

Abstract
The Na+/H+ exchanger isoform 1 is a ubiquitously expressed integral membrane protein that regulates intracellular pH in mammals by extruding an intracellular H+ in exchange for one extracellular Na+. We characterized structural and functional aspects of the critical transmembrane (TM) segment XI (residues 449-470) by using cysteine scanning mutagenesis and high resolution NMR. Each residue of TM XI was mutated to cysteine in the background of the cysteine-less protein and the sensitivity to water-soluble sulfhydryl reactive compounds MTSET ((2-(trimethylammonium) ethyl)methanethiosulfonate) and MTSES ((2-sulfonatoethyl) methanethiosulfonate) was determined for those residues with at least moderate activity remaining. Of the residues tested, only proteins with mutations L457C, I461C, and L465C were inhibited by MTSET. The activity of the L465C mutant was almost completely eliminated, whereas that of the L457C and I461C mutants was partially affected. The structure of a peptide representing TM XI (residues Lys447-Lys472) was determined using high resolution NMR spectroscopy in dodecylphosphocholine micelles. The structure consisted of helical regions between Asp447-Tyr454 and Phe460-Lys471 at the N and C termini of the peptide, respectively, connected by a region with poorly defined, irregular structure consisting of residues Gly455-Gly459. TM XI of NHE1 had a structural similarity to TM XI of the Escherichia coli Na+/H+ exchanger NhaA. The results suggest that TM XI is a discontinuous helix, with residue Leu465 contributing to the pore.
The mammalian Na+/H+ exchanger isoform 1 (NHE1)4 is a ubiquitous integral membrane protein that regulates intracellular pH. It mediates removal of a single intracellular proton in exchange for an extracellular sodium ion (1). NHE1 has many functions aside from protection of cells from intracellular acidification (2). It promotes cell growth and differentiation (3), regulates sodium fluxes and cell volume after challenge by osmotic shrinkage (4), and has been demonstrated to be involved in modulating cell motility (5). In addition its activity is important in invasiveness of neoplastic breast cancer cells (6). NHE1 also plays critical roles in heart disease. It has a contributing role in heart hypertrophy and in the damage that occurs during ischemia and reperfusion. Inhibition of NHE1 with Na+/H+ exchanger inhibitors protects the myocardium during various disease states (7-10).
NHE1 is composed of two general regions, an N-terminal membrane domain of ∼500 amino acids and a C-terminal regulatory domain of ∼315 amino acids (1, 8). The membrane domain is responsible for ion movement and an analysis of topology by cysteine scanning accessibility suggested it has 3 membrane-associated segments and 12 integral transmembrane segments (11) (Fig. 1A). The mechanism of transport of the membrane domain is of great interest both from a scientific viewpoint and in the design of improved NHE1 inhibitors that may be necessary for clinical use (1). In this regard, we have recently characterized the functionally important residues and the structure of both TM IV and TM VII. Prolines 167 and 168 of TM IV were critical to NHE1 function (12) and cysteine-scanning mutagenesis was used to show that Phe161 is a pore lining residue critical to transport. Analysis of the structure of TM IV showed that TM IV is composed of one region of β-turns, an extended middle region including Pro167-Pro168, and a helical region (13). TM VII was much more typical of a transmembrane helix although it was interrupted with a break in the helix at the functionally critical residues Gly261-Glu262 (14).
FIGURE 1.

Models of the Na+/H+ exchanger. A, simplified topological model of the transmembrane domain of the NHE1 isoform of the Na+/H+ exchanger as described earlier (11). EL, extracellular loop; IL, intracellular loop. B, model of amino acids present in TM XI.
Another important TM segment of the Na+/H+ exchanger is TM XI (Fig. 1B). Several different lines of evidence have suggested that it is critical to NHE1 function. A recent study generated chimeras of NHE1 from various species and found that a region including TM XI was important in determining NHE1 inhibitor sensitivity (15). More specifically, mutagenesis of several amino acids of TM XI has shown that it is likely involved in either ion transport or proper targeting to the plasma membrane. Two mutants in TM XI, Y454C and R458C, are retained in the endoplasmic reticulum (16). In addition, mutation of Gly455 and Gly456 in TM XI shift the pHi dependence of the exchanger to the alkaline side, whereas mutation of Arg440 in intracellular loop 5 at the N-terminal end of TM XI shifts the pHi dependence to make it more acidic (17, 18). Also, the structure of the bacterial Na+/H+ exchanger NhaA has been elucidated. Both TM IV and TM XI play a critical role forming an assembly that cross, with each being a helix, an extended polypeptide and a short helix (19). We found that TM IV of NHE1 has a similar structure and function to that of TM IV of NhaA (2, 13), leaving open the possibility that TM XI of NHE1 is also similar in structure and function to TM XI of NhaA.
For these reasons, we undertook a systematic examination of the structural and functional aspects of TM XI of the NHE1 isoform of the Na+/H+ exchanger. The sequence of human TM XI of NHE1 is 449QFIIAYGGLRGAIAFSLGYLLD470. In this study we use cysteine scanning mutagenesis and site-specific mutagenesis to identify and characterize critical pore lining residues of the protein. We also use nuclear magnetic resonance (NMR) spectroscopy to characterize the structure of a synthetic peptide representing TM XI in dodecylphosphocholine (DPC) micelles. Evidence has suggested that TM segments of membrane proteins possess all the structural information required to form their higher order structures in their amino acid sequence (20). This has been demonstrated in earlier studies on membrane protein segments such as the cystic fibrosis transmembrane conductance regulator (21), a fungal G-protein-coupled receptor (22), bacteriorhodopsin (23, 24), and rhodopsin (25), where it was shown that isolated TM segments from membrane proteins had structures in good agreement with the segments of the entire protein. Also, the use of DPC micelles has been shown to be an excellent membrane mimetic environment for these studies (26, 27). Our study identifies Leu465 as contributing to the pore of the protein and shows that the structure of TM XI consists of two helices corresponding to Asp447-Tyr454 and Phe460-Lys471 at the N and C termini, respectively, connected by a flexible region at residues 455-459. The structure of TM XI was similar to the x-ray structure of TM XI of NhaA.
EXPERIMENTAL PROCEDURES
Materials—Lipofectamine™ 2000 reagent was from Invitrogen. PWO DNA polymerase was obtained from Roche Applied Science. MTSET and MTSES were from Toronto Research Chemicals (Toronto, Ontario, Canada). Sulfo-NHS-SS-biotin was from Pierce and immobilized streptavidin resin was from Thermo Scientific (Rockford, IL), Deuterated DPC was purchased from C/D/N Isotopes (Pointe-Claire, Quebec, Canada).
