

Abstract
The transducer of Cdc42-dependent actin assembly (Toca-1)-N-WASP complex was isolated as an essential cofactor for Cdc42-driven actin polymerization in vitro. Toca-1 consists of an N-terminal F-BAR domain, followed by a Cdc42 binding site (HR1 domain) and an SH3 domain, (the N-WASP interacting site). N-WASP is an activator of actin nucleation through the Arp2/3 complex. The aim of the present study was to investigate the cellular function of the Toca-1-N-WASP complex. We report that Toca-1 induces filopodia and neurites as does N-WASP in N1E115 neuroblastoma cells. Toca-1 requires the F-BAR domain, Cdc42 binding site, and SH3 domain to induce filopodia. Toca-1 and N-WASP both require each other to induce filopodia. The expression of Toca-1 and N-WASP affects the distribution, size, and number of Rab5 positive membranes. Toca-1 interacts directly with N-WASP in filopodia and Rab5 membrane as seen by Forster resonance energy transfer. Thus the Toca-1-N-WASP complex localizes to and induces the formation of filopodia and endocytic vesicles. Last, three inhibitors of endocytosis, Dynamin-K44A, Eps15Δ95/295, and clathrin heavy chain RNA interference, block Toca-1-induced filopodial formation. Taken together, these data suggest that the Toca-1-N-WASP complex can link filopodial formation to endocytosis.
Cell migration and axon guidance rely on morphological actin-based structures such as filopodia and lamellipodia/membrane ruffles (1). Cdc42 is a member of the Rho family of GTPases and plays a prominent role in cell signaling pathways that control cell migration as well as cell shape and polarity. Cdc42 was first identified as a yeast protein involved in cell division that affects budding of daughter cells (2). In mammalian cells, one specific function of Cdc42 is to activate filopodial formation (3, 4). Filopodia are dynamic actin-based structures that protrude from the plasma membrane and are thought to play a role in a diverse number of cellular and developmental processes such as synaptogenesis, dorsal closure in embryos, and viral uptake (5-7). Cdc42-mediated filopodial formation involves a number of proteins including IRSp53, Mena, Eps8, and N-WASP (8-11). A potential mechanism by which Cdc42 generates filopodia is by recruiting IRSp53, which then recruits Mena, Eps8, and N-WASP (11). IRSp53 can induce membrane deformation via its I-BAR2,3 domain (12) and its SH3 domain partners, Mena, Eps8, and N-WASP, are likely to regulate actin dynamics.
N-WASP is a member of the WASP/WAVE family proteins that binds to (via the WA or VCA region) and regulates the Arp2/3 complex-mediated actin nucleation (for a recent review, see Ref. 13). N-WASP-Arp2/3 interaction is regulated by transducer of cdc42-dependent actin assembly (Toca-1), Cdc42, and phosphatidyl inositol,4,5-biphosphate (14). N-WASP-mediated actin nucleation is most closely linked with vesicle motility (15). N-WASP plays a role in filopodial formation but this is not directly connected with its ability to activate actin nucleation/polymerization via the Arp2/3 complex (11).
Toca-1 is a Cdc42 effector first identified through in an in vitro assay for actin polymerization. Ho et al. (16) using reconstitution and immunodepletion experiments demonstrated that the Toca-1-N-WASP interaction was essential for Cdc42-dependent actin polymerization driven from cell extracts. Toca-1 is composed of at least three prominent domains: an F-BAR4 domain, a Cdc42 binding site (HR1 domain), and an SH3 domain. The SH3 domain interacts specifically with N-WASP. Toca-1 is a member of a family of proteins that includes, Cdc42 interacting protein 4 (CIP4) and formin binding protein 17 (FBP17). The F-BAR domain of these proteins can deform membranes in vitro and this is linked with vesicle formation in vivo (17).3,4 There is some redundancy among Toca-1, CIP4, and FBP17 as knockdown (KD) of protein by RNAi of all three proteins is required to inhibit endocytosis substantially (17, 18).
The aim of the present study was to investigate the cellular function of the Toca-1-N-WASP complex in N1E115 neuroblastoma cells. We find that expression of Toca-1 and/or N-WASP induces filopodial and neurite formation in N1E115 neuroblastoma cells. For filopodial formation Toca-1 requires its F-BAR domain, Cdc42 binding site, SH3 domain, and N-WASP. N-WASP requires Toca-1 for filopodial formation. Using Forster resonance energy transfer (FRET) we show that Toca-1 interacts directly with N-WASP in filopodia and Rab5 membrane. Expression of the Toca-1-N-WASP complex affects the distribution, size, and number of Rab5 membrane. Last, Toca-1-induced filopodial formation was inhibited by Dynamin-K44A5 and Esp15-Δ95/295,6 and clathrin heavy chain (CHC) RNAi, three inhibitors of endocytosis. Taken together, these data suggest that the Toca-1-N-WASP complex links filopodial formation with endocytosis.
MATERIALS AND METHODS
Subcloning and Site-directed Mutagenesis—Human Toca-1 (a gift from Prof. Marc W. Kirschner, Harvard University, Boston, MA) was amplified by PCR and subcloned from pCS+ vector into the pXJ40-mRFP vector between the HindIII and NotI sites. Myc-Toca-1W518K, mRFP-Toca-1W518K, mRFP-Toca-1-K33Q/R35Q, mRFP-Toca-1-K51Q/K52Q, mRFP-Toca-1-R112Q/K113Q, and mRFP-Toca-1-M383I/G384S/D385T were generated by mutated primer pairs using the site-directed mutagenesis kit (Stratagene). The mutations were confirmed by DNA sequencing. The primer sequences are listed below: Toca-1/pXJ40-mRFP, FP, 5′-CCCAAGCTTAGCTGGGGCACGGAGCTGTG-3′ and RP, 5′-TTTTCCTTTTGCGGCCGCTCAGGAACCTTTACTGTTTTTCTC-3′; Myc-Toca-1-W518K/pCS+ and mRFP-Toca-1W518K/pXJ40, FP, 5′-GACAAAGGTGACGGAAAGACAAGAGCTCGGAG-3′ and RP, 5′-CTCCGAGCTCTTGTCTTTCCGTCACCTTTGTC-3′; mRFP-Toca-1-M383I/G384S/D385T/pXY40, FP, 5′-GAAGAATCCACAAATAAGCACTCCAGGGAGTTTGCAG-3′ and RP, 5′-CTGCAAACTCCCTGGAGTGCTTATTTGTGGATTCTTC-3′; mRFP-Toca-1-K33Q/R35Q/pXJ40, FP, 5′-GATATGCCAAATTTGTTCAAGAGCAGATAGAAATTGAACAG-3′ and RP, 5′-CTGTTCAATTTCTATCTGCTCTTGAACAAATTTGGCATATC-3′; mRFP-Toca-1-K51Q/K52Q/pXJ40, FP: 5′-GAGAAATCTGGTTCAGCAGTACTGCCCCAAACG-3′ and RP, 5′-CGTTTGGGGCAGTACTGCTGAACCAGATTTCTC-3′; mRFP-Toca-1-R112Q/K113Q/pXJ40, FP, 5′-GATCTGAAAACTGAACAACAAATGCATCTGCAAG-3′ and RP, 5′-CTTGCAGATGCATTTGTTGTTCAGTTTTCAGATC-3′.
Mammalian Cell Culture—N1E115 cells were cultured in Dulbecco's modified Eagle's medium (4500 mg/ml glucose) supplemented with 10% fetal bovine serum in a humidified 37 °C incubator with 5% CO2. N-WASP WT (Flox) and N-WASP KO (1h51) (51) cells were cultured in Dulbecco's modified Eagle's medium (1000 mg/ml glucose) supplemented with 10% fetal bovine serum and 1% antibiotics and incubated in a humidified 32 °C incubator with 5% CO2.
