At this stage, some patches of densities in the |Fo| - |Fc| map were visible extending from the third hinge that could be assigned to the α helices F and B of the third FMN domain, and thus the third FMN domain could be modeled. However, this FMN domain was not included in the refinement. The final R-factors were 27.9% for R-free and 21.9% for R-crystal. Data collection and the refinement statistics are given in Table 1.
FIGURE 2.

Omit |Fo| - |Fc| map of the ΔTGEE hinge regions for all three molecules, Mol A, Mol B, and Mol C. The last helix (helix F) of the FMN domain and the first β strand (β6) of the connecting/FAD domain are depicted with thin lines, and the hinge sections are depicted with thick lines. Maps are contoured at the 2.5 σ level. Although side chains of some residues in the hinge regions are not visible, the main chains of all three hinges are clearly defined.
TABLE 1.
Data collection and refinement statistics
| Sample | ΔTGEE |
|---|---|
| Data collection | |
| Resolution (Å) | 50-3.4 (3.52-3.40)a |
| Total measured reflections | 142,069 |
| Unique reflections | 33,940 |
| Completeness (%) | 97.4 (83.3) |
| Redundancy | 4.2 (2.4) |
| I/σ(I) | 11.0 (1.1) |
| Unit cell dimensions a, b, c (Å); β (°) | 109.1, 93.0, 125.7; 100.0 |
| Space group | P 21 |
| Rsym | 0.134 (0.652) |
| No. of monomers in an asymmetric unit | 3 |
| Vm (Da/Å3) | 3.0 |
| Refinement | |
| Resolution (Å) | 30-3.4 |
| Rcrystal/Rfree | 0.219/0.279 |
| r.m.s.d. bond (Å)/angle (°) | 0.009/1.5 |
| B-factor analysis (Å2) | |
| Wilson B | 86.0 |
| Mol A: FAD/FMN domain | 98.6/95.4 |
| Mol B: FAD/FMN domain | 93.8/160.0b |
| Mol C: FAD domain | 117.9 |
| Ramachandran analysis | |
| Most favored (%) | 71.6 |
| Allowed (%) | 26.0 |
| Generously allowed (%) | 1.8 |
| Disallowed (%) | 0.6 |
Numbers in parentheses are for the highest resolution shell
Only 78 of a total of 172 residues were included in the refinement
Docking of CYPOR and Cyt P450 2B4—Two docking methods were employed: GRAMM-X (34) and Z-DOCK (35). In the GRAMM-X docking, Mol A was assigned as the receptor molecule and cyt P450 2B4 (PDB code, 1SUO) as the ligand. Glu-208 of Mol A and any four of the nine residues (Arg-122, Lys-126, Arg-133, Phe-135, Met-137, Lys-139, Arg-422, Lys-433, and Arg-443) of cyt P450 were required to be in the interface area. Mutagenesis studies have identified Glu-208 of CYPOR as being involved in the interaction with cyt P450 (22). The nine residues of cyt P450 2B4 have been shown by site-directed mutagenesis to be involved in the interaction with CYPOR (26). In using ZDOCK, again, Glu-208 of Mol A and Arg-133 and Lys-433 of cyt P450 were included as required residues in the contact area. Among the top 10 solutions obtained from each method, the only common solution (fifth in both methods) was selected. No further refinement of the docked structure was attempted.
RESULTS AND DISCUSSION
Rationale for Mutations in the 12-Amino Acid Hinge Joining the FMN Domain to the Remainder of the Protein—To determine whether the flexible random-coil hinge joining the FMN to the connecting domain of CYPOR functioned in orienting the FMN and FAD domains of the protein for electron transfer from FAD via FMN to the acceptor protein cyt P450, the hinge was mutated. To identify the boundaries of the hinge a multiple sequence alignment of the 45 known CYPOR sequences and a structural alignment of two CYPOR structures (rat and yeast) were performed (17, 18, 36) (supplemental Fig. S1). The alignments indicated the length of the linker was conserved and that it spanned 12 residues from Gly-232 to Arg-243 in the rat protein. Although the sequence identity between rat and yeast reductase is only 25%, the structures were highly conserved (r.m.s.d. for backbone atoms, 1.4 Å) (36). Rat neuronal nitric-oxide synthase (NOS) reductase has an analogous, 24-amino acid flexible hinge, which influences the electron flow through the NOS reductase to an acceptor molecule (37–39). The NOS hinge sequence is not conserved with respect to the hinge of CYPOR. To investigate whether the length of the CYPOR hinge altered electron flow through the reductase and/or modulated the ability to reduce its electron transfer partner, cyt P450, the length of the hinge was increased by two and four alanine residues and shortened by two and four amino acids, using protein engineering techniques. The hinge also contains two charged residues (Glu-238 and Glu-239), which form salt bridges with the guanidinium side chain of Arg-104 in the FMN domain. To test the influence of these salt bridges on the stability of the closed conformation, Glu-238 and Glu-239 were mutated to alanine. In addition, a mutant with four amino acid changes in the middle of the hinge (Thr-236 → Ala, Gly-237 → Ala, Glu-238 → Ala, and Glu-239 → Ala) was constructed to determine if these particular amino acids and the sequence were required for normal function.
Activity of the Mutant Reductases with Cyt P450 2B4, Cytochrome c, and Ferricyanide—The ability of the CYPOR variants to support catalysis by the physiological redox partner, full-length cyt P450 2B4, was measured by quantitating the N-demethylation of benzphetamine to formaldehyde (Table 2). The 2- and 4-residue substitution mutants (E238A/E239A and T236A/G237A/E238A/E239A) and the 2- and 4-alanine addition mutants exhibited normal or slightly increased activity. The 2-amino acid deletion mutant, ΔTG 236–237 (hereafter, simply referred to as ΔTG), supported a wild-type rate of catalysis, whereas the activity of the 4-amino acid deletion ΔTGEE 236–239 (hereafter referred to as ΔTGEE) was undetectable.
TABLE 2.