Site-directed Mutagenesis—Mutations were made to an expression plasmid (pYN4+) containing a hemagglutinin (HA)-tagged human NHE1 isoform of the Na+/H+ exchanger. The plasmid contained the cDNA for the entire coding region of the Na+/H+ exchanger and we previously demonstrated that the tagged expressed protein functions normally (12). The mutations changed the indicated amino acids to cysteine (Table 1), using the functional cysteine-less NHE1 protein that we have described earlier (13). An additional series of mutations was done to change Leu465 to Ala, Lys, or Asp (Table 2). Site-directed mutagenesis was done using amplification by PWO DNA polymerase (Roche Molecular Biochemicals) and using the Stratagene (La Jolla, CA) QuikChange™ site-directed mutagenesis kit. Mutations created or deleted a restriction enzyme site and the fidelity of DNA amplification was confirmed by DNA sequencing.
TABLE 1.
Oligonucleotides used for site-directed mutagenesis of TM XI
Mutated nucleotides are underlined, restriction sites are bold. Restriction sites deleted are indicated in parentheses. Oligonucleotides used for cysteine scanning mutagenesis are shown. All mutations were to Cys.
| Mutation | Oligonucleotide sequence | Restriction site |
|---|---|---|
| Gln449 | 5′-GTATCGTGAAGCTTACCCCCAAGGACTGCTTCATCATCGCC-3′ | HindIII |
| Phe450 | 5′-CCCCCAAGGACCAATGCATCATCGCCTATGG-3′ | SstI |
| Ile451 | 5′-CCCAAGGACCAGTTCTGCATCGCATATGGGGGCCTGCGAG-3′ | NdeI |
| Ile452 | 5′-GGACCAGTTCATCTGCGCATATGGGGGCCTGC-3′ | FspI |
| Ala453 | 5′-CCAGTTCATCATCTGCTATGGAGGCCTGCGAGGGGC-3′ | StuI |
| Tyr454 | 5′-CAGTTCATCATCGCATGCGGGGGCCTGCGAGG-3′ | SphI |
| Gly455 | 5′-CATCATCGCCTATTGCGGCTTAAGAGGGGCCATCGCC-3′ | AflII |
| Gly456 | 5′-CATCGCCTATGGGTGCTTAAGAGGGGCCATCGCC-3′ | AflII |
| Leu457 | 5′-GCCTATGGGGGCTGCCGCGGGGCCATCGCCTTC-3′ | SacII |
| Arg458 | 5′-CATCGCCTATGGAGGCCTGTGTGGGGCCATCGCC-3′ | StuI |
| Gly459 | 5′-GGGGGCCTGCGATGCGCAATCGCCTTCTCTC-3′ | FspI |
| Ala460 | 5′-GGGGCCTGCGAGGATGCATCGCCTTCTCTC-3′ | NsiI |
| Ile461 | 5′-GGCCTGCGAGGGGCATGCGCCTTCTCTCTGGG-3′ | SphI |
| Ala462 | 5′-CTGCGAGGGGCCATATGCTTCTCTCTGGGC-3′ | NdeI |
| Phe463 | 5′-GAGGGGCCATCGCATGCTCTCTGGGCTACC-3′ | SphI |
| Ser464 | 5′-GGCCATCGCCTTCTGCCTAGGCTACCTCCTGG-3′ | AvrII |
| Leu465 | 5′-CATCGCCTTCTCTTGCGGGTACCTCCTGGACAAG-3′ | KpnI |
| Gly466 | 5′-CGCCTTCTCTCTGTGCTACCTTCTAGA CAAGAAGCACTTC-3′ | XbaI |
| Tyr467 | 5′-CCTTCTCTCTGGGCTGTCTACTGGACAAGAAGCAC-3′ | AccI |
| Leu468 | 5′-CTCTCTGGGCTACTGTCTAGACAAGAAGCACTTC-3′ | XbaI |
| Leu469 | 5′-GCCTTCTCTCTGGGATATCTCTGC GACAAGAAGCACTTC-3′ | EcoRV |
| Asp470 | 5′-GGGCTACCTCCTGTGCAAAAAGCACTTCCCCATG-3′ | (Xmn I) |
TABLE 2.
Synthetic oligonucleotides used for mutagenesis of Leu465 to Ala, Lys, or Asp
| Mutation | Oligonucleotide sequence | Restriction site |
|---|---|---|
| L465A | 5′-GCCATCGCCTTCTCTGCGGGGTACCTCCTGGACAAG-3′ | KpnI |
| L465K | 5′-GCCATCGCCTTCTCTAAGGGGTACCTCCTGGACAAG-3′ | KpnI |
| L465D | 5′-GCCATCGCCTTCTCTGATGGGTACCTCCTGGACAAG-3′ | KpnI |
Cell Culture and Transfections—Control and mutant Na+/H+ exchanger cDNA was transfected into AP-1 cells that lack an endogenous Na+/H+ exchanger (28). Stable cell lines of all mutants were made as described earlier using transfection with Lipofectamine™ 2000 reagent (Invitrogen) (12). Transfected cells were selected using 800 μg/ml geneticin (G418) and stable cell lines for experiments were regularly re-established from frozen stocks between passages 5 and 9.
SDS-PAGE and Immunoblotting—Cell lysates were made from AP-1 cells as described earlier (12). For Western blot analysis equal amounts of up to 100 μg of each cell lysate sample was resolved on 10% SDS-polyacrylamide gels. Nitrocellulose transfers were immunostained using anti-HA monoclonal antibody, which recognized the tag on the Na+/H+ exchanger protein (Roche Applied Science). The second antibody was peroxidase-conjugated goat anti-mouse antibody (Bio/Can, Mississauga, ON, Canada). The Amersham Biosciences enhanced chemiluminescence Western blotting and detection system was used to visualize immunoreactive proteins. NIH Image 1.63 software (National Institutes of Health, Bethesda, MD) was use for densitometric analysis of x-ray films.
Cell Surface Expression—Cell surface expression was measured essentially as described earlier (12). Briefly, the cell surface was labeled with sulfo-NHS-SS-biotin (Pierce) and immobilized streptavidin resin was used to remove plasma membrane Na+/H+ exchanger. Equal amounts of total and unbound proteins were analyzed by SDS-PAGE followed by Western blotting and densitometry measuring immunoreactive (HA-tagged) Na+/H+ exchanger protein. It was not possible to efficiently and reproducibly elute proteins bound to immobilized streptavidin resin reproducibly. The relative amount of NHE1 on the cell surface was calculated by comparing both the 110- and 95-kDa HA-immunoreactive species in Western blots of the total and unbound fractions.
