

Abstract
AIMS
To illustrate the use of pharmacokinetic–pharmacodynamic (PK–PD) models to select rational starting doses in clinical trials within the minimum anticipated biological effect level (MABEL) principle using literature data and through simulations.
METHODS
The new European Medicines Agency guidance on starting dose selection of high-risk biologics was analysed considering the basic pharmacological properties and preclinical testing limitations of many biologics. The MABEL approach to dose selection was illustrated through simulations and through literature-reported examples on the selection of starting doses for biologics such as antibodies based on in vitro biomarker data, in vivo PK and PK–PD data.
RESULTS
Literature reports indicating the use of preclinical pharmacological and toxicological data to select successfully safe starting doses in line with the MABEL principle are summarized. PK–PD model-based simulations of receptor occupancy for an anti-IgE antibody system indicate that the relative abundance of IgE in animal models and patients and the turnover rate of the IgE–antibody complex relative to the off-rate of the antibody from IgE are important determinants of in vivo receptor occupancy.
CONCLUSIONS
Mechanistic PK–PD models are capable of integrating preclinical in vitro and in vivo data to select starting doses rationally in first-in-human trials. Biological drug–receptor interaction dynamics is complex and multiple factors affect the dose–receptor occupancy relationship. Thus, these factors should be taken into account when selecting starting doses.
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Recent regulatory guidance has highlighted the importance of using pharmacokinetic–pharmacodynamic (PK–PD) modelling in the selection of starting doses in first-in-human trials of high-risk biologics.
However, limited examples exist in literature illustrating this procedure.
WHAT THIS STUDY ADDS
An interpretation of the recommended dose-selection methodology and the minimum anticipated biological effect level (MABEL) principle, contained in the updated European Medicines Agency guidance on risk-mitigation strategies for first-in-human studies, is presented.
Some literature and simulation-based examples of the application of PK–PD modelling principles to starting dose selection using in vitro and in vivo data under the MABEL paradigm are highlighted, along with the advantages and limitations of this approach.
Introduction
Severe adverse events seen in a first-in-human (FIH) clinical trial of a CD28 agonist antibody TGN1412 [1] have highlighted the importance of choosing safe starting doses in FIH trials. New guidance from the European Medicines Agency (EMEA) [2] has identified the dose selection process as a key risk-mitigation strategy in FIH trials, especially for compounds perceived to be of high risk, including biologics. Even though many methods are followed to calculate the starting doses in FIH trials [3–5], the Food and Drug Administration guidance on starting dose selection [3] is widely applied across the industry. Briefly, the no adverse event level (NOAEL) obtained from the most sensitive toxicological test species is allometrically scaled to obtain a human equivalent dose (HED). A safety factor, estimated based on multiple considerations including the previously known toxicity of the mechanism, is applied to the HED to obtain the maximum recommended starting dose (MRSD). The limitation of this method is that it relies on somewhat arbitrary safety factors to ensure safety of the starting dose [6, 7]. The pharmacokinetic–pharmacodynamic (PK–PD) predictions-guided approach [8] provides a more mechanistic rationale for starting dose selection by considering the human predicted PK and PD. However, neither of these methods is easily applicable to biologics in cases where there is no relevant animal species for PK and toxicological testing. The dose selection approach in the new EMEA guidance document attempts to address these limitations through the integration of all pharmacology, safety and efficacy testing data gathered during preclinical evaluation of the candidate in a PK–PD modelling framework, so that a starting dose can be chosen that would result in minimum anticipated biological effect level (MABEL) [2]. The use of predicted receptor occupancy (RO) to ensure minimum biological activity has been suggested [1], and a simple formula to calculate RO based on the equilibrium dissociation constant (Kd) of the drug–receptor interaction is also presented.
Even though the ability of PK–PD modelling and simulation to combine diverse data generated in different test systems during preclinical development has been recognized [9], limited examples of use of PK–PD modelling in FIH study design of biologics have been reported in the literature. This has led to a lack of clarity around the interpretation of the MABEL and the application of PK–PD principles for estimating a MABEL dose for biologics. This study illustrates, through literature examples and simulations, the application of the MABEL principle for FIH dose selection. While this discussion is, in general, applicable to any biologic, monoclonal antibodies (MABs) are used to illustrate many of the concepts because of their unique PK–PD characteristics and emerging importance within the human therapeutics industry [10].
Points to consider in the selection of FIH doses for biologics
While the rationale for starting dose selection that is applied to small molecules is, in general, applicable to large molecules such as MABs, some unique characteristics of these candidates pose further challenges in the design of FIH trials. Because of the human-specific nature of many MABs, adequate preclinical in vivo toxicological testing may not be possible due to lack of cross-reactivity in commonly accepted toxicological test species such as rats and dogs. Even for cross-reactive MABs, due to differences in the pharmacology between test species and humans, the NOAEL obtained in test species may not be relevant to human testing in some cases [11]. Furthermore, toxicity for many biologics is typically due to exaggerated pharmacology [12]. Therefore, characterizing the preclinical pharmacological response is critical to understanding potential clinical safety implications for these compounds.