Activity of the wild-type and CYPOR mutants with redox partners The activity of reductases with their redox partners cytochrome c, ferricyanide, and cyt P450 2B4 was determined at 30 °C as described under “Experimental Procedures.” The activity of CYPOR with cyt P450 2B4 is reported in units of nmol of formaldehyde produced/s/nmol of CYPOR.
| Reductase mutants | cyt P450 | Fe3+ cyt c | 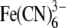 |
|
|---|---|---|---|---|
| nmol/s/nmol CYPOR | nmol reduced/s/nmol CYPOR | |||
| Wild type | 0.67 ± 0.07 | 70.6 ± 5.1 | 135 ± 6.4 | |
| ΔTG-(236–237) | 0.77 ± 0.083 | 11 ± 0.64 | 170 ± 9 | |
| ΔTGEE-(236–239) | NDa | 0.33 ± 0.026 | 136 ± 3.9 | |
| E238A/E239A | 0.95 ± 0.1 | 78 ± 6.4 | 141 ± 6.4 | |
| T236A/G237A/E238A/E239A | 0.83 ± 0.08 | 77 ± 6.4 | 167 ± 10 | |
| G237/+AA/E238 | 0.83 ± 0.1 | 109 ± 4 | 137 ± 5 | |
| G237/+AAAA/E238 | 0.82 ± 0.07 | 106 ± 2.6 | 160 ± 6.5 | |
ND, not detected
Cytochrome c is not a physiological redox partner for CYPOR. Nevertheless, it is often used to assess the reductase activity because of the simplicity and rapidity of the assay (24). Moreover, there is evidence that the binding site for cyt P450 and cytochrome c, both basic proteins, on the acidic FMN domain of the reductase is either identical or overlapping (40, 41). The data in Table 2 demonstrate that the substitution mutants (E238A/E239A and T236A/G237A/E238A/E239A) exhibit wild-type activity with cytochrome c, indicating that the two salt bridges do not influence the activity of the reductase. In contrast, the 2- and 4-alanine addition mutants exhibit a ∼50% increase in activity compared with the wild-type enzyme, suggesting that the rapid reduction of cytochrome c may be somewhat restricted by the length of the hinge in the wild-type CYPOR. In contrast, the two, ΔTG, and four, ΔTGEE, amino acid deletion mutants reduced cytochrome c 6.4- and 214-fold slower than wild type, respectively. The lower activity of the deletion mutants with cytochrome c could not be attributed to a diminished affinity of the mutant enzymes for either cytochrome c or NADPH. The Km values, of cytochrome c for the wild-type and 2- and 4-residue deletion mutants were 24 ± 3, 13.5 ± 3, and 15 ± 2 μm, respectively. NADPH bound to wild-type, and the 2- and 4-amino acid deletion mutants with a Km of 6.2 ± 0.75, 3 ± 0.4, and 2.7 ± 0.46 μm, respectively. If anything, the mutants had a slightly enhanced affinity for cytochrome c and NADPH. Thus, neither lengthening nor altering the sequence of the hinge interfered with the function of the reductase. Shortening the hinge by two amino acids was not deleterious to its function with the slow acting cyt P450 (kcat = 0.77 s-1 versus 0.67 s-1 of wild-type); however, it did diminish its more rapid activity with cytochrome c 7-fold from a kcat of 79 to 11 s-1 The 4-amino acid deletion in the hinge was a major blow to the reductase activity with both cyt P450 and cytochrome c. The activity with cyt P450 was undetectable, and cytochrome c reduction activity was <0.5% of the wild-type activity.
The reductase domain in a homologous protein, NOS, has a hinge between its FMN and connecting domain which, when shortened by two amino acids, leads to diminished nitric oxide formation due to a diminished rate of electron transfer to the heme (37). Apparently a minimum length of the hinge in CYPOR and NOS reductase is essential to enable the enzymes to transfer electrons to their physiological redox partners. Our observation with CYPOR is consistent with the notion that flexible hinges in multidomain redox proteins frequently regulate the activity of the enzymes, presumably by allowing large interdomain rearrangements (42).
Three Extended Structures of ΔTGEE Cyt P450 Reductase—The dramatic reduction in the activity of ΔTGEE with cyt P450 and cytochrome c, while retaining the full activity in ferricyanide reduction (Table 2), prompted us to determine the crystal structure of this deletion mutant. Fig. 1 illustrates the crystal structure of ΔTGEE in the three different conformations (Mol A, Mol B, and Mol C) found in an asymmetric unit. Initially, the entire polypeptide chains of Mol A and the FAD domains of Mol B and Mol C were easily traced. However, only helices and parts of β-strands of the FMN domain of Mol B were visible, and most of the polypeptide of the Mol C FMN domain was not visible. Although the FMN domains of Mol B and C FMN domains are not well ordered, it is clear that the polypeptide folds and the relative positions of cofactors in each of the five visible domains (three FAD- and two FMN-domains) are the same as those of the corresponding domains in the wild-type structure. At the final stages of the refinement, even the FMN domain of Mol C and its FMN cofactor could be placed, although parts of their electron densities are discontinuous. However, unlike the wild-type CYPOR structure in which the hinge is partially disordered, those in all three ΔTGEE molecules are relatively well ordered. Portions of the electron density map corresponding to the hinges are shown in Fig. 2. In Fig. 1, the FAD domains of the wild-type and three extended structures are superimposed showing that the relative positions and orientations of the four FMN domains are markedly different. The fact that the structure of each of the three FAD domains of the mutant CYPOR are the same as that of wild-type is consistent with the observation that the ferricyanide reduction activity of ΔTGEE is the same as that in wild type (Table 2).
FIGURE 1.

Crystal structure of three conformations of ΔTGEE. Three conformations of ΔTGEE and the wild-type structure are compared. The relative orientations of FMN domains of Mol A (magenta; FMN is green), Mol B (blue; FMN is red), and Mol C (gray; FMN is yellow) of ΔTGEE, when their FAD domains are superimposed onto the wild-type CYPOR structure (gold; FMN is blue, and FAD is black). For clarity, only the FAD domain of wild-type is shown. The hinges are depicted as thick tubes in their respective colors, and the flavins are shown as stick models. The FMN domain moves away from the FAD domain by extending the hinge and simultaneously rotating about a pivot point, which is located on the backbone carbonyl of the C-terminal residue of the hinge (Arg-243 in wild-type numbering). Note that the FMN isoalloxazine ring is exposed to solvent in all three conformations such that its dimethyl group can interact with cyt P450.
The Structure of ΔTGEE Is Flexible—Mol A is the most compact of the three structures, yet it is much more open than the wild-type structure (Fig. 1). The contact area between the FMN- and FAD-domains in Mol A is 395 Å2, whereas that of the wild-type structure is 966 Å2. The orientations of the FMN domain with respect to the FAD domain in the two molecules are very different. The FMN domain in Mol A has moved away from the FAD domain and rotated so that the dimethyl group is exposed to solvent and able to interact with its electron transfer partner, cyt P450. The movement of the FMN domains occurs essentially by pivoting on the backbone carbonyl carbon atom of Arg-239 of ΔTGEE (Arg-243 of wild type) at the C-terminal end of the hinge. For visualization of a plausible FMN domain movement between the conformation of the wild type and Mol A, see Refs. 43 and 44 and http://www.molmovdb.org/cgi-bin/morph.cgi?ID=234385-8941.