Na+/H+ Exchange Activity—Na+/H+ exchange activity was measured essentially as described earlier (14) using a PTI Deltascan spectrofluorometer. Briefly, the initial rate of Na+-induced recovery of cytosolic pH (pHi) was measured after ammonium chloride-induced acute acid load using 2′,7-bis(2-carboxyethyl)-5(6)-carboxyfluorescein-AM (Molecular Probes Inc., Eugene, OR). Recovery was in the presence of 135 mm NaCl. There was no difference in the buffering capacities of stable cell lines and the recovery was from the equivalent initial pH. Where indicated, Na+/H+ exchanger activity was corrected for the level of protein expression and targeting of the protein to the cell surface. For some experiments cells were treated with MTSET or MTSES. A two-pulse acidification assay was used. Cells were treated with ammonium chloride two times and allowed to recover in NaCl containing medium (13). One pulse was in the absence of inhibitor and one in the presence of either 10 mm MTSET or MTSES for 10 min. The starting pH of recovery was equivalent in the two pulses. The rate of recovery from acid load was compared ± inhibitor. The calculation used for residual activity was according to Equation 1.
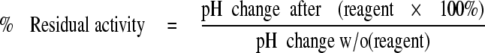 |
(Eq.1) |
All results are shown as mean ± S.E. and statistical significance was determined using the Mann-Whitney U test.
Peptide Synthesis and Purification—Peptide representing TM XI (sequence KDQFIIAYGGLRGAIAFSLGYLLDKK, acetyl-capped N terminus, amide-capped C terminus) was purchased from GI Biochem (Shanghai) Ltd. at >95% purity. Peptide purity was assessed with high performance liquid chromatography. The identity of the peptide was confirmed by using matrix-assisted laser desorption ionization mass spectrometry and sequential assignment of the NMR spectra. An ∼5% impurity was identified in the 1H NMR spectra (see below).
NMR Spectroscopy and Structure Calculation—Samples for NMR spectroscopy were prepared by dissolving ∼2 mm TM XI peptide and ∼150 mm DPC-d38 in 95% H2O, 5% D2O containing 0.25 mm deuterated 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS-d6) (Chenomx Inc.) as an internal NMR standard. The pH was adjusted to 4.9 without consideration of the deuterium effects on the glass electrode.
One-dimensional 1H, and two-dimensional 1H-1H DQCOSY, TOCSY (60 ms mix), and NOESY (250 ms mix) spectra were taken on Varian INOVA 600 and 800 MHz spectrometers. A natural abundance 1H-13C HSQC spectrum was taken on a Varian INOVA 500. Experiments were used as configured in the Varian Biopack package and taken at 30 °C. Spectra were processed using NMRPipe (29) and analyzed in NMRView (30). Sequential assignment was done using the DQCOSY, TOCSY, and NOESY spectra to assign and connect the amino acid spin systems. Assignment of peaks was done manually in NMRView.
Structure calculations were performed using the Python scripting interface in XPLOR-NIH 2.19 (31). NOE restraints for structure calculation were generated within NMRView by calibrating restraints by peak intensity and sorting them into distance ranges of strong (1.8-2.8 Å), medium (1.8-3.4 Å), or weak (1.8-5.0 Å). Some restraints were extended into a very weak category (1.8-6.0 Å) as needed during refinement. A soft square-well potential was used for the restraining function in structure calculation. In each round of structure calculation, an extended polypeptide was generated and subjected to simulated annealing, with 50 structures generated for each round. After each round, violated restraints were examined and either lengthened or removed. Initially, violations >0.5 Å in >50% of structures were examined. Refinement of restraints gradually increased in stringency, to where violations >0.1 Å in >10% of structures were examined. Eleven rounds of structure calculation and refinement were performed, after which a further 10 rounds were performed adding dihedral angle restraints. Weighting on the dihedral restraints was increased from 5 to 100, then decreased to 25 over successive rounds. The lowest total energy 40 of 50 structures from the final round were kept for further analysis. The set of restraints used and the ensemble of NMR structures have been deposited in the Protein Data Bank (Research Collaboratory for Structural Bioinformatics Protein Data Band code 2KBV, BMRB number 16056).
RESULTS
Fig. 1A shows a general model of the NHE1 isoform of the Na+/H+ exchanger based on the topology deduced by cysteine-scanning accessibility studies (11). Fig. 1B shows a schematic model illustrating the amino acids of TM XI. To examine which amino acids of TM XI are pore-lining, we used the cysteine-less Na+/H+ exchanger (cNHE). Each residue in TM XI of cNHE1 was mutated to a cysteine residue. Initial experiments examined whether these mutant forms of the Na+/H+ exchanger were active enough for further study. Fig. 2, A-C, examines the expression, targeting, and activity of the wild type and mutant TM XI mutants. Expression levels of several of the mutants were greatly decreased in comparison to the controls. Changing Ile452, Gly455, or Ala460 to Cys decreased expression to less than 20% of that of cNHE1. Eight other mutant Na+/H+ exchangers, I451C, A453C, Y454C, G456C, R458C, G459C, A462C, and L469C, also had expression reduced to levels 22-45% that of cNHE1. All the mutants with reduced expression of NHE1 showed a great reduction in the level of mature, fully glycosylated protein. In some of these mutants (I451C, Y454C, G455C, G456C, R458C, G459C, and L469C) expression was predominantly of the lower molecular weight form of the Na+/H+ exchanger, which represents protein with reduced levels of glycosylation. We have earlier shown that unglycosylated Na+/H+ exchanger may be functional (32), therefore the unglycosylated protein was included in analysis of the levels of protein expression.
FIGURE 2.

Analysis of wild type and mutant NHE1 proteins. A, Western blot of cell extracts of AP-1 cells containing stably transfected Na+/H+ exchanger mutants or control. In all mutations the amino acid indicated was changed to cysteine. 100 μg of total protein was loaded in each lane. Numbers below the lanes indicate the values obtained from densitometric scans of both the 110- and 95-kDa bands relative to wild type NHE. Results are typical of three to five measurements. AP1 refers to mock transfected AP-1 cells. Wt, refers to cells stably transfected with wild type Na+/H+ exchanger protein. B, subcellular localization of control and TM XI mutants in AP-1 cells. Sulfo-NHS-SS-biotin-treated cells were lysed and streptavidin-agarose beads were used to bind labeled proteins as described under “Experimental Procedures.” Equal amounts of total cell lysate (T) and unbound intracellular lysate (I) were run on SDS-PAGE and Western blotting with anti-HA antibody identified NHE1 protein. Wt, refers to cells stably transfected with wild type Na+/H+ exchanger protein. Ct refers to a control experiment in which nonspecific binding to streptavidin-agarose beads was carried out following the standard procedure but without labeling cells with biotin. The percent of the total NHE1 protein found within the plasma membrane is indicated for each mutant. For Ct the numbers indicate the amount of nonspecific binding to streptavidin-agarose beads. Results are the mean ± S.E. of at least three determinations. C, rate of recovery from an acid load by AP-1 cells stably transfected with cNHE, and TM XI Na+/H+ exchanger mutants. Na+/H+ exchanger activity was measured after transient induction of an acid load as described under “Experimental Procedures.” The activity of cNHE1 stably transfected with NHE1 was 0.036 Δ pH/s, and this value was set to 100%. All mutations to cysteine were done in the background of the cysteine-less NHE1 and activities are percent of those of cNHE. Mutations were in the cNHE1 and results are expressed as mean ± S.E. of 8-16 determinations. Results are shown for mean activity of both uncorrected (black) and normalized for surface processing and expression levels (hatched). * indicates mutants with uncorrected activity that is less than 20% of cNHE1.