Transfection—N1E115 cells were seeded onto laminin (10 μg/ml) coated coverslips at a density of 1.5 × 105 cells/40-mm dish. N1E115 cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol after overnight growth. Briefly plasmid DNA and Lipofectamine 2000 (1:3 ratio) were separately diluted in serum-free medium. After DNA and Lipofectamine were combined, the transfection mixture was incubated at room temperature for 45 min. After that, the mixture was added into the cells and incubated at 37 °C in a incubator for 4 h. Then the medium was changed to fresh growth medium and the cells were incubated for protein expression. Transfection of N-WASP WT and KO fibroblasts was as described in Ref. 11.
Immunofluorescence—24 h after transfection, the cells were fixed in 4% paraformaldyhyde for 15 min followed by three washes with PBS for 5 min each. Then the cells were permeabilized in 0.2% Triton X-100 for 5 min. After three washes with PBS, the cells were blocked in 5% normal goat serum for 30 min, washed, and incubated in PBS-diluted primary antibody (anti-Myc (1:300), anti-Rab5 (1:50), all from Santa Cruz) overnight at 4 °C. Following the washing steps, the cells were incubated with Alexa 488- or Alexa 647-conjugated secondary antibody (Molecular Probes, Invitrogen). Finally the cells were washed and mounted onto the glass slides using Hydromount (National Diagnostics).
Blockers of Endocytosis—EGF phosphorylation substrate 15 (Eps15) is a key component for assembling functional clathrin-coated pits (19, 20). Plasmid constructs of Eps15 (GFP-EΔ95/295 dominant negative and GFP-D3Δ2) were kindly provided by Alice Dantry (Pasteur Institute, Paris, France). The GTPase Dynamin is required for both clathrin- and caveolae-dependent endocytosis (21). Plasmid constructs of Dynamin, HA-Dynamin 1-K44A dominant negative, and HA-Dynamin 1 wild type, were used as endocytosis blocker and control, respectively. Alexa Fluor 488-/647-transferrin (TF; Molecular Probes, Invitrogen) were used as markers for clathrin-mediated endocytosis.
Two independent RNAi were used to investigate the effects of CHC KD. The target sequences were: GGAAAUGGAUCUCUUUGAA (CHC oligo I) and UAAUCCAAUUCGAAGACCA (CHC oligo II) (Dharmacon, Lafayette, CO). Sequences were submitted to BLAST searches against the mouse genome sequence to ensure that only the desired mRNA was targeted. Stealth RNAi negative control duplexes (Invitrogen) were used as non-silencing control. Based on individual and pooled KD efficiency, pooled CHC oligos (oligo I + II) were used for subsequent experiments. N1E115 cells were seeded at a density of 150,000 per well in 6-well plates. 1 h after seeding, N1E115 were transfected with 10 nm CHC or negative control duplexes using HiPerfect (Qiagen, Valencia, CA) according to the manufacturer's instructions. At 24 h, cells were transfected with 0.5 μg of myc-Toca-1 using Lipofectamine 2000 (Invitrogen). N1E115 cells were then processed at 48 h for Western blotting or immunofluorescence. CHC was visualized by rabbit anti-CHC (Abcam) at a dilution of 1:100 during immunofluorescence. CHC was detected in whole cell lysates by mouse anti-CHC (Clone TD.1, Sigma) at a dilution factor of 1:1000 during Western blotting. Equal loading in SDS-PAGE was ensured by protein quantitation using the Bradford assay (Bio-Rad). Mouse anti-glyceraldehyde-3-phosphate dehydrogenase (Chemicon) was used to normalize for protein loading in each lane.
Transferrin Labeling—24 h after transfection, cells grown on coverslips to 70-80% confluency were rinsed twice with Dulbecco's PBS containing 1 mm CaCl2 and 0.5 mm MgCl2 (D-PBS) (Invitrogen) and serum-starved for 2 h in serum-free Dulbecco's modified Eagle's medium. Cells were then incubated with 25 μg/ml Alexa-transferrin (Molecular Probes) in serum-free Dulbecco's modified Eagle's medium for 10 min at 37 °C and 5% CO2. Subsequently, coverslips were placed on ice and rinsed twice with ice-cold D-PBS.
Determination of Endocytosis Inhibition Using Labeled Transferrin—Quantification of fluorescence intensities was performed using Metamorph software (Universal Imaging, PA), as previously described (22). Briefly, fluorescence intensities were measured by thresholding and outlining whole individual cells, followed by determination of integrated fluorescence intensities, which were then normalized to cell area. Average values were expressed as a ratio relative to control cells without endocytosis blockers.
Filopodial Assay—Toca-1 and N-WASP-induced filopodia were scored on neurites of live N1E115 cells, as previously described (8, 11). N1E115 cells were transfected with myc-Toca-1, GFP-Eps15, or HA-Dynamin 1 and corresponding mRFP- or GFP-actin. Twenty-four to thirty-six hours after transfection, time-lapse microscopy was carried out on a heated stage at 37 °C and 5% CO2. Cells were imaged under phase-contrast and fluorescence with Axiovert 200 (Zeiss) with frame captures every 10 s for 5 min, driven by Metamorph software.
Confocal Microscopy—Immunofluorescence was examined on an Olympus FV 1000 confocal microscope (Olympus, Japan) using a ×63 oil immersion objective with a 1.4 numerical aperture and a pinhole at airy 1. GFP-tagged proteins were excited by a 488-nm laser line and the emission was selected using a band-pass filter between 500 and 550 nm. mRFP-tagged proteins were excited by a 561-nm laser line and the emission was selected using a band-pass filter between 580 and 680 nm. The images were taken at a speed 2 μs/pixel sequentially at a size of 512 × 512.
Intensity Profiling—Cells were transfected with the relevant constructs and stained with anti-Rab5 antibody 24 h post-transfection. The cells were then fixed and examined by confocal microscopy. A line was drawn through the region of interest and the intensity profile was measured using FV10-ASW software of the Olympus FV-1000 confocal microscope. We used Metamorph software to determine the co-localization correlation coefficients.
FRET Measurement—FRET was measured by acceptor photobleaching method (8, 11, 23) by making appropriate settings in a Zeiss LSM 510 confocal microscope with the objective of C-Apochromat 63 × 1.2 W objective. The fusion proteins of GFP/mRFP were excited using 488 and 561 nm laser lines as excitation source, by selecting 405/488/561 dichoric mirror and 490,565 secondary dichoric mirrors for GFP and mRFP emission, respectively. The emission was monitored by selecting GFP (band-pass 505-550) and red (long-pass 575) emission filters to record the fluorescence intensity. Regions of interest were selected and photobleached using 70% of 561 nm laser power by selecting 50 iterations. Bleaching was performed following pre- and post-scan images. The increase in GFP fluorescence intensity followed by mRFP bleaching was measured as FRET. FRET efficiency was calculated using the change in background subtracted fluorescence intensity as 100 × ((post-bleach intensity) - (pre-bleach intensity)/(post-bleach intensity)).
To verify the increase in GFP intensity due to any possible artifact we obtained the Pearson product moment correlation coefficient r, a dimensionless index that ranges from -1.0 to 1.0 inclusive and reflects the extend of a linear relationship between the two fluorescence intensity data of GFP and mRFP while bleaching. In our case we expect -1.0 as the perfect fitting of the linear relation. However, we selected the range of -0.7 to -1.0 as the best range of index.
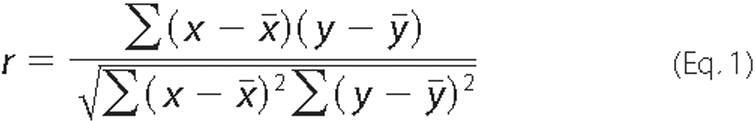
Where x and y are the sample means average (array 1, GFP intensity) and average (array 2, mRFP intensity). For complete details of AP-FRET method see Ref. 11.