On the other hand, Mol B and Mol C have completely extended conformations without any contact between their respective FAD- and FMN-domains and with their hinges completely stretched (Fig. 1). In the crystal, the FMN domain of Mol A has contacts with the FAD domain of Mol B, and these contacts are probably responsible for the relatively well ordered electron densities for the Mol A FMN domain compared with that of Mol C. Because only the intact ΔTGEE protein was observed by SDS-PAGE analysis of the crystalline protein (supplemental Fig. S2), the lack of density for Mol C is not due to the absence of the FMN domain as a result of proteolysis during the crystallization procedure. Rather, the FMN domain of Mol C must be highly disordered in the crystal lattice, indicating that the ΔTGEE molecule is completely flexible in solution.
In the wild-type structure, FAD and FMN are juxtaposed and the C-8 methyl groups of their benzoid rings are in van der Waals contact, suggesting that the electron transfer between the cofactors is relatively rapid (17). However, the distance between the two C-8 methyl groups of the two flavins in Mol A is ∼29 Å, in Mol B is ∼59 Å, and that in Mol C is ∼60 Å, predicting that electron transfer between the two flavins in any of these three ΔTGEE conformations would be extremely inefficient. This is reflected in our kinetic studies. The activity for cytochrome c reduction in ΔTGEE is only 0.5% of that in wild type. A similar value was observed in a reconstituted system containing the separate FMN and FAD domains. Smith and coworkers reported that reconstitution of the separate FMN and FAD domains of human CYPOR gave 1.6% of the wild-type activity in cytochrome c reduction (45). Thus, the observed activity in ΔTGEE could be due to a bimolecular interaction rather than the electron transfer from FAD to FMN in a same molecule. On the other hand, it is also possible that, in certain conformations of ΔTGEE in solution, electrons could be transferred from FAD to FMN in the same molecule but very inefficiently. These two possibilities will be discussed further in the next section.
ΔTGEE Is Monomeric and Its Conformation Is Elongated in Solution—A third possibility for ΔTGEE having a small but significant rate of cytochrome c reduction activity is that ΔTGEE forms a dimer in solution as observed in the studies of human CYPOR, in which seven residues of the hinge were deleted (46). This seven-residue deletion mutant of human CYPOR was found to form a dimer in solution, and its electron transfer path was postulated to be from the FAD domain of one monomer to the FMN domain of the other monomer in the dimeric molecule. Although the crystal structure of ΔTGEE showed no indication of oligomer formation, we investigated the hydrodynamic nature and oligomeric state of the ΔTGEE protein in solution. Thus, the protein was subjected to gel filtration chromatography. Fig. 3 shows that the ΔTGEE protein is monomeric but runs slightly faster than the wild-type CYPOR, indicating that the mutant adopts a slightly more elongated conformation compared with wild-type. This is also consistent with the results of the limited trypsin digestion (supplemental Fig. S3). Although the wild-type protein is rather resistant to trypsin treatment, the mutant is more susceptible to proteolysis, indicating that the mutant protein has a more open conformation, again consistent with the crystal structure.
FIGURE 3.

Quaternary structure of the ΔTGEE protein. HPLC gel filtration profiles of wild-type (dotted curve) and ΔTGEE (solid curve). For column calibration, bovine serum albumin (BSA, 66kDa) and alcohol dehydrogenase (ADH, 150 kDa) were used. The mutant protein eluted slightly faster than the wild type, indicating that the mutant is monomeric, but slightly more elongated than wild type.
Electron Transfer from NADPH to FAD Occurs at the Same Rate in the Wild-type and Mutant CYPORs—There are three possible explanations for the inability of the ΔTGEE mutant to catalyze the oxygenase activity of the cyt P450 and to reduce cytochrome c. One is that hydride transfer from NADPH to FAD has been disrupted. This unlikely possibility, in light of the structure of the mutant CYPOR, was eliminated by demonstrating that the steady-state reduction of potassium ferricyanide by the mutants was similar to that of the wild-type protein (Table 2). The reduction of ferricyanide has been extensively used to probe the integrity of hydride transfer from NADPH to FAD, because it has been shown previously that FAD is responsible for the reduction of ferricyanide in the intact protein (10, 11, 24). Moreover, our results are also consistent with the findings of other investigators that a 7-amino acid deletion in the hinge of human CYPOR did not perturb the binding of 2′,5′-ADP and presumably NADPH (46). The second and most likely explanation for the poor activity of the ΔTGEE mutant, in view of its open structure, is that electron transfer from FAD to FMN has been disrupted. The third possibility, which was considered improbable due to the structure of ΔTGEE, is that the FMN domain exists in a conformation that can receive electrons from FAD but is incapable of donating them to the acceptor protein.
Electron Transfer from FAD to FMN Is Moderately Retarded in ΔTG but Severely Inhibited in ΔTGEE—To determine whether FAD to FMN or FMN to cyt P450 electron transfer was disrupted in the mutants, the kinetics of electron transfer from the FAD hydroquinone to FMN were investigated by stopped-flow spectrophotometry under anaerobic conditions following their reduction by a stoichiometric amount of NADPH. In the wild-type protein, there is a rapid (30–70 s-1), reversible exchange of electrons between the two flavins (20, 21). At equilibrium, the distribution of the electrons is governed by the redox potentials of the individual flavin half-reactions (FMNox/sq =-110 mV, FMNsq/hq =-270 mV, FADox/sq = -290 mV, and FADsq/hq =-365 mV) (7–9).
Kinetics of CYPOR Reduction by 1 Equivalent of NADPH—Table 3A and Fig. 4B reveal that, when followed at 450 nm, the reduction of CYPOR by 1 molar equivalent of NADPH is biphasic. The first rapid phase represents reduction of FAD by NADPH. In the wild-type protein, ∼20% of this decrease in absorbance also reflects the rapid electron transfer from FADH2 to FMN. As expected from the structures, the rate constant for the first phase is similar for all three proteins, indicating that reduction of FAD by NADPH has not been modified in the mutant proteins. The ability of the FAD domains of the mutant proteins to reduce ferric cyanide at the wild-type rate supports the notion that reduction of FAD by NADPH proceeds in a normal manner (Table 2). The amplitude of the second phase of the wild-type protein is small (10%), whereas in the mutants, the amplitude of the second phase is of approximately the same magnitude as the first phase. Because the rate constants of the mutant's second phase are within experimental error, similar to the rate constants for blue semiquinone formation observed at 590 nm, the second phase was proposed to represent transfer of an electron from FADhq to FMN.