We have previously (13) demonstrated that mutation of amino acids of transmembrane segments can affect plasma membrane targeting of the Na+/H+ exchanger. Therefore, we examined intracellular targeting of the NHE1 TM XI expressing cell lines. Cells were treated with sulfo-NHS-SS-biotin and then lysed and solubilized as described under “Experimental Procedures.” Labeled proteins were bound to streptavidin-agarose beads and equal amounts of total cell lysates and unbound lysates (representing intracellular protein) were separated by size using SDS-PAGE. Western blotting with anti-HA antibody identified the tagged NHE1 protein. Fig. 2B illustrates examples of the results and a summary of quantification of both the 106- and 97-kDa bands. The immature form of the protein was included because we previously noted that there can be some leak of this protein to the cell surface (33). A number of the mutants had reduced targeting to the cell surface, in comparison to cNHE1. Less than 10% of G456C, R458C, and G459C were targeted to the cell surface, and because the nonspecific was ∼5%, the results suggest that these proteins were mostly, if not all, retained within the cell. A second class of mutant was targeted about half as well as the cysteine-less NHE1, with ∼20-30% targeted to the cell surface. This consisted of I451C, G455C, F463C, Y467C, and L469C. The balance of the mutants were similar to cNHE1 in their targeting efficiency.
We determined how mutations of TM XI affected the activity of the protein. The rate of recovery from an acute acid load was determined as described earlier (33). Fig. 2C illustrates the rate of recovery from an acute acid load by stable cell lines transfected with either wild type Na+/H+ exchanger or mutants of TM XI. The rate of recovery is also shown when corrected for both the level of expression and surface targeting for most mutants. The mutants fell into several general categories. One mutant A453C had less than 50% of the cNHE activity, but the activity was near normal when correcting for expression levels and surface targeting, suggesting that the protein itself was fully functional but the mutations were affected for expression levels and surface targeting. L465C was similar to A453C but with higher uncorrected NHE1 activity. In both cases this was due to a combination of effects on expression levels and surface targeting. Mutants L457C and I461C retained over 50% of cNHE1 activity and had relatively normal surface targeting and expression levels. The balance of the mutants had quite reduced activity, less than 50% of the cNHE1 level. Q449C, F450C, S464C, and G466C had 25-45% of cNHE1 activity and this was only slightly improved by correcting for expression levels and surface targeting. This indicated that these proteins themselves were at least partially defective in Na+/H+ exchange activity. A number of other mutants had quite low activity relative to cNHE1, less than 20% of uncorrected values. This included, I451C, I452C, Y454C, G455C, G456C, R458C, G459C, A460C, A462C, F463C, Y467C, L468C, L469C, and D470C. For most of these mutants correcting for expression levels and surface targeting increased activity although this was always well below that of cNHE1, suggesting that the effects were at least in part due to direct effects on the activity of the protein. It was not possible to accurately determine the corrected activity for many of these mutants because of their variability, low expression levels, and poor surface targeting. This included G456C, R458C, and G459C.
We examined the sensitivity to MTSET or MTSES of the mutant Na+/H+ exchangers that had greater than 20% residual activity of cNHE1 (Fig. 3, A and B). Of the active Na+/H+ exchangers, only L465C was greatly affected by treatment with positively charged MTSET. This resulted in elimination of most of the activity of the protein. Two other mutants, L457C and I461C, were partially inhibited by MTSET. Negatively charged MTSES had no effect on any of the mutant proteins. Mutants G459C and A460C are not illustrated in the inhibition results due to their low activity, but were also tested. Experiments indicated that they were not inhibited by addition of MTSES or MTSET.
FIGURE 3.

Effect of MTSET and MTSES on activity of cNHE1 and single cysteine TM XI mutant NHE1 containing cell lines. A, summary of results of Na+/H+ exchanger assays of active TM XI mutants. Activity was measured after two pulses of transient induction of an acid load as described under “Experimental Procedures.” The first activity was in the absence of MTSES or MTSET, the second was when cells were treated with 10 mm reagent before transient acidification. Results illustrated represent the % of activity of the second acid load, in comparison to the first. * indicates that the second recovery from acid load was significantly lower than the first at p < 0.01, Mann-Whitney U test. Solid filled bars represent MTSES treatments and hatched bars represent MTSET treatments. B, example of results of effect of MTSET on activity of wild type and L465C mutant. Wild type (Wt) and L465C NHE1 protein activity was assayed in stably transfected AP-1 cells as described under “Experimental Procedures.” Activity was measured after two acid pulses as described in A. The first pulse in the absence of MTSET is shown. For ease of viewing, only the recovery from acidosis is shown for the second pulse, in which cells were treated with MTSET. NH4Cl, treatment with ammonium chloride; sodium free, treatment with Na+ free buffer to induce acidosis; NaCl, recovery from acidosis in NaCl containing buffer, for the second pulse this contained MTSET and cells were pretreated with MTSET for 10 min prior to NH4Cl-induced acid load.
Because of the strong effect that we observed of MTSET on L465C, we conducted a series of experiments in which we mutated that amino acid to a neutral residue (Ala), to a negatively charged residue (Asp), and to a positively charged residue (Lys). We then determined effects on activity, surface processing, and expression of the intact NHE1 protein. The results are shown in Fig. 4. Western blotting (Fig. 4A) against the anti-HA tag of the NHE1 mutant proteins showed varying effects dependent on the mutation. Mutation of Leu465 to Asp resulted in a large decrease in the amount of protein expressed and in addition, virtually all of the protein expressed was of the unglycosylated or partially glycosylated form. Similarly, mutation to Ala reduced the amount of protein expressed and reduced the amount of fully glycosylated protein, but not to as great an extent. Mutation of Leu465 to Lys did not reduce the amount of NHE1 expressed. There was also a significant amount of fully glycosylated protein although the amount relative to the de-glycosylated protein was somewhat reduced. In all three cases, targeting to the cell surface was low (Fig. 4B). This was especially true for the L465D mutant. Activity of the three mutants was greatly reduced (Fig. 4C). In all cases the raw activity we observed for the mutants was only a small fraction of the control level. It was of interest that for the L465A and L465D mutants, there was a significant amount of activity present, when correcting for aberrant surface targeting and expression levels. In contrast, the L465K protein was well expressed and targeted to the cell surface, but the change to a positively charged residue completely eliminated activity of the protein.
FIGURE 4.