Predicting human pharmacological response from preclinical data also presents unique challenges in the case of biologics compared with small molecules. An example is the prediction of in vivo RO based on in vitro binding affinity. Interaction of MABs with their target is, in many cases, different from that of small molecules: (i) because of their high affinity, MABs are typically dosed at equal molar ratios to their targets [13]; (ii) the on- and off-rates of MABs at their receptors are, in general, slower than those of small molecules [14]; (iii) binding of target by MAB may change the natural kinetics of the receptor, e.g. trigger internalization or stabilization of the receptor [15–17]; and (iv) due to the relatively slow distribution to the site of action and target-mediated elimination of MABs, unbound MAB concentrations at the biophase after single doses and at steady state may be one to three orders of magnitude below unbound MAB concentrations in plasma [18, 19]. Because of these characteristics, simple equilibrium calculations of in vivo RO based on affinity that are applied for many small molecules (e.g. a concentration of Kd corresponds to 50% RO) may not be applicable for MAB therapeutics.
An added note of caution with FIH dose selection with MABs is that any toxic effects in humans are likely to be sustained for long durations due to the long half-lives commonly associated with them. Conversely, selection of too low a starting dose could substantially increase the duration of clinical studies through the investigation of non-informative low doses increasing the cost and duration of development with limited additional safety benefit. Therefore, rational selection of starting doses is of added importance for MAB programmes.
Proposed approach by EMEA
The dose selection approach in the EMEA guidance [2] attempts to address some of the limitations outlined above through the integration of all pharmacology, safety and efficacy testing data gathered during preclinical evaluation of a candidate in a PK–PD framework, so that a starting dose can be chosen that would result in minimum biological activity. The process is illustrated in Figure 1. Stepwise, this overall methodology could be interpreted as consisting of the following steps.
Figure 1.

Schematic representation of the process of selecting starting doses in a first-in-human trial of a MAB. Eff1–3 could be predicted human effects such as biomarkers, toxicological effects, etc. HED, human equivalent no adverse event level dose; RO, receptor occupancy
(a)Obtain the predicted human dose–response for all measured biological effects
These biological effects would typically include standard pharmacological and toxicological tests, in vivo biomarker (either target and/or mechanism biomarkers) studies, and dose–response studies in disease models. Routine in vitro pharmacological testing includes receptor binding affinity and occupancy experiments with human receptors or receptor-bearing cells and other mechanism-specific assays such as antibody-dependant cell cytotoxicity (ADCC) assay [20]. In vivo testing may include PK–PD experiments to characterize the relationship between dose, exposure, and biomarker changes, and efficacy studies in appropriate disease models, when available. Toxicology testing includes general toxicity and safety pharmacology testing. In addition, other safety tests such as cytokine release assay [21] and cell proliferation assay [11] may also be performed, if deemed relevant.
All relevant biological effects, measured in the preclinical setting, are scaled to obtain human dose–response (Eff1–3 in Figure 1). For pharmacological effects, the human scale-up methods depend on the measured biomarker. Using mechanism-based PK–PD models [22] that account for differences in target affinity, target abundance and turnover between species, human RO can be predicted from preclinical in vivo data (see Example 1 and [23]), from preclinical PK data (Example 2 and [23]) and from in vitro data (Example 3 and [9, 24]). Scale-up of preclinical in vivo mechanism biomarker study data to obtain human dose–response may present substantial difficulties due to differences in biomarker response in preclinical species and humans [25]. In such cases, prior clinical experience with other similarly acting compounds can be used to predict human dose–response for the candidate (Example 4 and [9]). For toxicological assays such as cytokine release assay, the human predicted PK, obtained by allometric scaling or through PK–PD model-based extrapolation, can be coupled with in vitro concentration–effect relationship to predict clinical effect profile (see Example 5 and [26]).
(b)Select the dose that would result in minimal biological activity – MABEL dose
In the set of dose–response curves generated in step (a), the left-most curve represents the most sensitive measured biological effect. A minimal response on this curve (e.g. ED10) could be interpreted as the MABEL dose for this compound. For most blocking and neutralizing biologics, the left-most curve is typically the RO curve. In these cases, a dose targeting a low predicted RO is typically the MABEL dose. However, targeting a low predicted RO may not be appropriate or sufficient to ensure safety where (i) RO is not the most sensitive biological effect and/or (ii) the relationship between RO and downstream biological effects is not known. For agonist biologics such as erythropoietin receptor agonists (ERA), maximum effect could occur at submaximal RO [27]. For MABs that work through effector mechanisms such as ADCC [28] and for antibody–drug conjugates [29], the cytotoxic effect may not be directly related to RO, but instead to the number of cell membrane-bound MAB molecules. In such cases, understanding the dose–mechanism biomarker response may be necessary to ensure low pharmacological activity at the starting clinical dose. Thus, for a typical neutralizing or blocking MAB, the human dose–RO relationship may be sufficient to guide starting dose selection. For other cases, prediction of human dose–biomarker response may also be necessary.