Fluorescence Lifetime Imaging Microscopy (FLIM)—FLIM experiments were performed with the LIFA system (Lambert Instruments, The Netherlands) on an inverted wide-field fluorescence microscope (Olympus IX71, Center Valley, PA) with 60 × 1.35 oil immersion objective. Fluorescence lifetime was measured using software provided by Lambert Instruments. GFP was excited by a sinusodially modulated 4-milliwatt 470-nm light-emitting diode at 40 MHz. The GFP filter set was used for excitation and emission signals. The emission was collected by an intensified CCD camera. Fluorescein isothiocyanate was used as a standard lifetime reference of 4 ns. 12 phase and modulation shifted images were taken, fitted with a sinus function and used for the calculation of lifetime (for further details see Ref. 24). The lifetimes from 50 regions of interest were taken from different cells are averaged to give the average lifetime and standard deviation. The experiments were repeated three times and data from a single representative experiment are shown. The lifetime (nanoseconds) is shown in pseudocolors.
Scoring of Filopodia and Neurites—Filopodia were scored as protrusions that were dynamic with a lifetime in the range of minutes and length (as defined in Ref. 11). Filopodial lifetime was the time interval measured between appearance and disappearance of individual filopodial. Filopodial length was measured from the base of the structure to the tip (under DIC) and scored when the filopodia reached its maximum length. Neurites (or cell extensions) were scored as processes that protruded from the cell body and were at least one cell body length. Each experiment was repeated at least three times and the data presented are one of the representative experiments.
Characterization of Rab5 Membranes—(i) Distribution of vesicles was measured by placing squares with the size 1.5 × 1.5 μm (9 × 9 pixel) from the cell periphery (region 1) to the perinuclear region (region 5) within individual cells and the average fluorescence intensity of each square was measured by Meta-morph. (ii) Vesicle size was analyzed by randomly picking 20 vesicles/cell measuring their diameter using Metamorph software. (iii) Vesicle number was analyzed by selecting fixed areas within each cell and counting the number of vesicles present. Vesicle numbers were then normalized to the non-transfected cell. The experiments were repeated three times, with n = 20, as the mean ± S.D. and a representative experiment is presented.
Statistical Analysis—In general, readings were obtained from at least three independent experiments, and expressed as the mean ± S.D. Experimental data were analyzed by Student's t test.
Toca-1 KD by RNA Interference—Toca-1 short interference RNA sequences were synthesized by Invitrogen according to the published work (25). The Toca-1 RNAi sequence was GCGAGAAGUUGUAGCAGAAGAAAUG and the control RNAi sequence was CCAUCAGCAAGAAGUAAGUACAUUA. HeLa cells were transfected with the RNAi twice (day 0 and day 2) as described in the manufacture's manual. The cells were lysed 48 h after the second transfection. Immunoblot was performed to determine the protein level in the cell lysate. Anti-Toca-1 antibody was a kind gift from Prof. Marc W. Kirschner (Harvard). To determine the effect of Toca-1 KD on the N-WASP phenotype, the cells were transfected with HA-N-WASP and GFP-actin. The filopodial number per cell was counted 24 h after transfection.
Liposome Binding Assay—F-BAR domain (amino acid residues 2-293) of Toca-1 and three F-BAR domain mutants (K33Q/R35Q, K51Q/K52Q, R112Q/R113Q) were amplified by PCR and cloned into pGEX-4T-1 vector between BamHI and XhoI sites. The mutant clones were confirmed by DNA sequencing. The glutathione S-transferase-tagged protein was expressed in BL21 and purified using standard protocol. The protein concentration was determined by the Bradford protein assay. Liposome binding assays were performed as previously described (18, 25, 26). Lipids were from Sigma. The liposomes were either made from total brain lipids (Folch fraction I) or phosphatidylethanol (65%), phosphatidylcholine (20%), phosphatidylserine (5%), and phosphatidylinositol 4,5-bisphosphate (10%). Briefly, 5 μg of proteins were incubated with 100 μg of liposome at room temperature for 15 min. The mixture was then subjected to centrifugation at 16,000 × g for 15 min at 25 °C. SDS-PAGE was performed for both supernatants and pellets. The gel was stained by Coomassie Blue and the protein bands were quantified using Image J software (National Institutes of Health).
RESULTS
Toca-1 and N-WASP Induce Filopodial and Neurite Formation—We have previously used mouse neuroblastoma N1E115 cells as a model neuronal cell to examine the morphological role of Ras, phosphatidylinositol 3-kinase, and the effectors of Rho GTPase (3, 27-29). In particular, we have shown that Cdc42 and one of its effectors, IRSp53, induces filopodia and neurites in these cells (8). Because Toca-1 is also a Cdc42 effector we decided to use N1E115 cells to understand Toca-1 function. We expressed Toca-1 with GFP-actin in N1E115 cells and monitored the phenotype. Toca-1 induced filopodial and neurite formation in N1E115 cells (Fig. 1). For time lapse see supplemental Movie 1.7 Under these conditions Toca-1 did not affect lamellipodial formation or membrane ruffling significantly (data not shown). Toca-1 also induced filopodia (and a loss of stress fibers) in other cell types including: HeLa cells and Swiss 3T3 fibroblasts (data not shown). To show the specificity of the Toca-1 phenotype we used a mutant Toca-1W518K (with a defective SH3 domain). Toca-1W518K failed to induce filopodia but was still competent to induce neurites (Fig. 1A). Toca-1 induced multiple neurites that were highly branched. In addition, the neurites were longer than that observed after serum starvation of N1E115 cells (8). N-WASP expression in N1E115 cells had a similar phenotype to Toca-1; N-WASP induced filopodia and neurites (Fig. 1C). However, the neurites generated by N-WASP were shorter than those induced by Toca-1.
FIGURE 1.

Toca-1 and N-WASP induce filopodia and neurites in N1E115 cells. A, N1E115 cells were transfected with GFP-actin, GFP-actin/Myc-Toca-1, or GFP-actin/Myc-Toca-1W518K and GFP-actin/HA-N-WASP. GFP fluorescence was monitored by confocal microscopy. The panels below the arrows show enlarged neurites to allow monitoring of complexity and filopodial formation. Bar = 50 μm. B, bar charts show statistical analysis (for at least three independent experiments, n = 15 cells). Cells were scored for neurites and filopodia as described under “Materials and Methods.” Data are presented as mean ± S.D. C, to measure filopodial characteristics we used DIC time-lapse microscopy. Individual filopodia were followed from appearance to disappearance and the time required defined as the lifetime. Length of filopodia is the maximum length as measured from the base to the tip under DIC. All these experiments include sequential GFP-actin imaging to ensure that the structures contain actin. To assess the characteristics of the Toca-1-induced filopodia we carried out DIC time-lapse experiments of transfected cells. The DIC image shows a typical neurite with associated Toca-1-induced filopodia over a time course as indicated in individual frames. Arrowheads (empty, white, and black) follow three independent filopodia over the time course. Bar = 4 μm.
Using sequential fluorescence/DIC time lapse microscopy with GFP-actin-transfected cells we were able to show that Toca-1 induced bona fide filopodia.8 The Toca-1-induced filopodia had an average lifetime of 127.5 s and average length of 6.6 μm (Table 1). These filopodial characteristics were similar to endogenous structures as well as those induced by Cdc42/RacN17, IRSp53, and N-WASP (Table 1).
TABLE 1.