Characterization of site-specific mutants of Leu465. A, Western blot of cell extracts of AP-1 cells containing stably transfected Na+/H+ exchanger mutants or control. Samples were analyzed as described in the legend to Fig. 2A. Numbers below the lanes indicate the values obtained from densitometric scans of both the 110- and 95-kDa bands relative to wild type NHE. Results are typical of three to five measurements. cNHE refers to cysteine-less NHE1. A, D, and K refer to mutant cell lines containing cNHE1 with the Leu465 mutations to Ala, Asp, and Lys respectively. B, subcellular localization of TM XI mutants in AP-1 cells. Mutants contained the NHE1 Leu465 mutations to Ala, Asp, and Lys as indicated. Sulfo-NHS-SS-biotin-treated cells were lysed and streptavidin-agarose beads were used to bind labeled proteins as described under “Experimental Procedures.” Equal amounts of total cell lysate (T) and unbound intracellular lysate (I) were run on SDS-PAGE and Western blotting with anti-HA antibody identified the NHE1 protein. The percent of the total NHE1 protein found within the plasma membrane is indicated for each mutant. Results are the mean ± S.E. of at least three determinations. C, rate of recovery from an acid load by AP-1 cells stably transfected with cNHE, and TM XI Na+/H+ exchanger Leu465 mutations to Ala, Asp, and Lys. Na+/H+ exchanger activity was measured after transient induction of an acid load as described in the legend to Fig. 2C. All mutations to cysteine were done in the background of the cysteine-less NHE1 and activities are percent of those of cNHE. Mutations were in the cNHE1 and results are expressed as mean ± S.E. of 8-16 determinations. Results are shown for mean activity of both uncorrected (black) and normalized for surface processing and expression levels (hatched).
We used NMR spectroscopy to determine the structure of a TM XI peptide. Cationic residues at the termini of peptides have been shown to aid in their purification and maintenance of transbilayer orientation (34). For this reason, the ends of the peptide were chosen to be Lys447 and Lys472, which provides cationic residues at both ends of the peptide and include the predicted TM XI sequence. The N- and C-terminal acetyl and amide caps, respectively, remove charges at the peptide termini that would not be present in the full protein.
The peptide was readily solubilized in the presence of DPC, which acts as a membrane mimetic, and provided a sample that was stable for months at room temperature. Previous structural studies of transmembrane helices from NHE1 also found DPC micelles to be effective for solubilization and sample stability (35, 36). An examination of the two-dimensional 1H-1H NMR spectra showed additional peaks that could not be assigned to the major conformation of the TM XI peptide. Adjusting sample conditions such as peptide concentration and the peptide-to-lipid ratio did not affect intensity of these peaks in the DQCOSY NMR spectra, and therefore they were assumed to be an impurity (∼5% based upon the area of the NMR peaks) in the synthetic TM XI peptide and not a minor conformation in slow exchange. The sample for structure determination contained ∼2 mm TM XI peptide and ∼150 mm DPC in 95% H2O, 5% D2O with 0.25 mm deuterated 2,2-dimethyl-2-silapentane-5-sulfonic acid as a chemical shift and intensity standard. Concentrations of 1 mm peptide and 75 mm DPC were used in previous studies (14, 36); however, we decided to use concentrations of 2 and 150 mm for an increased signal to noise ratio in the NMR spectra. There were no noticeable differences in the spectra at the higher concentration. Spectra were taken at 30 °C, which gave good one-dimensional 1H NMR spectral characteristics.
Sequential assignment using the two-dimensional 1H-1H DQCOSY, TOCSY, and NOESY NMR spectra allowed all of the hydrogen atoms in the peptide to be assigned except for Phe Hζ and Tyr Hη. Assignments for carbon atoms using the natural abundance 13C-1H HSQC NMR spectrum were incomplete because of the limited chemical shift dispersion. The predominantly α-helical nature of the peptide, resulting in limited chemical shift dispersion, and the relatively broad line widths in the spectra, meant some of the NOE assignments were ambiguous. The impurities were ∼5% of the concentration of the TM XI peptide, and any impurities from the NOE, similarly reduced, were unlikely to be a concern for assignment. Nonetheless, a soft-well potential function was used during structure calculation to allow for any ambiguous or erroneous assignments. Dihedral angle restraints were not initially applied during structure calculation to allow the structure to be based mainly on the observed NOE distance restraints. Chemical shift prediction of secondary structure (37, 38) (supplemental Fig. S1) and NOE connectivities (i+3, i+4) (Fig. S2) suggested helical regions at approximately residues 449-453 and 461-469. Therefore, the phi and psi angles in these regions were restricted to -60 ± 30° and -40 ± 40°, respectively, for additional rounds of annealing and refinement. After refinement, a total of 548 unique restraints were used for the final structure calculation. The 40 lowest energy structures of the 50 calculated were retained, which satisfied restraints with minimal NOE and dihedral angle violations. Structure calculation statistics are summarized in Table 3 and supplemental Fig. S2.
TABLE 3.
NMR structural statistics for the 40 structures retained out of 50 structures calculated
| Unique NOE restraints | |
| Total | 548 |
| Intraresidue | 195 |
| Sequential | 195 |
| Medium range (i+2 to i+4) | 156 |
| Long range (≥i+5) | 2 |
| Ambiguous | 47 |
| Ramachandran plot statistics | |
| Core | 72.0% |
| Allowed | 26.0% |
| Generously allowed | 1.0% |
| Disallowed | 1.0% |
| XPLOR-NIH energies (kcal/mol) | |
| Total | 16.78 ± 2.44 |
| NOE | 0.89 ± 1.03 |
| NOE violations | |
| 0.1-0.2 Å | 6 |
| 0.2-0.3 Å | 2 |
| >0.3 Å | 1 |
It was not possible to superimpose the entire ensemble of NMR structures over the full-length of the peptide. The pair wise backbone r.m.s. deviation for each of the ensemble members compared with the average structure ranged from 1.81 to 4.36. Superposition of the ensemble members was done using the method of Kabsch (39), as implemented in LSQKAB in the CCP4 suite. To look for regions of fixed structure, segments of the peptide 4 to 15 residues long were superimposed, and contiguous segments of residues obtained from the superimpositions with an r.m.s. deviation less than 1.0 were examined based on the length and r.m.s. deviation of the segments.