TABLE 3.
Reduction of wild-type and mutant CYPOR by 1 (A) and 10 (B) molar equivalents of NADPH The reactions were carried out anaerobically in 100 mm potassium phosphate, pH 7.4, plus 15% glycerol buffer at 25 °C, as described under “Experimental Procedures.” The final concentration of the CYPOR after mixing was 10 μm, whereas NADPH concentration was 10 μm in A and 100 μm in B. A is the amplitude of the phase.
| A) Reductase | λobs | A1 | kobs1 | A2 | kobs2 |
|---|---|---|---|---|---|
| nm | % | s–1 | % | s–1 | |
| Wild type | 450 | 90 ± 6 | 42 ± 4 | 10 ± 2 | 3.8 ± 0.5 |
| 590 | 45 ± 6 | ||||
| ΔTG-(236–237) | 450 | 53 ± 5 | 26 ± 5 | 47 ± 4 | 3.5 ± 0.7 |
| 590 | 6.5 ± 2 | ||||
| ΔTGEE-(236–239) | 450 | 43 ± 5 | 38.7 ± 5 | 56 ± 3 | 0.13 ± 0.03 |
| 590 | 0.11 ± 0.02 |
| B) Reductase | λobs | A1 | kobs1 | A2 | kobs2 | A3 | kobs3 |
|---|---|---|---|---|---|---|---|
| nm | % | s–1 | % | s–1 | % | s–1 | |
| Wild type | 450 | 74 ± 5 | 47 ± 5 | 26 ± 2 | 4.8 ± 0.5 | ||
| 590 | 49 ± 6 | 3.6 ± 0.3(decay)1 | |||||
| ΔTG-(236–237) | 450 | 38 ± 6 | 23 ± 5 | 46 ± 5 | 4.7 ± 2 | 16 ± 3 | 0.145 ± 0.03 |
| 590 | 5.8 ± 2 | 0.34 ± 0.05(decay)1 | |||||
| ΔTGEE-(236–239) | 450 | 29 ± 3 | 55 ± 6 | 20 ± 3 | 3.1 ± 0.3 | 51 ± 6 | 0.036 ± 0.006 |
| 590 | 0.06 ± 0.01 | 0.0043 ± 0.001(decay)1 |
This phase represents a decrease in absorbance
FIGURE 4.

Kinetics of reduction of wild-type, ΔTG, and ΔTGEE CYPOR by 1 molar equivalent of NADPH. The final concentration after mixing the CYPOR- and NADPH-containing solutions in the anaerobic stopped-flow spectrophotometer was 10 μm for the wild-type and ΔTG protein and 8.5 μm for ΔTGEE. The kinetic traces have been normalized. A, the absorbance increase at 590 nm indicates blue semiquinone formation; B, the decrease in absorbance at 450 nm reflects flavin reduction. The experiments were conducted at 25 °C, as described under “Experimental Procedures.”
Formation of the blue disemiquinone (FADsq/FMNsq), as indicated by the absorbance at 590 nm, occurred rapidly in the wild-type protein (k = 45 s-1), but was moderately (7-fold) and dramatically (410-fold) slower with the 2- and 4-amino acid deletion mutants (k = 6.5 s-1 and 0.11 s-1, respectively). Semiquinone formation increased exponentially, reaching a maximum in 1–3 s for wild-type, compared with the ΔTGEE mutant, where the maximum is merely approached at 100 s (Fig. 4A) (8). We suggest that the prominent, slower second phase at 450 nm (Fig. 4B) in the mutants reflects electron transfer from FADH2, which transiently accumulates in the mutants, to FMN to ultimately form an equilibrium mixture of two forms of the reductase (∼70% FMNhq/FADox and ∼30% FMNsq/FADsq).
Kinetics of CYPOR Reduction by 10 Equivalents of NADPH—Further characterization of the electron transfer properties of the mutants was undertaken by investigating the kinetics of reduction of the mutants by a 10-fold excess of NADPH. With 10 molar equivalents of NADPH, the reduction of CYPOR proceeds by the mechanism in Reaction 1.
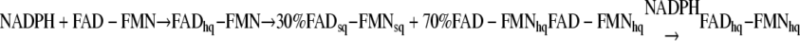 |
REACTION 1 |
Because the FAD-FMNhq is converted to the dihydroquinone, the FADsq-FMNsq will form more of the FAD-FMNhq to reestablish the equilibrium mixture. Due to the fact that NADPH is an obligate hydride ion donor, only the FMNH2/FAD form of the reductase is competent to receive a second hydride ion from NADPH.
The data in Table 3B and Fig. 5 illustrate that the kinetics of reduction by a 10-fold molar excess of NADPH are considerably more complicated than the reduction kinetics by a one molar equivalent of NADPH. Nevertheless, they also demonstrate that interflavin electron transfer is moderately impaired in ΔTG and severely damaged in ΔTGEE. In the wild-type protein, semiquinone formation (590 nm), which reflects electron transfer from FAD to FMN, occurs rapidly with a rate constant of 49 ± 6 s-1, its concentration peaking at 69 ms (Fig. 5A). The semiquinone then monophasically decays (k = 3.6 ± 0.3 s-1) as the reductase is reduced by a second molecule of NADPH, with a net increase in hydroquinones at the expense of semiquinones (see Reaction 2). The rate of reduction by the second equivalent of NADPH is slower than with the first equivalent for two reasons. The first is because of a decreased thermodynamic driving force. The FAD hydroquinone can no longer rapidly transfer its electrons to a FMN, which has already been reduced by the first NADPH. The second contributing factor is the decrease (∼30%) in FAD concentration compared with the completely oxidized protein. Semiquinone formation by ΔTGEE (k = 0.06 ± 0.01 s-1) is delayed for ∼1 s, and its level reaches a maximum much later, at ∼100 s. Moreover, the maximum amount of semiquinone formed is ∼20% greater than with that of wild type, consistent with accumulation of semiquinone secondary to slower interflavin transfer, which limits further reduction by the excess NADPH. The conversion of the ΔTGEE semiquinones to the hydroquinones is approximately three orders of magnitude slower (k = 0.0043 versus 3.6 s-1) than in the wild-type protein. The kinetics of the 2-residue deletion mutant was intermediate between those of wild type and ΔTGEE.