Characteristics of endogenous and induced filopodia in N1E115 cells
The table shows a comparison of the lifetime and length of endogenous filopodia with those induced by cDNA transfections Cdc42/RacN17, IRSp53, N-WASP (11), and Toca-1. Details of the measurements are described under “Materials and Methods.”
| Treatment | Filopodia length | Filopodia lifetime | Ref. |
|---|---|---|---|
| μm | s | ||
| None (endogenous) | 15 ± 7.68 | 142 ± 101 | 11 |
| Cdc42V12/RacN17 | 8.37 ± 1.59 | 157 ± 30 | 11 |
| IRSp53 | 6.83 ± 1.88 | 187 ± 38 | 11 |
| N-WASP | 7.35 ± 0.97 | 154 ± 20 | 11 |
| Toca-1 | 6.6 ± 1.5 | 127.5 ± 41.4 | This study |
Domains of Toca-1 Required for Filopodial and Neurite Formation—Next we generated Toca-1 F-BAR domain mutations (K33Q/R35Q, R51Q/K52K, and R112Q/K113Q) and a Cdc42 binding site mutant (M383I/G384S/D3385T). The F-BAR domain mutants were based on CIP4 mutants that fail to induce tubulation in cells and have reduced lipid binding ability (18). The ability of these Toca-1 mutants to bind brain lipids and phosphatidylinositol 4,5-bisphosphate was also substantially reduced compared with wild-type (Fig. 2D). Toca-1 mutants were expressed in N1E115 cells independently and phenotypes were scored. Filopodial formation by Toca-1 required its F-BAR domain, Cdc42 binding site, and the SH3 domain (Fig. 2). However, only the Cdc42 binding site was essential for neurite formation.
FIGURE 2.

Effect of mutations in Toca-1 on filopodial formation. A, a schematic of the domain structure of Toca-1 is shown with positions of the mutations used. Three distinct domain mutations were generated: F-BAR domain, HR1 domain (Cdc42 binding site), and SH3 domain (N-WASP interacting site). B, mutant Toca-1 genes were transfected into N1E115 cells as mRFP fusions with GFP-actin. The main panel shows three images: mRFP, GFP, and the merge with the different version of Toca-1 transfected. Three F-BAR domain mutants, K33Q/R35Q, K51Q/K52Q, and R112Q/K113 were used. The Cdc42 binding mutant was M383I/G384S/D385T. The SH3 domain mutant was W518K. C, bar charts show statistical analysis of the experiments. Three independent experiments were performed (with n = 15-20). Transfected cells were scored for neurite and filopodial formation. The data are presented as mean ± S.D. Bar = 10 μm. D, Toca-1 F-BAR domain and mutants were purified as glutathione S-transferase fusion proteins and lipid sedimentation assays carried out as described under “Materials and Methods.” S, supernatant; P, pellet. The data are presented as mean ± S.D. (n = 6) from two experiments. PI(4,5)P2, phosphatidylinositol 4,5-bisphosphate.
Toca-1 Requires N-WASP for Filopodial Formation—The inability of the Toca-1W518K mutant to induce filopodia suggested that interaction with N-WASP was important for this phenotype. To determine the role of N-WASP in Toca-1 phenotypes we expressed Toca-1 in N-WASP WT and KO cells and compared the phenotype obtained with wild-type and control cells. Toca-1 failed to induce filopodia (or cell extensions) in N-WASP KO cells (Fig. 3A). Toca-1W518K failed to induce filopodia in either wild-type cells or N-WASP KO cells (Fig. 3B).
FIGURE 3.

Toca-1 does not generate filopodia in N-WASP KO cells. A, Toca-1 was transfected into N-WASP WT (flox) and N-WASP KO cells (1h51) and filopodial number per cell was scored as described under “Materials and Methods.” Flox and 1h51 N-WASP KO cells were from Lommel et al. (51). Panels a and b, WT cells; panel c, N-WASP KO cells. Three independent experiments were performed (n = 20) and one representative example is shown. Data shown are mean ± S.D. Bar = 10 μm. B, Toca-1W518K was transfected into WT and N-WASP KO cells and morphology monitored. C, N-WASP KO cells were transfected with Toca-1 and N-WASP to allow reconstitution of the cells. The right panel shows a DIC time-lapse sequence (in seconds: 0, 50, 100, 150, 200, 250, and 300) of the three combinations. Toca-1 in N-WASP WT cells, Toca-1 in N-WASP KO cells, and Toca-1 with N-WASP and N-WASP KO cells. Tracings show structures generated: red, filopodia and blue, lamellipodia. In parts a-c, the bar charts show a statistical analysis of the experiments. Three independent experiments were performed (withn=15-20). Data shown are mean ± S.D. Bar= 10 μm.
Next we carried out reconstitution experiments by including low amounts of N-WASP cDNA with Toca-1 and Toca-1 W518K and measured filopodial formation. Filopodial formation could be reconstituted in N-WASP KO cells by co-injection of Toca-1/N-WASP (Fig. 3C) but not in the case of Toca-1 W518K/N-WASP (Fig. 3, B and C).
N-WASP Requires Toca-1 for Filopodial Formation—Having established that Toca-1 does not induce filopodia in the absence of N-WASP we now asked the reverse question. Does N-WASP require Toca-1 for filopodial formation? When N-WASP and Toca-1 were co-transfected there was a synergistic induction of filopodial formation with the average cell possessing ∼44 filopodia/cell (Fig. 4A). In contrast, when N-WASP was cotransfected with dominant negative versions of Toca-1 (mutants of the F-BAR domain (K33Q/R35Q, K51Q/K52Q, or R112Q/K113Q) filopodial formation was dramatically reduced (Fig. 4A). We also examined the effect of Toca-1 KD on N-WASP-induced filopodial formation. In support of the data obtained with dominant negative Toca-1 mutants, KD of Toca-1 to negligible levels eliminated N-WASP-induced filopodial formation (Fig. 4B). Interestingly, in the absence of Toca-1 N-WASP expression did not lead to a breakdown of stress fibers (Fig. 4B). Thus Toca-1 is essential for N-WASP-induced filopodial formation.
FIGURE 4.

N-WASP-induced filopodial formation requires and synergizes with Toca-1. A, N1E115 cells were transfected with N-WASP alone, N-WASP with Toca-1 mutants, and N-WASP with Toca-1 (a). All transfections were carried out with GFP-actin. Panels show GFP and DIC images with a merge. The bar chart shows a statistical analysis of the experiments (b). Neurites and filopodial formation were scored as described under “Materials and Methods.” Toca-1 F-BAR domain and Cdc42 binding mutants were as described in the legend to Fig. 2. Three independent experiments were performed (with n = 10-15). Data shown are mean ± S.D. Bar = 10 μm. B, Toca-1 was KD using RNAi and the affect of N-WASP on filopodial formation examined. a, Toca-1 protein levels with control RNAi and Toca-1 RNAi. b, left panel, N-WASP transfection with control RNAi. Right panel, N-WASP transfection with Toca-1 RNAi. Lower panel shows tracing of filopodia in red. c, statistical analysis of experiments similar to those presented in b of single cells. Data shown are mean ± S.D. (n = 20).
Localization of the Toca-1 and N-WASP—Because N-WASP is associated with endocytosis we decided to investigate Toca-1/N-WASP localization markers associated with membrane trafficking. Of the markers (Mitotracker, endoplasmic reticulum tracker, GM130 (Golgi), lysotracker, and Rab7 (late endosomes) (data not shown)) for vesicular structures tested Rab5 was identified as the only marker that colocalized with Toca-1. Next we transfected cells with N-WASP, Toca-1, or Toca-1W518K alone or N-WASP/Toca-1 and followed localization of the proteins with anti-Rab5 antibodies. N-WASP and Toca-1W518K did not colocalize with Rab5 to any significant degree (Fig. 5, A and B). In contrast, Toca-1 partially colocalized with Rab5. The coexpression of Toca-1/N-WASP gave the clearest phenotype with strong colocalization of these proteins in vesicular structures (Fig. 5B). To determine whether the membranes where Toca-1/N-WASP was localized were positive for Rab5 we carried out a co-localization analysis on the images (Fig. 5B, parts b and c). We examined individual vesicles for Toca-1/Rab5 co-localization. A representative group of 12 vesicles, with differing Toca-1 intensities, each stained positive for Rab5 (Fig. 5B, part b). Fig. 5B, part c, compares intensity profiles of selected Rab5 membranes in the presence of Toca-1/N-WASP or Toca-1W518K. The intensity traces show that Toca-1 but not Toca-1W518K co-localizes with Rab5 (Fig. 5B, part c) with correlation coefficients of 0.72 ± 0.09 (n = 12) and 0.04 ± 0.03 (n = 10), respectively.