Residues Lys447-Gly454 at the N terminus and Ala460-Lys471 at the C terminus best indicated a region of fixed structure from the r.m.s. deviation analysis and are mainly helical. The relative orientation of these two regions vary with respect to each other among the ensemble members around a structurally variable central region consisting of residues Gly455-Gly459. The dihedral order parameters (35) (Fig. 5) for the N- and C-terminal regions are close to unity, indicating strongly conserved dihedral angles in these regions across the ensemble, whereas the order parameter for the central region shows large variation in the dihedral angles for those residues. A plot of the averaged backbone r.m.s. deviations per residue versus the average structure, for superimposition of residues 447-454 and 460-471, is shown in supplemental Fig. S3. The two superimposed regions show low r.m.s. deviations, which increase in toward the extended region. The superimposition of the C-terminal helix (supplemental Fig. S3, diamonds) shows an increase in r.m.s. deviation at residues Leu465-Gly466. This is reflected in the structures as a slight bend in the helix at these residues. Superimposition of residues Ala460-Leu465 and Gly466-Lys471 separately give better r.m.s. deviation values than for the entire C-terminal helix, and show that there is some slight variability between the two halves of the helix at this bend. Residues Phe461-Lys471 and Asp448-Tyr453 fall within the helical region of the Ramachandran plot and show good clustering of the dihedral angles, and so can be assigned as α-helical. Superimposition of the structurally conserved regions at the N and C termini are shown in Fig. 6, A and B. Fig. 6C shows a schematic of an ensemble member from two views, which shows the general structure of the peptide: two helices connected by an extended segment.
FIGURE 5.

Dihedral angle order parameters for the final 40 ensemble members. Order parameters are calculated as in Ref. 35. An order parameter of 1 indicates the particular angle is the same for all members in the ensemble, and is 0 when the angles are completely random.
FIGURE 6.

NMR structure of TM XI in DPC micelles. Superimposition of structurally conserved regions of the TM XI peptide structure. Superimposition of the backbone atoms of the structurally conserved regions (A) Lys447-Gly454 and (B) Gly460-Lys471. The backbone is shown in black, side chains are shown in color. C, two views of a single ensemble member with MTSET-sensitive residues labeled.
The region between the two helices does not superimpose well. At best, Gly455-Arg458 superimposes poorly, with an r.m.s. deviation of 0.8, and shows decreased dihedral angle order parameters compared with the helical regions. The large variation in dihedral angles over this segment and variation in the relative orientation of the helices in the ensemble could suggest that this region is flexible. It should be noted, however, that variability seen in the structure could be a result of a lack of NOE restraints to constrain the structure of that region, instead of actual flexibility in the structure. Glycine residues have fewer NOE restraints compared with other residues (supplemental Fig. S3) due to the lack of side chain contacts, and could explain the variability in the structures because much of the flexibility in the structure occurs around Gly residues, 455, 456, 459, and 466.
A comparison of the NMR structure of TM XI of NHE1 in DPC micelles with the structure of TM XI in the x-ray structure of NhaA is presented in Fig. 7. Fig. 7A shows an alignment of the TM XI sequences from NhaA and NHE1 using SEQSEE (40), which shows reasonable sequence similarity but a poor alignment of the helical and central regions that are predicted by the x-ray and NMR structures. Attempting to align the structures of the extended region to the x-ray structure of TM XI of NhaA results in reasonable backbone superimposition of residues 455-459 of NHE1 on to residues 335-339 of NhaA, with the orientations of both the N- and C-terminal helices around this segment varying. Using this central region to align the sequences of NHE1 and NhaA TM XI (Fig. 7B) suggests that the MTSET-sensitive residues Leu457, Ile461, and Leu465 identified in this article would correspond to Ile337, Met341, and Ile345, respectively. Fig. 7C shows a comparison of structures of NhaA and NHE1 TM XI with arbitrary rotation of the helices around the variable Gly455 and Gly459 residues in the NMR structure to better match the orientation of the NhaA helical regions. Clearly the general structure of the two peptides is similar for the two proteins; although this cannot be taken as proof that the NMR structure in DPC micelles is equivalent to that of the full-length protein in vivo.
FIGURE 7.

Comparison of TM XI of NHE1 with TM XI of NhaA. A, SEQSEE-based (40) alignment of the amino acid sequences of TM XI of NhaA and NHE1. B, alignment of the sequences based on optimal structural alignment of the extended regions (highlighted) of NHE1 TM XI (Gly455-Gly459) and NhaA TM XI. Colon indicates replacement by similar amino acid and asterisk indicates amino acid identity. MTSET-sensitive residues, and the corresponding residues in NhaA are shown in bold. C, comparison of the NMR structure of TM XI of NHE1 in DPC micelles with that of TM XI in the x-ray structure of NhaA. Amino acids 447-472 of NHE1 and amino acids 327-352 of NhaA are included. The ensemble member of the NMR structures chosen for comparison had the lowest backbone r.m.s. deviation compared with the corresponding region in NhaA for the superimposition of the extended regions, and with arbitrary rotation of the two helices around the flexible Gly455 and Gly459 to match the helices of NhaA.
DISCUSSION
In this study we examined the structural and functional characteristics of TM XI of the NHE1 isoform of the Na+/H+ exchanger. A number of studies (15, 17, 18) have suggested that this transmembrane segment is critical in activity of the protein. When we replaced amino acids of TM XI with cysteines we found that it was quite sensitive to this substitution. Of the 22 amino acids mutated, 14 had activity severely reduced. Several others also had activity reduced to a lesser degree. We found earlier that the susceptibility to mutation was high in some transmembrane segments such as TM IV, whereas other TM segments such as TM IX appear more tolerant of changes. We noted this variability earlier (36) and it has been found in other proteins (41). This could reflect a more critical role of a particular TM segment, in function. Of note, amino acids 454-463 were all greatly reduced in activity, with the exception of Leu457 and Ile461. Most of the mutations made resulted in decreased activity of the product protein that were often caused at least partially by effects on targeting and expression levels. Arg458 and Tyr454 of this region have previously (16) been suggested to be retained in the endoplasmic reticulum when mutated to cysteine. In our hands, proteins with these mutations showed little activity. We found that Arg458 was retained in an intracellular location, however, targeting of Tyr454 was only slightly reduced below that of the cysteine-less protein. It was not possible to test either mutant protein for the effects of sulfhydryl reactive reagents on activity.
When we examined the remaining active mutants for sensitivity to sulfhydryl reactive reagents, we found that only the L465C mutant was strongly affected by positively charged MTSET. It was unaffected by negatively charged MTSES. This suggested that this residue is likely a pore lining residue of the protein. That the positively charged compound would inhibit activity, but not the negatively charged one, could be due to direct electrostatic repulsion of a transported cation. MTSES would not provide any such repulsion. This concept is supported by site-specific mutagenesis studies of this amino acid. The L465K mutant was totally inactive despite being well expressed and targeted to the plasma membrane. The L465D mutant, whereas reduced in activity and not well targeted or expressed, was still functional. Mutation of Leu465 to Ala resulted in a protein with minimal activity, possibly due to structural effects of expression of the smaller amino acid. The L465A protein was poorly expressed and targeted.