FIGURE 5.

Kinetics of reduction of wild-type, ΔTG, and ΔTGEE CYPOR by 10 molar equivalents of NADPH. The experiment was performed at 25 °C as described in the legend to Fig. 4 and under “Experimental Procedures.” The final reductase concentrations were 15 μm (590 nm) and 10 μm (450 nm). NADPH concentrations were 150 μm and 100 μm to provide a 10-fold molar excess. The absorbance was followed at 590 nm to observe blue semiquinone formation (A) and at 450 nm to estimate flavin reduction (B).
The complexity of the kinetics of reduction of the mutants at 450 nm by a 10-fold molar excess of NADPH was due to multiple, sequential interflavin electron transfer processes as the reductase became progressively reduced (Table 3B and Fig. 5B). The trend, however, was similar: reduction of ΔTG was moderately depressed, whereas reduction of ΔTGEE was severely diminished. At high NADPH concentrations, the amplitude of the slower phase of reduction of wild-type CYPOR measured at a 450 nm increases, whereas its rate constant (k = 4.8 ± 0.5 s) was the same as that at one molar equivalent of NADPH (k = 3.8 ± 0.5 s-1) and within experimental error, similar to the decay of the semiquinone species to hydroquinones (k = 3.6 ± 0.3) measured at 590 nm. This slower phase of bleaching at 450 nm for the mutants was therefore tentatively assigned to reduction of the FAD-FMNH2 hydroquinone form of the reductase by a second molecule of NADPH.
Kinetics of Electron Transfer in Dilute and Concentrated Solutions of CYPOR—Electron transfer from FAD to FMN and semiquinone formation in the ΔTG (k = 6.5 ± 2 s-1) and ΔTGEE (k = 0.11 ± 0.02 s-1) mutants occurs at a rate approximating the rate of cytochrome c reduction (kcat = 11 ± 0.64 s-1 and 0.33 ± 0.026 s-1) (Tables 2 and 3). The former reaction was measured at a reductase concentration of 10 μm, whereas the rate of cytochrome c reduction was measured at 10 nm. Because a 1000-fold dilution of protein did not alter the apparent rate of electron transfer from FAD to FMN, we suggest an intramolecular electron transfer process is being observed.
ΔTGEE Reduces Ferric Cyt P450 2B4 Slowly in the Presence of NADPH—To elucidate whether the FMN domain of ΔTGEE exists in a conformation that is capable of delivering electrons to cyt P450, the kinetics of reduction of ferric cyt P450 was investigated with stopped-flow spectrophotometry, by following formation of ferrous CO cyt P450. Under our experimental conditions CO rapidly associates with cyt P450, at a rate of ∼1000 to 2000 s-1, and does not contribute to the rate-limiting step (47, 48).
Fig. 6 illustrates the biphasic kinetic traces of reduction of ferric cyt P450 by wild-type and ΔTGEE in the absence and presence of benzphetamine. The reduction of cyt P450 by the 2-residue deletion mutant was slightly slower than that of wild-type (6.7 s-1 versus 4.2 s-1, Table 4) and occurred with a rate constant similar to the rate of electron transfer from FAD to FMN. Cyt P450 reduction was faster in the presence of benzphetamine, because the substrate raises the redox potential of the heme, which increases the thermodynamic driving force of the reaction (49). Fig. 6 also demonstrates that the substrate concomitantly enhanced the rate constant and the amplitude of the fast phase of the wild-type reductase (Table 4). In a striking contrast, ΔTGEE reduced cyt P450, with a lag of ∼1 s after the addition of NADPH, which reflects the time required for FMN hydroquinone formation. The reduction of cyt P450 by ΔTGEE was independent of the absence or presence of benzphetamine, demonstrating that the rate-limiting step with ΔTGEE was not the potential of the ferric heme but rather the reduction of FMN. The fitting of kinetics traces of cyt P450 reduction by ΔTGEE yielded two rate constants, 0.06 and ∼0.005 s-1 (Table 4), which correspond to the rate constants for electron transfer to FMN in the presence of excess NADPH (Table 3B).
FIGURE 6.

Kinetics of reduction of ferric cytochrome P450 by wild-type and mutant CYPOR in the presence of CO. The ΔAN (normalized absorbance) represents the ratio between 450 nm A∞ - At and 450 nm A∞ - At = 0, where t = time and ∞ is infinity. The final cyt P450 and reductase concentration after mixing with NADPH was 5 μm. Final NADPH concentration was 50 μm. Experiments were conducted as described under “Experimental Procedures” with a saturated solution of CO in both syringes. BZ, benzphetamine.
TABLE 4.
Reduction of substrate-bound and substrate-free ferric cyt P450 2B4 by CYPOR in the presence of a 10-fold excess of NADPH The reactions were carried out in 100 mm potassium phosphate, pH 7.4, plus 15% glycerol buffer at 25 °C. The final concentration after mixing was 5 μm for reductase and cyt P450 2B4 and 50 μm for the NADPH. A is the amplitude of the phase.
| Reductase | Benzphetamine | A1 | kobs1 | A2 | kobs2 |
|---|---|---|---|---|---|
| mm | % | s–1 | % | s–1 | |
| Wild type | 0 | 56 ± 5 | 1.32 ± 0.3 | 44 ± 4 | 0.16 ± 0.025 |
| 1 | 88 ± 8 | 6.7 ± 0.7 | 12 ± 2 | 0.8 ± 0.1 | |
| ΔTG-(236–237) | 0 | 54 ± 6 | 0.62 ± 0.05 | 46 ± 5 | 0.13 ± 0.02 |
| 1 | 77 ± 8 | 4.2 ± 0.7 | 23 ± 4 | 0.38 ± 0.1 | |
| ΔTGEE-(236–239) | 0 | 24 ± 3 | 0.053 ± 0.006 | 76 ± 8 | 0.005 ± 0.00045 |
| 1 | 30 ± 3 | 0.06 ± 0.004 | 70 ± 5 | 0.0046 ± 0.0005 |
Two-electron Reduced ΔTGEE Catalyzes Product Formation by Cyt P450 under Single Turnover Conditions at the Same Rate as the Native CYPOR—Cyt P450 requires two electrons to catalyze the oxidation of substrate. The first electron reduces the ferric substrate-bound cyt P450. The ferrous cyt P450 rapidly binds oxygen and then accepts a second electron from the FMN hydroquinone to form the reduced oxyferrous cyt P450 intermediate. In the previous section, reduction of ferric cyt P450 but not oxyferrous cyt P450 was observed. To investigate whether the FMNH2 of ΔTGEE was competent to deliver the second electron to oxyferrous cyt P450 2B4 and catalyze product formation, its ability to metabolize the two substrates, cyclohexane and benzphetamine, was measured under single turnover conditions.