FIGURE 5.

Localization of N-WASP and Toca-1 with Rab5. A, N1E115 cells were transfected with N-WASP, Toca-1, or Toca-1W518K and stained for Rab5 with anti-Rab5 antibody. Images follow the sequence, N-WASP or Toca-1 (green), Rab5 (red) and merge (a). Bar = 10 μm. Magnification of the images presented in A show potential co-localization and vesicle morphology (b). B, cells were transfected with Toca-1/N-WASP and then stained for Rab5. a, shows a series of three images of a typical cell (in the merge panel; red, Toca-1; green, Rab5). The lower set of image panels are enlarged (zoom) areas of the box in the merge panel. b, 12 randomly picked vesicles were placed in a grid pattern to allow direct side-by-side comparison and co-localization analysis. c, a group of Rab5 membrane from either Toca-1/N-WASP or Toca-1W518K transfection were analyzed for co-localization by intensity tracing through the vesicle. First two panels in c following intensity tracing are the actual vesicles that were examined. The schematic of the vesicles (third panel) shows the intensity line. Intensity analysis was carried out as described under “Materials and Methods.”
Next we transfected cells with Toca-1 and GFP-actin to determine where Toca-1 localized in relation to the filopodia and neurites that were being induced. Actin and Toca-1 colocalized in vesicular/punctuate structures along the length of neurites and at the base of filopodia but not within filopodia (Fig. 6B, parts a-c). The correlation coefficient obtained for Toca-1 actin co-localization was 0.90 ± 0.02 (n = 10). In contrast, N-WASP was found in filopodia and co-localized with actin (Fig. 6A). When N-WASP and Toca-1 were expressed together there was a synergistic effect on filopodial and neurite formation (Fig. 4). Under these conditions Toca-1 was found in filopodia (Fig. 6A, part b).
FIGURE 6.

Localization of N-WASP and Toca-1 with actin. N1E115 cells were transfected with Toca-1 with GFP-actin, Toca-1 with N-WASP, or N-WASP with mRFP-actin, and filopodia and vesicles followed. Images are in a series of three: GFP, mRFP, and overlay. A, a, localization of Toca-1/actin; b, Toca-1/N-WASP; and c, N-WASP/actin in filopodia. B, GFP-actin/mRFP-Toca-1 expression in N1E115 cells. Lower panels of a are duplicates that have tracings showing the relationship between filopodia and Toca-1 localization. Toca-1 is found at the base of filopodia. Localization of Toca-1 with actin, at the leading edge (a) and in neurites (b) (the inset is an enlargement of the box area). c, intensity profile of two of the vesicles from b showing that Toca-1 and actin co-localize. Bar = 10 μm.
N-WASP and Toca-1-N-WASP Expression Affects the Distribution, Size, and Number of Rab5 Membranes—Having established that Toca-1/N-WASP co-localize with Rab5 we next tried to determine whether expression of N-WASP, Toca-1, and Toca-1W518K had any affect on the size and distribution of Rab5 membranes. In control cells Rab5 membranes were primarily found around the perinuclear area. Expression of N-WASP or N-WASP/Toca-1 changed the distribution of Rab5 membranes such that they were found evenly between cell periphery and nucleus (Fig. 7A). Interestingly, Toca-1W518K expression with N-WASP blocked the change in vesicle distribution suggesting that N-WASP interaction with Toca-1 was important. Next we compared the size of 14 randomly picked Rab5 membranes from control (non-transfected) cells with Toca-1-N-WASP-transfected cells (Fig. 7B). The size of the Rab5 membranes in Toca-1/N-WASP-transfected cells (shown in the grid) was 0.59 ± 0.07 μm (n = 14), whereas those from control cells was 0.28 ± 0.05 μm (n = 14). Thus there was a 2-fold increase in vesicle size. The size of the vesicles was also affected by N-WASP expression and Toca-1W518K blocked the N-WASP-induced change (Fig. 7B). The vesicles where Toca-1/N-WASP was localized were motile with rates of movement similar to those reported for endocytic vesicles (data not shown). Last, the total number of vesicles was dramatically increased by expression of N-WASP/Toca-1. N-WASP alone also increased vesicle number. In other cell types including: Chinese hamster ovary, HeLa, and COS7, and Toca-1/N-WASP coexpression also gave clear phenotype with strong colocalization of these proteins in vesicular structures.9 Taken together with the activities associated with N-WASP (Arp2/3 activation) and Toca-1 (F-BAR-mediated membrane deformation) these data on Rab5 membranes suggest that the Toca-1-N-WASP complex plays a role in their formation and motility.
FIGURE 7.

Effect of N-WASP and Toca-1 on the distribution, size, and number of Rab5 membrane. A, N1E115 cells were transfected with constructs, N-WASP, Toca-1/N-WASP, Toca-1, Toca-1W518K, and Toca-1W518K/N-WASP, and then cells were stained with Rab5 antibody. Intensity tracing below images follows the region of interest, open white arrow, from perinuclear area to the cell membrane, of the individual cell. Distribution of Rab5 membranes was determined by creating five zones (perinuclear to cell membrane) within the cell and counting the fluorescence intensity within each zone (details can be found under “Materials and Methods”). The bar charts show a statistical analysis of the intensity distributions in the five zones. A schematic of a cell showing the five zones used in the statistical analysis is shown at the bottom of the figure. Bar = 10 μm. Please note that the bar in the Toca-1/N-WASP transfection panel is smaller than the other panels. B, 14 randomly picked vesicular structures from either control or Toca-1/N-WASP-transfected cells were placed in a grid to allow side by side comparison. Vesicle size was determined by enlarging images so that the intensity boundary could be seen and then using Metamorph software to measure diameters. A schematic of the vesicle size is shown below the actual images to reflect the observed boundary. The bar chart shows statistical analysis of the vesicle size with different transfections. C, the bar chart shows a statistical analysis of vesicle number with different transfections. At least three independent experiments were carried out (with n = 20). Data shown are mean ± S.D. Bar = 2 μm.
Toca-1 Interacts with N-WASP in Filopodia and Rab5 Positive Membranes—Previous work has shown that Toca-1 and N-WASP form a complex in cell lysates but the spatial distribution of the complex in cells has not been investigated. To determine where in cells Toca-1 and N-WASP interact with each other we used acceptor photobleaching-FRET (AP-FRET) and FLIM. Expression of GFP-N-WASP and mRFP-Toca-1 induced a clear phenotype of filopodia and apparent vesicle formation, with the vesicles representing the major site of Toca-N-WASP interaction as seen by acceptor photobleaching-FRET (Fig. 8A). As controls we used unfused cytosolic GFP and mRFP (negative) and a fused mRFP-GFP tandem protein (positive). The correlation coefficient is a measure of whether there is a relationship between rates of increase of donor signal on bleaching the acceptor. If FRET is occurring we predict the correlation coefficient value to be between -0.7 and -1.0. For full details of AP-FRET and controls see “Materials and Methods” and Ref. 11.
FIGURE 8.

Toca-1 and N-WASP interact directly in filopodia and vesicles. A, N1E115 cells were transfected with mRFP-Toca-1 with GFP-N-WASP, or mRFP-Toca-1W518K with GFP-N-WASP, mRFP-Toca-1 with GFP-actin, and mRFP-Toca-1W518K with GFP-actin. FRET was measured by acceptor photobleaching as described under “Materials and Methods.” Images show a series of three panels: mRFP, GFP, and merge. Boxes within the image show areas where bleaching was carried out. Panels in the extreme right show signal intensity from GFP (green line) and mRFP (red line) channels, pre- and post-bleach, as a function of time. Table shows statistical analysis of the data generated in A. FRET efficiency and the Pearson correlation coefficient were calculated as described under “Materials and Methods.” Three independent experiments were performed and one representative example is shown. Data are presented as mean ± S.D. (n = 10). Bar = 10 μm. B, cells were generated as described above and GFP lifetime measurements were made using the frequency domain method (see “Material and Methods” and Clayton et al. (24)). GFP lifetimes were determined using Lambert Instruments (Netherlands) software. Images were acquired sequentially over a period of 4-5 s through 12 phase settings. Data are presented as a mean ± S.D. (n = 10).