We also found that MTSET treatment resulted in reduction of activity of two other amino acids, Leu457 and Ile461. We suggest that their side chains may be near and partially accessible to the protein translocation pore, but likely are not directly pointing to the pore. This is supported by the structural analysis of the protein. TM XI was found to be comprised of two helical segments, with an intervening more extended region. The location of the pore lining residues in the peptide structure is shown in Fig. 6C. Both Leu465 and Ile461 are on the same side of the C-terminal helix. Looking down the helical axis, the two residues are ∼40° offset from each other, such that Leu465 could be pointing into the pore, whereas Ile461 is not directly pointing toward the pore, but is at an orientation in agreement with the idea that it could be close to the pore. The side chain of Leu457 is located in the structurally variable region, so no conclusion can be drawn about its orientation from the peptide structure. Further insights into its orientation must await elucidation of the structure of the intact protein and its presence in a variable region is likely a limitation in the present analysis imposed by examining the peptide in DPC micelles. In the full protein, Leu457 would likely be more rigid and possibly in a different position due to the environment provided by the surrounding TM helices in the intact protein.
It is important to note that recently, there has been disagreement on the proposed topology of the NHE1 protein. An earlier model (11) used substituted cysteine accessibility studies to predict a 12-TM protein with an internal C terminus and several membrane-associated segments (Fig. 1A). More recently, Landau et al. (42) proposed a different topology using the crystal structure of Escherichia coli NhaA as a template for bioinformatics and computational analysis for fold alignment algorithms to propose a new topology. Although several of the TM segments differed between the two models, TMs X-XII were essentially unaltered. This supports the assignment of these amino acids as TM XI in our study.
We found that TM XI consisted of two helical regions, comprising amino acids 447-453 and 460-471 connected by an extended region from amino acids 455 to 459. In their study, Landau et al. (42) made several predictions about the structure of TM XI. They suggested that Gly455 and Gly456 would facilitate unwinding of that region of the TM XI helix. Our results agree with their predictions as these two amino acids were within the extended region from amino acids 455 to 459. The fact that there were only two amino acids that retained good functional activity within the 454-463 sequence supports the concept that this is a domain critical to function of the protein. Unfortunately mutant proteins containing these amino acids did not retain enough activity to be examined functionally.
Gly455 and Gly456 have been shown to be critical in affecting the set point of the NHE1 protein and have been suggested to constitute important elements of the pH sensor of NHE1 (17, 18). Their presence in a conserved extended region of the protein would support this hypothesis. It should be noted, however, that in NhaA Arkin et al. (43) favor a model of pH regulation by which protononation-deprotonation of a single carboxyl of an acidic residue affects protein structure and regulates pH sensitivity. If this were true for NHE1, then changes to Gly455 and Gly456 would likely affect NHE1 function through alterations in structure, which secondarily alter the pH sensor of the protein or regulation by the pH sensor. We found that mutation of these two residues resulted in both reduced expression and glycosylation of the protein. In addition, targeting of the protein was mostly aberrant. This, plus their presence in, and possible mediation of the structure of the extended region supports the concept that they are critical for the structure of the protein.
Because TM IV of NHE1 has a similar structure and function to TM IV of NhaA (2), we examined the possibility that TM XI of NHE1 has a similar structure and function to TM XI of NhaA. Although the two proteins have poor primary amino acid sequence conservation, we have earlier suggested that they may have similar three-dimensional structures and might conserve key residues important in function (44). We therefore compared our structure of TM XI of NHE1 with the NhaA crystal structure (19). The equivalent of the MTSET-sensitive residues Leu465 and Ile461 of NHE1 in the NhaA structure would be Ile345 and Met341, respectively, based on the alignment shown in Fig. 7B. Residues 345 and 341 in NhaA are on the C-terminal helix of TM XI, on the periplasmic side of the pore. If the corresponding residues in NHE1 are in a similar location, near the extracellular pore, this would account for them being MTSET accessible. Residue Ile337 of NhaA, equivalent to residue Leu457 in NHE1, does not appear to be accessible from the pore and points toward the cytoplasmic side. The crystal structure is of an inactive state of NhaA, however; conformational changes suggested to occur with activation (45, 46) may allow access to this region.
In summary, we have shown that TM XI of NHE1 is critical to function of the protein and possesses a pore lining residue at amino acid Leu465. The structure of TM XI is that of a helix with an intervening interrupted irregular structure consisting of residues Gly455-Gly459. It has an overall similarity to the structure of TM XI of the E. coli antiporter NhaA. Future experiments will further compare the structure and function of NHE1 with that of NhaA in an effort to further elucidate the mechanism of function of the mammalian Na+/H+ exchanger. Solving the structure of the full-length protein will provide more details of transmembrane segment overall structure and elucidate the precise location of any residues that are in variable regions of peptide segments.
Supplementary Material
Acknowledgments
We thank the National High Field Nuclear Magnetic Resonance Centre (NANUC) for assistance and use of the 800 MHz spectrometer.
The atomic coordinates and structure factors (code 2KBV) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported in part by NANUC, Canadian Institutes of Health Research, Natural Science and Engineering Research Council of Canada, the University of Alberta, and a grant from the Canadian Institutes of Health Research (to L. F. and B. D. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S3.
Footnotes
The abbreviations used are: NHE1, Na+/H+ exchanger isoform 1; TM, transmembrane; DPC, dodecylphosphocholine; MTSET, (2-(trimethylammonium) ethyl)methanethiosulfonate; MTSES, (2-sulfonatoethyl)methanethiosulfonate; cNHE1, cysteine-less NHE1; HA, hemagglutinin; r.m.s., root mean square; HSQC, heteronuclear signal quantum coherence.