The strategy of this experiment is to bypass the first electron transfer step and focus on the molecular events, which occur subsequent to transfer of the second electron to oxyferrous cyt P450 and its catalytic turnover to yield ferric cyt P450 and the product. The rate of product formation was measured in a rapid chemical quench apparatus by mixing a 1:1 complex of the preformed dithionite reduced Fe2+ cyt P450:2-electron reduced ΔTGEE with oxygen (49, 28). The reaction was quenched, and the product was quantified by high-pressure liquid chromatography-mass spectrometry. The kinetics of cyclohexanol and norbenzphetamine formation by cyt P450 in the presence of wild-type and ΔTGEE are identical and occurred in a single phase (Table 5). It is noteworthy that product formation (k = ∼0.1 s-1 for benzphetamine at 15 °C and k =∼ 3 s-1 for cyclohexane at 25 °C) was slower than reduction of ferric cyt P450 (k = 6.7 s-1 for wild type) (Table 4). This is because catalysis by cyt P450 2B4 proceeds via a long lived intermediate in the presence of CYPOR (28, 49).
TABLE 5.
Rate of product formation by cyt P450 2B4 under single turnover conditions in the presence of wild-type and ΔTGEE CYPOR
| Substrate | Reductase | k (s–1) | Yielda |
|---|---|---|---|
| Benzphetamine | Wild type | 0.15 ± 0.05 | 0.7 |
| ΔTGEE | 0.1 ± 0.014 | 0.52 | |
| Cyclohexane | Wild type | 2.5 ± 0.05 | 0.21 |
| ΔTGEE | 3.3 ± 0.3 |
Expressed in units of nmol of product, norbenzphetamine or cyclohexanol, per nmol of cyt P450. The reactions were conducted as described under “Experimental Procedures”
To summarize the results of the preceding experiments characterizing the function of the mutant CYPOR, an outline of the kinetics of intramolecular and intermolecular electron transfer in wild-type and ΔTG and ΔTGEE follows.
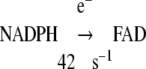 |
REACTION 2 |
Reaction 2 was normal in ΔTG and ΔTGEE (Table 3A).
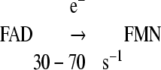 |
REACTION 3 |
Reaction 3 decreased 7-fold in ΔTG (k = 6.5 s-1) and 410-fold in ΔTGEE (k = 0.11 s-1) (Table 3A) (20, 21).
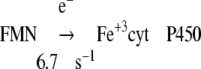 |
REACTION 4 |
Reaction 4 was normal in ΔTG and ΔTGEE if provided with sufficient electrons. In the presence of NADPH, reduction of cyt P450 was limited due to slow electron transfer from FAD to FMN (Table 4).
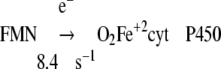 |
REACTION 5 |
Reaction 5 was inferred to be normal, because the rate of product formation was the same in the wild-type and mutant proteins. The rate constant under each arrow is the rate for wild-type CYPOR (49).
These data unambiguously demonstrate that the 4-amino acid deletion in the hinge did not affect the functional interaction of the FMN domain with cyt P450, provided that the FMN hydroquinone was present in the reductase.
Model of the CYPOR·P450 Complex—To determine whether the extended reductase could bind to cyt P450, we have constructed a model complex between these electron transfer partners, using the structures of ΔTGEE Mol A and cyt P450 2B4 (PDB code, 1SUO) (12). Mol A is the least extended of the three molecules. The common solution obtained from the two docking methods (GRAMM-X and Z-DOCK) is displayed in Fig. 7. The CYPOR molecule (Mol A) has an open conformation such that the concave basic proximal surface of the cyt P450 molecule could snuggly fit into the acidic, convex-exposed FMN side of the molecule, which has been implicated by mutagenesis experiments to be involved in binding cyt P450 (22). The total contact area between the two molecules is ∼1500 Å2, of which 870 Å2 is located between the FMN domain and cyt P450.
FIGURE 7.

Model of a complex between cyt P450 2B4 and Mol A of ΔTGEE. A, surface representation of the model complex: pink, cyt P450; yellow, FMN domain; and green, FAD domain. B and C, electrostatic surface of the interfaces of cyt P450 (B) and CYPOR (C) where blue represents a positively charged surface and red is a negative surface. The heme and flavin are shown below the surface of cyt P450 and CYPOR, respectively. D, stick representation of heme (red) and FMN (yellow) cofactors in the complex showing their relative orientations and the two residues (Phe-429 and Glu-549) of cyt P450 that lie in between the two redox cofactors. E and F, interfaces of docked CYPOR and cyt P450. Five pairs of salt bridges are shown at the interface between cyt P450 and CYPOR. The salt-bridge pairs are marked with the same letters, i.e. R122(a) of cyt P450 pairs with E92(a) of CYPOR. Arg-443 of cyt P450 forms an H-bond with Tyr-178 of the CYPOR. The views are the same as those in panels B and C, respectively.
The contacts between the FMN domain and cyt P450 are mostly electrostatic (Fig. 7, B and C). There are five possible salt bridges (Glu-92, Glu-93, Asp-113, Glu-142, and Asp-208 from the FMN domain and Arg-122, Arg-126, Lys-433, Arg-422, and Arg-133 of cyt P450, respectively) and one hydrogen bond at the interface (Arg-443 of cyt P450 and Tyr-178 of ΔTGEE) (Fig. 7E). The FMN domain interacts with the concave basic proximal face of the cyt P450 so that the planes of the heme and FMN are almost perpendicular to each other (Fig. 7D). The shortest distance between the heme and the flavin (the vinyl group of the B ring and the C8 methyl group of FMN) is ∼12 Å. However, in between them, lie the side chains of Phe-429 and Glu-439 of cyt P450, suggesting that these residues might serve as an electron transfer conduit between the FMN and heme cofactors.