To confirm further the protein-protein interaction as seen by AP-FRET we used donor lifetime (GFP) values to follow FRET between N-WASP and Toca-1. GFP lifetimes were measured using the phase modulation method (see “Materials and Methods” for details). GFP lifetimes in mammalian cells are around 2.37 ns. GFP-N-WASP in the presence of mRFP-Toca-1 reduced the lifetime to 1.80 ns and in the presence of the mutant W518K, the lifetime was 2.30 ns (Fig. 8B). Spatial resolution of this frequency domain FLIM measurement was limited and did not allow us to distinguish filopodia and vesicles (Fig. 8B).
Inhibitors of Endocytosis Inhibit Toca-1-induced Filopodial Formation—Toca-1 induces filopodial formation and localizes to endocytic vesicles (and associated actin) and interacts with N-WASP in the filopodia and vesicles. The increased formation of vesicles observed with coexpression of Toca-1 with N-WASP suggested that Toca-1 uses N-WASP to drive actin polymerization and pinching off of vesicles. Taken together, this data suggests that filopodial formation and endocytosis may be linked through the Toca-1-N-WASP complex. To investigate this possibility we examined the affect of two inhibitors of endocytosis on Toca-1-induced filopodial formation. Cells were transfected with Toca-1 to induce filopodia and then with either Dynamin (wild-type and a DN mutant K44A) or Eps15 (D3Δ2 and DN mutant Δ95/295) under conditions where endocytosis was significantly inhibited (as monitored by transferrin uptake; Fig. 9, A and B). Toca-1 was coexpressed with the Dynamin or Eps15 proteins and filopodial number scored. Both DN Dynamin and DN Eps15 proteins significantly inhibited filopodial formation (Fig. 9B). To examine whether clathrin-mediated endocytosis was involved with Toca-1-induced filopodial formation we used RNAi-mediated KD of CHC. Using RNAi (Dharmacon) we are able to reduce CHC by over 85% (Fig. 9C). Under these conditions there was a significant reduction in Toca-1-mediated filopodial formation. However, Toca-1-mediated neurite formation was not affected (data not shown). Thus three different inhibitors of endocytosis blocked Toca-1-induced filopodial formation.
FIGURE 9.

Effect of inhibitors of endocytosis on filopodial formation. A, N1E115 cells were transfected with Toca-1, GFP, or mRFP-actin, and either Eps15 (inactive variant or dominant negative) or Dynamin (wild-type or dominant negative). Cells were then imaged in the GFP and DIC channels. Imaging was carried out using Olympus FV1000 confocal microscope. B, endocytosis and filopodial formation were measured as described under “Materials and Methods.” Bar charts (a and b) show transferrin (TF) uptake. Bar charts (c and d) show filopodial number per cell. Three independent experiments were carried out (with n = 10-15) and one representative example is presented. C, protein expression of CHC was analyzed by Western blotting of cell extracts obtained with or without RNAi (a) as described under “Materials and Methods.” b, Toca-1-mediated filopodial formation was measured in cells transfected with Toca-1, GFP-actin, and +/- RNAi (first panel). Second panel shows an enlargement of the neurite of the cell. Third panel shows a tracing with filopodia indicated in red. Statistical analysis of the experiment is presented as a bar chart in c. Three independent experiments were carried out (with n = 10-15) and one representative example is presented. Data are presented as mean ± S.D. Experimental data were analyzed by Student's t test. Difference was significant when p < 0.05 (* stands for p < 0.05; ** for p < 0.01; *** for p < 0.001). Bar = 10 μm.
DISCUSSION
Work in the mid-1990s linked signaling via Cdc42 to filopodial formation (3, 4). A large number of Cdc42 effectors, and their interacting partners, have been isolated over the intervening years (30, 31). The exact role of Cdc42 interacting protein kinases (such as myotonic dystrophy-related Cdc42 binding kinase, Cdc42-associated kinase, and p21 activated kinase) in filopodial formation is unclear. Certainly there are possible roles for these kinases in modulating actin dynamics and focal complex turnover (32). Cdc42 controls filopodial formation primarily by regulating the formation of protein complexes at the plasma membrane through the recruitment of proteins. The Cdc42 effector IRSp53 seems to play a dominant role in protein complex formation connected to filopodial formation (8-10, 12). Following Cdc42 recruitment of IRSp53 to the plasma membrane, IRSp53 recruits Mena, Eps8, and N-WASP. We have recently presented evidence that IRSp53 generates filopodia by coupling membrane protrusion (through its I-BAR domain) with actin dynamics through interaction with N-WASP, Mena, and Eps8 (11). The Rho family GTPases have also been linked to membrane trafficking events (33). Specifically, Cdc42 is connected to endocytic trafficking in polarized mammalian cells, Caenorhabditis elegans and yeast cells (34-36). The mechanism(s) by which Cdc42 regulates endocytosis in mammalian cells is unclear.
N-WASP has been linked to filopodial formation and endocytosis. Initial work with N-WASP found that it could drive filopodial formation and dominant negatives could block Cdc42-dependent filopodial formation (37). N-WASP plays an important role in filopodial formation downstream of Cdc42 and IRSp53 and this is independent of its ability to activate the Arp2/3 complex (11). N-WASP has been found associated with endocytic vesicles in Xenopus ocytes (15). Furthermore, Benesch et al. (38) demonstrated that N-WASP is essential for phosphatidylinositol phosphate 5-kinase-induced vesicle motility. Toca-1 has also been linked with endocytosis. Tsujita et al. (18) investigated the biology of the F-BAR domains in CIP4, FBP17, and Toca-1 and showed that they could tubulate membranes. The SH3 domains of CIP4, FBP17, and Toca-1 recruited N-WASP and Dynamin facilitating the coupling of membrane changes to actin polymerization. Furthermore, they showed that KD of Toca-1, CIP4, and FBP17 inhibited endocytosis significantly, whereas the effects of individual KD of these proteins were less effective. These data suggest that there is significant redundancy and this makes it difficult to investigate the individual roles of Toca-1, CIP4, and FBP17 in endocytosis.
Toca-1 and N-WASP are both Cdc42 interacting proteins and therefore the Toca-1-N-WASP complex could play roles in filopodial formation and/or endocytosis. In the present study we investigated the cellular function of the Toca-1-N-WASP complex in mouse neuroblastoma N1E115 cells. Expression of either Toca-1 or N-WASP alone induced filopodial and neurite formation. A Toca-1 mutant unable to bind N-WASP failed to induce filopodial formation suggesting that the interaction of the two proteins was important. Toca-1 failed to induce filopodia in N-WASP KO cells. N-WASP failed to induce filopodia in the presence of Toca-1 dominant negative proteins or when Toca-1 was KD with RNAi. Taken together, these data suggest that Toca-1-N-WASP can regulate filopodial formation. In contrast to N-WASP, Toca-1 did not localize to filopodia. However, expression of Toca-1 together with N-WASP induced strong filopodial formation and Toca-1 could now be seen in filopodia. This result suggests that N-WASP can recruit Toca-1 to filopodia. So what is the potential mechanism by which the Toca-1-N-WASP complex regulates filopodial formation? The phenotype of Toca-1 is reminiscent of work reported a few years ago on Syndapins/PACSIN by Qualmann and Kelly (39). Syndapin has a domain structure similar to Toca-1, F-BAR and SH3 domains (but no Cdc42 interaction site). Like Toca-1, Syndapin interacts with N-WASP and is involved in endocytosis. Syndapin was also found to induce actin microspike formation, structures that resemble filopodia (39). With Toca-1 we show conclusively that it generates filopodia by carrying out time-lapse experiments using GFP-actin-transfected cells. The lifetime and length of Toca-1-generated filopodia is similar to that of endogenous filopodia and those generated by Cdc42/RacN17, IRSp53, and N-WASP. More recent work with Syndapin shows that it dimerizes (and oligomerizes) and these events are required for its function (40). Similarly it is likely that CIP4 and FBP17 (and by analogy Toca-1) deform membranes by forming dimers and oligomers on the membrane surface (17). The potential consequence of Toca-1 and/or Syndapin oligomers on the membrane is the generation of a localized concentration or “hotspots” of specific SH3 domains. These SH3 domains could recruit polyproline containing proteins such as N-WASP, Mena, and Eps8; proteins that are directly implicated in filopodial formation. The hypothesis is stronger for Toca-1, CIP4, and FBP17, than for Syndapins, as these former proteins possess Cdc42-binding sites and would thus also recruit Cdc42, creating hotspots, of SH3 domains and Cdc42 binding sites on the membrane. Subsequently, Cdc42 may recruit proteins such as IRSp53 that directly activate filopodial formation.