References
- 1.Karmazyn, M., Sawyer, M., and Fliegel, L. (2005) Curr. Drug Targets Cardiovasc. Haematol. Disord. 5 323-335 [DOI] [PubMed] [Google Scholar]
- 2.Slepkov, E. R., Rainey, J. K., Sykes, B. D., and Fliegel, L. (2007) Biochem. J. 401 623-633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grinstein, S., Rotin, D., and Mason, M. J. (1989) Biochim. Biophys. Acta 988 73-97 [DOI] [PubMed] [Google Scholar]
- 4.Shrode, L., Cabado, A., Goss, G., and Grinstein, S. (1996) in The Na+/H+ Exchanger (Fliegel, R. G., ed) pp. 101-122, Landes Company, Austin, TX
- 5.Denker, S. P., and Barber, D. L. (2002) J. Cell Biol. 159 1087-1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paradiso, A., Cardone, R. A., Bellizzi, A., Bagorda, A., Guerra, L., Tommasino, M., Casavola, V., and Reshkin, S. J. (2004) Breast Cancer Res. 6 R616-R628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karmazyn, M., Liu, Q., Gan, X. T., Brix, B. J., and Fliegel, L. (2003) Hypertension 42 1171-1176 [DOI] [PubMed] [Google Scholar]
- 8.Fliegel, L. (2001) Basic Res. Cardiol. 96 301-305 [DOI] [PubMed] [Google Scholar]
- 9.Mentzer, R. M., Jr., Lasley, R. D., Jessel, A., and Karmazyn, M. (2003) Ann. Thorac. Surg 75 S700-S708 [DOI] [PubMed] [Google Scholar]
- 10.Lang, H. J. (2003) in The Na+/H+ Exchanger, From Molecular to Its Role in Disease (Karmazyn, M., Avkiran, M., and Fliegel, L., eds) pp. 239-253, Kluwer Academic Publishers, Boston
- 11.Wakabayashi, S., Pang, T., Su, X., and Shigekawa, M. (2000) J. Biol. Chem. 275 7942-7949 [DOI] [PubMed] [Google Scholar]
- 12.Slepkov, E. R., Chow, S., Lemieux, M. J., and Fliegel, L. (2004) Biochem. J. 379 31-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Slepkov, E. R., Rainey, J. K., Li, X., Liu, Y., Cheng, F. J., Lindhout, D. A., Sykes, B. D., and Fliegel, L. (2005) J. Biol. Chem. 280 17863-17872 [DOI] [PubMed] [Google Scholar]
- 14.Ding, J., Rainey, J. K., Xu, C., Sykes, B. D., and Fliegel, L. (2006) J. Biol. Chem. 281 29817-29829 [DOI] [PubMed] [Google Scholar]
- 15.Pedersen, S. F., King, S. A., Nygaard, E. B., Rigor, R. R., and Cala, P. M. (2007) J. Biol. Chem. 282 19716-19727 [DOI] [PubMed] [Google Scholar]
- 16.Wakabayashi, S., Pang, T., Su, X., and Shigekawa, M. (2000) FEBS Lett. 487 257-261 [DOI] [PubMed] [Google Scholar]
- 17.Wakabayashi, S., Hisamitsu, T., Pang, T., and Shigekawa, M. (2003) J. Biol. Chem. 278 43580-43585 [DOI] [PubMed] [Google Scholar]
- 18.Wakabayashi, S., Hisamitsu, T., Pang, T., and Shigekawa, M. (2003) J. Biol. Chem. 278 11828-11835 [DOI] [PubMed] [Google Scholar]
- 19.Hunte, C., Screpanti, E., Venturi, M., Rimon, A., Padan, E., and Michel, H. (2005) Nature 435 1197-1202 [DOI] [PubMed] [Google Scholar]
- 20.Cunningham, F., and Deber, C. M. (2007) Methods 41 370-380 [DOI] [PubMed] [Google Scholar]
- 21.Oblatt-Montal, M., Reddy, G. L., Iwamoto, T., Tomich, J. M., and Montal, M. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 1495-1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naider, F., Khare, S., Arshava, B., Severino, B., Russo, J., and Becker, J. M. (2005) Biopolymers 80 199-213 [DOI] [PubMed] [Google Scholar]
- 23.Hunt, J. F., Earnest, T. N., Bousche, O., Kalghatgi, K., Reilly, K., Horvath, C., Rothschild, K. J., and Engelman, D. M. (1997) Biochemistry 36 15156-15176 [DOI] [PubMed] [Google Scholar]
- 24.Katragadda, M., Alderfer, J. L., and Yeagle, P. L. (2001) Biophys. J. 81 1029-1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeagle, P. L., Choi, G., and Albert, A. D. (2001) Biochemistry 40 11932-11937 [DOI] [PubMed] [Google Scholar]
- 26.Damberg, P., Jarvet, J., and Graslund, A. (2001) Methods Enzymol. 339 271-285 [DOI] [PubMed] [Google Scholar]
- 27.Henry, G. D., and Sykes, B. D. (1994) Methods Enzymol. 239 515-535 [DOI] [PubMed] [Google Scholar]
- 28.Murtazina, R., Booth, B. J., Bullis, B. L., Singh, D. N., and Fliegel, L. (2001) Eur. J. Biochem. 268 4674-4685 [DOI] [PubMed] [Google Scholar]
- 29.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) J. Biomol. NMR 6 277-293 [DOI] [PubMed] [Google Scholar]
- 30.Johnson, B. A., and Blevins, R. A. (1994) J. Biomol. NMR 4 603-614 [DOI] [PubMed] [Google Scholar]
- 31.Schwieters, C. D., Kuszewski, J. J., Tjandra, N., and Clore, G. M. (2003) J. Magn. Reson. 160 65-73 [DOI] [PubMed] [Google Scholar]
- 32.Haworth, R. S., Frohlich, O., and Fliegel, L. (1993) Biochem. J. 289 637-640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li, X., Ding, J., Liu, Y., Brix, B. J., and Fliegel, L. (2004) Biochemistry 43 16477-16486 [DOI] [PubMed] [Google Scholar]
- 34.Davis, J. H., Clare, D. M., Hodges, R. S., and Bloom, M. (1983) Biochemistry 22 5298-5305 [Google Scholar]
- 35.Rainey, J. K., Fliegel, L., and Sykes, B. D. (2006) Biochem. Cell Biol. 84 918-929 [DOI] [PubMed] [Google Scholar]
- 36.Reddy, T., Ding, J., Li, X., Sykes, B. D., Rainey, J. K., and Fliegel, L. (2008) J. Biol. Chem. 283 22018-22030 [DOI] [PubMed] [Google Scholar]
- 37.Wishart, D. S., Sykes, B. D., and Richards, F. M. (1992) Biochemistry 31 1647-1651 [DOI] [PubMed] [Google Scholar]
- 38.Wishart, D. S., Bigam, C. G., Holm, A., Hodges, R. S., and Sykes, B. D. (1995) J. Biomol. NMR 5 67-81 [DOI] [PubMed] [Google Scholar]
- 39.Kabsch, W. (1976) Acta Crystallogr. Sect. A 32 922-923 [Google Scholar]
- 40.Wishart, D. S., Boyko, R. F., Willard, L., Richards, F. M., and Sykes, B. D. (1994) Comput. Appl. Biosci. 10 121-132 [DOI] [PubMed] [Google Scholar]
- 41.He, M. M., Sun, J., and Kaback, H. R. (1996) Biochemistry 35 12909-12914 [DOI] [PubMed] [Google Scholar]
- 42.Landau, M., Herz, K., Padan, E., and Ben-Tal, N. (2007) J. Biol. Chem. 282 37854-37863 [DOI] [PubMed] [Google Scholar]
- 43.Arkin, I. T., Xu, H., Jensen, M. O., Arbely, E., Bennett, E. R., Bowers, K. J., Chow, E., Dror, R. O., Eastwood, M. P., Flitman-Tene, R., Gregersen, B. A., Klepeis, J. L., Kolossvary, I., Shan, Y., and Shaw, D. E. (2007) Science 317 799-803 [DOI] [PubMed] [Google Scholar]
- 44.Dibrov, P., and Fliegel, L. (1998) FEBS Lett. 424 1-5 [DOI] [PubMed] [Google Scholar]
- 45.Olkhova, E., Padan, E., and Michel, H. (2007) Biophys. J. 92 3784-3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kozachkov, L., Herz, K., and Padan, E. (2007) Biochemistry 46 2419-2430 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.