Interestingly, the arrangement of the FMN domain and cyt P450 and the relative orientations of FMN and the heme in this docked model are similar to those found in the crystal structure of the complex between the heme- and FMN-binding domains of bacterial cyt P450 BM3, except that the distance between the two cofactors in the cyt P450 BM3 structure is slightly longer (∼18 Å) (23). Because the FAD domain also makes contact with cyt P450 in the model (the remaining surface contact area was 630 Å2), Mol A is the minimum open conformation of CYPOR that can accommodate the cyt P450 molecule for a productive electron transfer between the two partners. All nine cyt P450 residues predicted by mutagenesis studies to be interacting with CYPOR are on the interface of our model between cyt P450 and the FMN domain (26).
In conclusion, the biochemical and structural data presented here demonstrate for the first time that the two flavin domains in CYPOR are able to move apart by pivoting on the C terminus of the hinge. We infer that a similar separation of domains occurs in the wild-type protein and that this movement enables the FMN domain to adopt a conformation capable of interacting with its physiological electron transfer partner, cyt P450. The previously determined wild-type CYPOR crystal structure, in which the two flavins are juxtaposed, represents an optimal conformation for the FAD to FMN electron transfer, whereas the currently observed open conformations represent those for electron transfer from the CYPOR FMN to the cyt P450 heme. Previous and current crystal structures elucidate the structural basis of the function of CYPOR and demonstrate that CYPOR undergoes significant rearrangements during its catalytic cycle (18). The first step in the CYPOR catalytic cycle is the binding of NADPH. In order for the nicotinamide ring of NADPH to bind to and reduce FAD, the indole ring of Trp-677, which stacks on the isoalloxazine ring of FAD, must move away from the FAD (18). Electron transfer from FAD to FMN is facilitated by the hinge joining the FMN and connecting domain, by properly aligning and orienting the two flavin domains in a “closed” CYPOR conformation for optimal electron transfer. This same hinge not only provides optimal alignment of the FAD and FMN cofactors, but it also allows the FMN domain to pivot on the C terminus of the hinge and thereby undergo a drastic structural rearrangement that separates the FMN and FAD domains to form an extended conformation capable of interacting with and reducing cyt P450. The hinge of CYPOR, therefore, regulates electron flow from FAD to FMN and from FMN to cyt P450 by adjusting the distance and orientation of the two flavin cofactors. The three CYPOR structures provide insight into the mechanism by which CYPOR undergoes a large conformational change in the course of electron transfer from FAD to FMN and from FMN to cyt P450, its physiological redox partner. Furthermore, because CYPOR is the prototype of the diflavin enzyme family that utilizes pyridine nucleotides, our findings can be applied to the mechanism of electron transfer in other members of the family, including NOS isozymes and methionine synthase reductase.
Supplementary Material
Acknowledgments
We thank the staff at the Advanced Photon Source beamlines SBC 19ID for their excellent assistance in data collection. Helpful discussions with Dr. David Ballou of the University of Michigan are gratefully acknowledged. The assistance of Launa Wakenhut in preparation of the manuscript is greatly appreciated.
The atomic coordinates and structure factors (code 3ES9) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grants GM35533 (to L. W.) and GM52682 (to J. J. K.). This work was also supported by a Veterans Affairs Merit Review Grant (to L. W.). Use of the Advanced Photon Source was supported by the U.S. Dept. of Energy, Basic Energy Sciences, Office of Science, under Contract W-31-109-Eng-38 and Office of Biological and Environmental Research under contract DE-AC02-06CH11357. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental text, references, Figs. S1–S3, and Table S1.
Footnotes
The abbreviations used are: CYPOR, NADPH-cytochrome P450 oxidoreductase; cyt, cytochrome; FADhq, FAD hydroquinone; FADsq, FAD semiquinone; FADox, oxidized FAD; FMNhq, FMN hydroquinone; FMNsq, FMN semiquinone; FMNox, oxidized FMN; NOS, nitric oxide synthase; r.m.s.d., root mean square deviation; ΔTG, the 2 (ΔThr-236, Gly-237) amino acid deletion mutant; ΔTGEE, the 4 (ΔThr-236, Gly-237, Gly-238, Gly-239) amino acid deletion mutant.
References
- 1.Masters, B. S., and Okita, R. T. (1980) Pharmacol. Ther. 9 227-244 [DOI] [PubMed] [Google Scholar]
- 2.Paine, M. J., Scrutton, N. S., Munro, A. W., Gutierrez, A., Robert, G. C. K., and Wolf, C. R. (2005) in Cytochrome P450, 3rd Ed. (Ortiz de Montellano, P. R., ed) pp. 115-148, Kluwer Academic/Plenum Publishers, New York
- 3.Schacter, B. A., Nelson, E. B., Marver, H. S., and Masters, B. S. (1972) J. Biol. Chem. 247 3601-3607 [PubMed] [Google Scholar]
- 4.Enoch, H. G., and Strittmatter, P. (1979) J. Biol. Chem. 254 8976-8981 [PubMed] [Google Scholar]
- 5.Horecker, B. L. (1950) J. Biol. Chem. 183 593-605 [Google Scholar]
- 6.Ortiz de Montellano, P. R., and De Voss, J. J. (2005) in Cytochrome P450, 3rd Ed. (Ortiz de Montellano, P. R., ed) pp. 183-245, Kluwer Academic/Plenum Publishers, New York
- 7.Iyanagi, T., Makino, N., and Mason, H. S. (1974) Biochemistry 13 1701-1710 [DOI] [PubMed] [Google Scholar]
- 8.Oprian, D. D., and Coon, M. J. (1982) J. Biol. Chem. 257 8935-8944 [PubMed] [Google Scholar]
- 9.Vermilion, J. L., and Coon, M. J. (1978) J. Biol. Chem. 253 8812-8819 [PubMed] [Google Scholar]