One could also visualize the reverse scenario with Cdc42-IRSp53 activation inducing filopodial formation, N-WASP and then Toca-1 recruitment to the membrane, and a subsequent stimulation of endocytosis. In this model N-WASP plays a prominent role in the coupling of filopodial formation to endocytosis.
Toca-1 localized with Rab5 positive membranes and associated actin. Expression of Toca-1 together N-WASP induced the formation of Rab5 positive membranes. N-WASP and N-WASP/Toca-1 affected the distribution, size, and number of Rab5 membranes but Toca-1 alone did not. The lack of effect of Toca-1 alone may be due to the redundancy in the system (with CIP4 and FBP17, as mentioned above). This result also suggest that the control of Rab5 membrane trafficking may lie with regulation of and by N-WASP. Using FRET we established that there are two major cellular sites of Toca-1 interaction with N-WASP; filopodia and Rab5 membranes. Taken together, these data suggest that the Toca-1-N-WASP complex can regulate the formation of both filopodia and endocytic vesicles.
The induction of filopodial formation by Toca-1 was inhibited by blockers of endocytosis. Is there a potential physiological function of linking filopodial formation to endocytosis? Lidke et al. (41) have proposed that filopodia act as sensing organelles probing for receptor ligands facilitating activation of cell signaling pathway events through receptor endocytosis. Examples of processes that link filopodia with endocytosis include (i) migrating border cells of Drosophila where polarized receptor cycling act as guidance cues (42); (ii) growth cone collapse driven by Sema3A where endocytosis is enhanced and this is linked with changes in F-actin (43); and (iii) growth cone guidance where cycling of adhesion molecules such as (CAM) L1 facilitates cell movement (44).
In Drosophila Rab5 KO leads to defects in endocytosis and cell polarity (45). Furthermore, a recent genome wide RNAi screen of proteins involved in membrane trafficking (including endocytosis) identified proteins linked with cell polarity (22). Prominent among the proteins implicated in endocytosis and cell polarity was the Cdc42-PAR3/6-PKC complex. In yeast Marco et al. (46) have recently modeled cell polarity using Cdc42Q61L expression and found that endocytosis is likely to play a crucial role. Thus there is evidence for a link between cell polarity and endocytosis. We show here for the first time that the Toca-1-N-WASP complex localizes to filopodia and Rab5 membranes and that this complex induces the formation of these important cellular structures. The linking through Cdc42 of filopodial formation/cell motility (via IRSp53-N-WASP, Mena, and Eps8), endocytosis (via Toca-1-N-WASP), as well as cell polarity (via PAR3-PAR6) may provide a mechanism to coordinate environmental cues, cell signaling events, and cellular/developmental processes.
Supplementary Material
The on-line version of this article (available at http://www.jbc.org) contains supplemental Movies 1 and 2.
Footnotes
The abbreviations used are: BAR, bin-amphiphysin-Rsv domain; CIP4, cdc42 interacting protein 4; FBP17, formin binding protein 17; CHC, clathrin heavy chain; I-BAR, inverse Bin-amphiphysin-Rsv domain; Mena, mammalian enabled; N-WASP, neuronal Wiskott Aldrich syndrome protein; SH3, Src homology domain 3; Toca-1, transducer of Cdc42-dependent actin polymerization; WAVE, Wasp family veprolin-homologous protein; FRET, Forster resonance energy transfer; FLIM, fluorescence lifetime imaging microscopy; mRFP, monomeric red fluorescence protein; GFP, green fluorescent protein; KO, knockout; KD, knockdown; RNAi, short interference RNA; AP, acceptor photobleaching; PBS, phosphate-buffered saline; HA, hemagglutinin; Eps15, epidermal growth factor phosphorylation substrate 15; DIC, differential interference contrast.
The BAR domain derives from a domain common to Bin-Amphysin-Rvs proteins. The BAR domain forms dimers/oligomers and deforms/tubulates membranes.
F-BAR is one of the variants of the BAR domain found in CIP4, FBP17, Toca-1, Sydapins, Nostrinm, and FER.
Receptor endocytosis is regulated by Dynamin, a GTPase that is targeted to clathrin-coated pits where it oligomerizes around the neck of budding vesicles (reviewed in Ref. 47)). Both clathrin- and caveolae-dependent endocytosis require the activity of GTPase Dynamin (21, 48). Dynamin with a point mutation in the nucleotide-binding site (K44A) interferes with the function of endogenous Dynamin by blocking vesicle internalization before membrane scission occurs (49). Both dyn1(K44A) and dyn2(K44A) were potent inhibitors of receptor-mediated endocytosis; however, neither mutant directly affected other membrane trafficking events, including transport mediated by four distinct classes of vesicles budding from the trans-Golgi network (50).
Eps15 is a key component for assembling functional clathrin-coated pits (20, 21). Eps15 is ubiquitously and constitutively associated with AP-2. The N-terminal of Eps15 contains three EH (Eps homology) domains that bind regulatory proteins containing the NPF amino acids motif, including epsin, AP180, and synaptojanin. The central domain allows for oligomerization of Eps15 into homodimers. The C-terminal domain contains an AP-2 binding site, which together with EH domains, is required for targeting of Eps15 to clathrin-coated pits. Dominant negative mutant Eps15-Δ95/295 lacks the second and third EH domains, whereas the inactive variant Eps15-D3Δ2 is a C-terminal domain construct lacking all AP-2 binding sites. Expression of dominant negative mutant Eps15-Δ95/295, but not inactive variant Eps15-D3Δ2, inhibits clathrin-mediated endocytosis of transferrin in many cell types (20).
See Movies 1 (Toca-1) and 2 (N-WASP).
Mammalian filopodia have been defined as cell protrusions that contain actin and have a lifetime of 142-187 s and a length of 7-15 μm (11).