- 10.Vermilion, J. L., Ballou, D. P., Massey, V., and Coon, M. J. (1981) J. Biol. Chem. 256 266-277 [PubMed] [Google Scholar]
- 11.Kurzban, G. P., and Strobel, H. W. (1986) J. Biol. Chem. 261 7824-7830 [PubMed] [Google Scholar]
- 12.Bredt, D. S., Hwang, P. M., Glatt, C. E., Lowenstein, C., Reed, R. R., and Snyder, S. H. (1991) Nature 351 714-718 [DOI] [PubMed] [Google Scholar]
- 13.Leclerc, D., Wilson, A., Dumas, R., Gafuik, C., Song, D., Watkins, D., Heng, H. H., Rommens, J. M., Scherer, S. W., Rosenblatt, D. S., and Gravel, R. A. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 3059-3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olteanu, H., and Benerjee, R. (2001) J. Biol. Chem. 276 35558-35563 [DOI] [PubMed] [Google Scholar]
- 15.Paine, M. J., Garner, A. P., Powell, D., Sibbald, J., Sales, M., Pratt, N., Smith, T., Tew, D. G., and Wolf, C. R. (2000) J. Biol. Chem. 275 1471-1478 [DOI] [PubMed] [Google Scholar]
- 16.Rooseboom, M., Commandeur, J. N., and Vermeulen, N. P. (2004) Pharmacol. Rev. 56 53-102 [DOI] [PubMed] [Google Scholar]
- 17.Wang, M., Roberts, D. L., Paschke, R., Shea, T. M., Siler-Masters, B. S., and Kim, J. J. P. (1997) Proc. Natl. Acad. Sci. U. S. A. 944 8411-8416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hubbard, P. A., Shen, A. L., Paschke, R., Kasper, C. B., and Kim, J. J. P. (2001) J. Biol. Chem. 276 29163-29170 [DOI] [PubMed] [Google Scholar]
- 19.Page, C. C., Moser, C. C., Chen, X. X., and Dutton, P. L. (1999) Nature 402 47-52 [DOI] [PubMed] [Google Scholar]
- 20.Bhattacharyya, A. K., Lipka, J. J., Waskell, L., and Tollin, G. (1991) Biochemistry 30 759-765 [DOI] [PubMed] [Google Scholar]
- 21.Gutierrez, A., Paine, M., Wolf, C. R., Scrutton, N. S., and Roberts, G. C. (2002) Biochemistry 41 4626-4637 [DOI] [PubMed] [Google Scholar]
- 22.Shen, A. L., and Kasper, C. B. (1995) J. Biol. Chem. 270 27475-27480 [DOI] [PubMed] [Google Scholar]
- 23.Sevrioukova, I. F., Li, H., Zhang, H., Peterson, J. A., and Poulos, T. L. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 1863-1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen, A. L., Porter, T. D., Wilson, T. E., and Kasper, C. B. (1989) J. Biol. Chem. 264 7584-7589 [PubMed] [Google Scholar]
- 25.Saribas, A. S., Gruenke, L., and Waskell, L. (2001) Protein Expr. Purif. 21 303-309 [DOI] [PubMed] [Google Scholar]
- 26.Bridges, A. G. L., Chang, Y. T., Vaksar, I. A., Loew, G., and Waskell, L. (1998) J. Biol. Chem. 273 17036-17049 [DOI] [PubMed] [Google Scholar]
- 27.Oprian, D. D., Vatsis, K. P., and Coon, M. J. (1979) J. Biol. Chem. 254 8895-8902 [PubMed] [Google Scholar]
- 28.Zhang, H., Im, S.-C., and Waskell, L. (2007) J. Biol. Chem. 282 29766-29776 [DOI] [PubMed] [Google Scholar]
- 29.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 30.Collaborative Computational Project Number 4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50 760-76315299374 [Google Scholar]
- 31.McCoy, A. J., Grosse-Kuntstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007) J. Appl. Crystallogr. 40 658-674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brunger, A. T. (2007) Nat. Protocols 2 2728-2733 [DOI] [PubMed] [Google Scholar]
- 33.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 34.Tovchigrenchko, A., and Vakser, I. A. (2006) Nucleic Acids Res. 22 W310-314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wiehe, K., Peterson, M. W., Pierce, B., Mintseris, J., and Weng, Z. (2007) Methods Mol. Biol. 413 283-314 [DOI] [PubMed] [Google Scholar]
- 36.Lamb, D. C., Kim, Y., Yermalitskaya, L. V., Yermalitsky, V. N., Lepesheva, G. I., Kelly, S. L., Waterman, M. R., and Podust, L. M. (2006) Structure 14 51-56 [DOI] [PubMed] [Google Scholar]
- 37.Haque, M. M., Panda, K., Tejero, J., Aulak, S., Fadalla, M. A., Mustovich, A. T., and Stuehr, D. J. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 9254-9259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcin, E. D., Bruns, C. M., Lloyd, S. J., Hosfield, D. J., Tiso, M., Gachhui, R., Stuehr, D. J., Tainer, J. A., and Getzoff, E. D. (2004) J. Biol. Chem. 279 37918-37927 [DOI] [PubMed] [Google Scholar]
- 39.Ilagan, R. P., Tiso, M., Konas, D. W., Hermann, C., Durra, D., Hille, R., and Stuehr, D. J. (2008) J. Biol. Chem. 283 19603-19615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamburini, P. P., and Schenkman, J. B. (1986) Mol. Pharmacol. 30 178-185 [PubMed] [Google Scholar]
- 41.Davydov, D. R., Darovsky, B. V., Dedinsky, I. R., Kanaeva, I. P., Bachmanova, G. I., Blinov, V. M., and Archakov, A. L. (1992) Arch. Biochem. Biophys. 297 304-313 [DOI] [PubMed] [Google Scholar]
- 42.Darrouzet, E., Moser, C. C., Dutton, L. P., and Daldal, F. (2001) Trends Biochem. Sci. 26 445-451 [DOI] [PubMed] [Google Scholar]
- 43.Krebs, W. G., and Gerstein, M. (2000) Nucleic Acids Res. 28 1665-1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flores, S., Echols, N., Milburn, D., Hespenheide, B., Keating, K., Lu, J., Wells, S., Yu, E. Z., Thorpe, M., and Gerstein, M. (2006) Nucleic Acids Res. 34 D296-D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith, G. C., Tew, D. G., and Wolf, C. R. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 8710-8714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grunau, A., Geraki, K., Grossmann, J. G., and Gutierrez, A. (2007) Biochemistry 46 8244-8255 [DOI] [PubMed] [Google Scholar]
- 47.Lange, R., Heiber-Langer, I., Bonfils, C., Fabre, I., Negishi, M., and Balny, C. (1994) Biophys. J. 66 89-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gray, R. D. (1982) J. Biol. Chem. 257 1086-1094 [PubMed] [Google Scholar]
- 49.Zhang, H., Gruenke, L., Arscott, D., Shen, A. L., Kasper, C. B., Harris, D. L., Glavanovich, M., Johnson, R., and Waskell, L. (2003) Biochemistry 42 11594-11603 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.