W. Bu and S. Ahmed, manuscript in preparation.
References
- 1.Bray, D. (2001) Cell Movement, From Molecules to Motility, 2nd Ed., Garland Publishing, New York, NY
- 2.Johnson, D. I., and Pringle, J. R. (1990) J. Cell Biol. 111 143-152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kozma, R., Ahmed, S., Best, A., and Lim, L. (1995) Mol. Cell. Biol. 15 1942-1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nobes, C. D., and Hall, A. (1995) Cell 81 53-62 [DOI] [PubMed] [Google Scholar]
- 5.Gupton, S. L., and Gertler, F. B. (2007) Sci. STKE 2007 re5. [DOI] [PubMed] [Google Scholar]
- 6.Mattila, P. K., and Lappalainen, P. (2008) Nat. Rev. Mol. Cell Biol. 9 446-454 [DOI] [PubMed] [Google Scholar]
- 7.Sherer, N. M., Lehmann, M. J., Jimenez-Soto, L. F., Horensavitz, C., Pypaert, M., and Mothes, W. (2007) Nat. Cell Biol. 9 310-315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Govind, S., Kozma, R., Monfries, C., Lim, L., and Ahmed, S. (2001) J. Cell Biol. 152 579-594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krugmann, S., Jordens, I., Gevaert, K., Driessens, M., Vandekerckhove, J., and Hall, A. (2001) Curr. Biol. 11 1645-1655 [DOI] [PubMed] [Google Scholar]
- 10.Disanza, A., Mantoani, S., Hertzog, M., Gerboth, S., Frittoli, E., Steffen, A., Berhoerster, K., Kreienkamp, H. J., Milanesi, F., Di Fiore, P. P., Ciliberto, A., Stradal, T. E., and Scita, G. (2006) Nat. Cell Biol. 8 1337-1347 [DOI] [PubMed] [Google Scholar]
- 11.Lim, K. B., Bu, W., Goh, W. I., Koh, E., Ong, S. H., Pawson, T., Sudhaharan, T., and Ahmed, S. (2008) J. Biol. Chem. 283 20454-20472 [DOI] [PubMed] [Google Scholar]
- 12.Mattila, P. K., Pykalainen, A., Saarikangas, J., Paavilainen, V. O., Vihinen, H., Jokitalo, E., and Lappalainen, P. (2007) J. Cell Biol. 176 953-964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takenawa, T., and Suetsugu, S. (2007) Nat. Rev. Mol. Cell Biol. 8 37-48 [DOI] [PubMed] [Google Scholar]
- 14.Rohatgi, R., Ho, H. Y., and Kirschner, M. W. (2000) J. Cell Biol. 150 1299-1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taunton, J., Rowning, B. A., Coughlin, M. L., Wu, M., Moon, R. T., Mitchison, T. J., and Larabell, C. A. (2000) J. Cell Biol. 148 519-530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho, H. Y., Rohatgi, R., Lebensohn, A. M., Le, M., Li, J., Gygi, S. P., and Kirschner, M. W. (2004) Cell 118 203-216 [DOI] [PubMed] [Google Scholar]
- 17.Shimada, A., Niwa, H., Tsujita, K., Suetsugu, S., Nitta, K., Hanawa-Suetsugu, K., Akasaka, R., Nishino, Y., Toyama, M., Chen, L., Liu, Z. J., Wang, B. C., Yamamoto, M., Terada, T., Miyazawa, A., Tanaka, A., Sugano, S., Shirouzu, M., Nagayama, K., Takenawa, T., and Yokoyama, S. (2007) Cell 129 761-772 [DOI] [PubMed] [Google Scholar]
- 18.Tsujita, K., Suetsugu, S., Sasaki, N., Furutani, M., Oikawa, T., and Takenawa, T. (2006) J. Cell Biol. 172 269-279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benmerah, A., Lamaze, C., Begue, B., Schmid, S. L., utry-Varsat, A., and Cerf-Bensussan, N. (1998) J. Cell Biol. 140 1055-1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benmerah, A., Bayrou, M., Cerf-Bensussan, N., and utry-Varsat, A. (1999) J. Cell Sci. 112 1303-1311 [DOI] [PubMed] [Google Scholar]
- 21.Sever, S., Damke, H., and Schmid, S. L. (2000) J. Cell Biol. 150 1137-1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balklava, Z., Pant, S., Fares, H., and Grant, B. D. (2007) Nat. Cell Biol. 9 1066-1073 [DOI] [PubMed] [Google Scholar]
- 23.Herrick-Davis, K., Weaver, B. A., Grinde, E., and Mazurkiewicz, J. E. (2006) J. Biol. Chem. 281 27109-27116 [DOI] [PubMed] [Google Scholar]
- 24.Clayton, A. H., Walker, F., Orchard, S. G., Henderson, C., Fuchs, D., Rothacker, J., Nice, E. C., and Burgess, A. W. (2005) J. Biol. Chem. 280 30392-30399 [DOI] [PubMed] [Google Scholar]
- 25.Itoh, T., Erdmann, K. S., Roux, A., Habermann, B., Werner, H., and De Camilli, P. (2005) Dev. Cell 9 791-804 [DOI] [PubMed] [Google Scholar]
- 26.Lee, E., Marcucci, M., Daniell, L., Pypaert, M., Weisz, O. A., Ochoa, G. C., Farsad, K., Wenk, M. R., and De Camilli, P. (2002) Science 297 1193-1196 [DOI] [PubMed] [Google Scholar]
- 27.Kozma, R., Ahmed, S., Best, A., and Lim, L. (1996) Mol. Cell. Biol. 16 5069-5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozma, R., Sarner, S., Ahmed, S., and Lim, L. (1997) Mol. Cell. Biol. 17 1201-1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarner, S., Kozma, R., Ahmed, S., and Lim, L. (2000) Mol. Cell. Biol. 20 158-172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bishop, A. L., and Hall, A. (2000) Biochem. J. 348 241-255 [PMC free article] [PubMed] [Google Scholar]
- 31.Cotteret, S., and Chernoff, J. (2002) Genome Biol. 3 REVIEWS0002 [DOI] [PMC free article] [PubMed]
- 32.Zhao, Z. S., and Manser, E. (2005) Biochem. J. 386 201-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qualmann, B., and Mellor, H. (2003) Biochem. J. 371 233-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kroschewski, R., Hall, A., and Mellman, I. (1999) Nat. Cell Biol. 1 8-13 [DOI] [PubMed] [Google Scholar]
- 35.Adamo, J. E., Moskow, J. J., Gladfelter, A. S., Viterbo, D., Lew, D. J., and Brennwald, P. J. (2001) J. Cell Biol. 155 581-592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fares, H., and Greenwald, I. (2001) Nat. Genet. 28 64-68 [DOI] [PubMed] [Google Scholar]
- 37.Miki, H., Sasaki, T., Takai, Y., and Takenawa, T. (1998) Nature 391 93-96 [DOI] [PubMed] [Google Scholar]
- 38.Benesch, S., Lommel, S., Steffen, A., Stradal, T. E., Scaplehorn, N., Way, M., Wehland, J., and Rottner, K. (2002) J. Biol. Chem. 277 37771-37776 [DOI] [PubMed] [Google Scholar]
- 39.Qualmann, B., and Kelly, R. B. (2000) J. Cell Biol. 148 1047-1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kessels, M. M., and Qualmann, B. (2006) J. Biol. Chem. 281 13285-13299 [DOI] [PubMed] [Google Scholar]
- 41.Lidke, D. S., Lidke, K. A., Rieger, B., Jovin, T. M., and rndt-Jovin, D. J. (2005) J. Cell Biol. 170 619-626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jekely, G., Sung, H. H., Luque, C. M., and Rorth, P. (2005) Dev. Cell 9 197-207 [DOI] [PubMed] [Google Scholar]
- 43.Luo, Y., Raible, D., and Raper, J. A. (1993) Cell 75 217-227 [DOI] [PubMed] [Google Scholar]
- 44.Kamiguchi, H., and Lemmon, V. (2000) J. Neurosci. 20 3676-3686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu, H., and Bilder, D. (2005) Nat. Cell Biol. 7 1232-1239 [DOI] [PubMed] [Google Scholar]
- 46.Marco, E., Wedlich-Soldner, R., Li, R., Altschuler, S. J., and Wu, L. F. (2007) Cell 129 411-422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmid, S. L., McNiven, M. A., and De Camilli, P. (1998) Curr. Opin. Cell Biol. 10 504-512 [DOI] [PubMed] [Google Scholar]
- 48.Henley, J. R., Krueger, E. W., Oswald, B. J., and McNiven, M. A. (1998) J. Cell Biol. 141 85-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Damke, H., Baba, T., Warnock, D. E., and Schmid, S. L. (1994) J. Cell Biol. 127 915-934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altschuler, Y., Barbas, S. M., Terlecky, L. J., Tang, K., Hardy, S., Mostov, K. E., and Schmid, S. L. (1998) J. Cell Biol. 143 1871-1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lommel, S., Benesch, S., Rottner, K., Franz, T., Wehland, J., and Kuhn, R. (2001) EMBO Rep. 2 850-857 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.