Furthermore, when a low RO is targeted at the starting dose, the expected safety of the dose will be likely to depend on the relationship between RO and pharmacological effect, which can be widely different for different compounds. For example, agonists can produce maximal effect at very low RO: β-agonists produce maximal bronchodilation at <5% RO [30], and ERAs produce maximum effect at 20–30% RO [27]. On the other hand, antagonists require generally higher RO to exert their effect: anti-IgE and anti-CCR5 therapeutics are clinically effective at >90% [31] and >99% RO [32], respectively. Therefore, based on target-mediated effects, a starting dose targeting 50% RO could be considered a MABEL dose for a CCR5 antagonist, whereas <3% RO is more appropriate for an ERA. Hence, the choice of starting dose should take into consideration all available information on the relationship between RO and biological effect for the relevant class of compounds. Furthermore, because of the nature of antibody interaction with receptors, nonmonotonic (U- and bell-shaped) concentration–occupancy–response may also be observed in some cases [11], so care should be taken to characterize the dose–RO–effect response over a wide concentration range. Other factors to consider in the selection of starting doses include the therapeutic area (a higher perceived safety risk might be acceptable in an area of critical therapeutic need such as oncology [33]) and observed preclinical toxicity profile (higher starting RO for a ‘clean’ candidate).
(c)Obtain the NOAEL from toxicological testing
The HED, calculated from the toxicological testing NOAEL [3], is included in the dose–response curve. A further safety factor to the MABEL dose may be applied to ensure that the starting dose is sufficiently lower than the NOAEL dose. This would ensure adequate safety margins for any nonmechanism-related toxicity seen during testing. It should be noted that applying a further safety factor is akin to targeting a lower biological effect. For many biologics, the HED is typically greater than the MABEL doses estimated from steps 1 and 2.
Illustration of the use of PK–PD modelling principles in FIH dose selection
The following examples illustrate the use of PK–PD modelling principles in the prediction of human pharmacological response based on preclinical in vivo and in vitro data and subsequently the FIH starting dose selection. All modelling and simulation was performed using Berkeley-Madonna software (v8.3.9; Berkeley, CA, USA). All plots were generated using S-Plus (Insight, Seattle, WA, USA). {For all simulations in this manuscript RO is calculated as: [1 − (concentration of free receptors at time t ÷ concentration of free receptors at time 0)] × 100}.
Example 1: calculation of human dose–RO response using preclinical in vivo PK–PD data
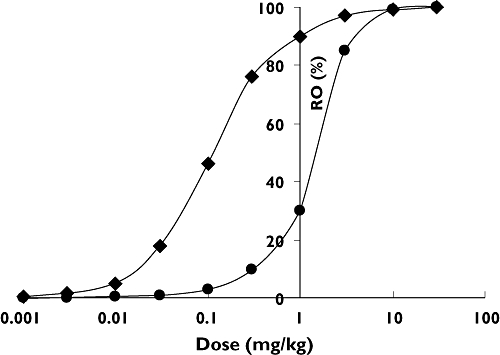
As mentioned above, human dose–RO response may be predicted using a mechanism-based PK–PD modelling approach applied to preclinical PK–PD data for many biologics. Literature PK–PD data were obtained on the use of an anti-IgE MAB in cynomolgus monkeys [34]. A target-mediated disposition (TMD) model ([22]; Figure 2) was used to describe the data; the model accounts for nontarget specific elimination of drug, receptor synthesis and turnover, drug-receptor binding, and complex turnover. Model parameters were: volume of distribution (Vc) = 0.2 l, MAB half-life =15 days (kel_mab = 0.044 day−1), baseline receptor concentration = 55 nM, IgE half-life = 1 day (kel_Ag = 0.7 day−1) and complex half-life = 3.3 days (kel_com = 0.21 days), and Kd = 1 nM (kon = 1 nM−1 day−1, koff = 1 day−1). Human model parameters were obtained through allometric scaling of monkey parameters (Vc, kel_mab, and kel_com) using standard exponents for volume (1) and elimination rate constants (−0.25), from literature (IgE half-life, baseline receptor concentration), and from in vitro experiments (kon and koff). Literature-reported human model parameters based on clinical data are similar to those obtained from the procedure described above [13, 16]: MAB half-life = 28 days (kel_mab = 0.025 day−1), Vc = 4 l, IgE halflife = 1 day (kel_Ag = 0.7 days), baseline Ag concentration = 4.5 nM, complex half-life = 3.5 days (kel_com = 0.02 days), and Kd = 1 nM (kon and koff both = 1 unit). Simulations were performed to obtain the dose–IgE occupancy relationship in cynomolgus monkeys and in humans using these parameters (Figure 3). As illustrated, the differences in the pharmacological components, primarily the baseline free IgE levels, lead to substantial differences in the in vivo RO at a given dose. For example, the simulations indicate that a 30-fold lower dose in humans vs. cynomolgus monkeys (0.03 vs. 1 mg kg−1) could result in a similar RO (approximately 30%). Therefore, obtaining the starting clinical dose as a factor of the NOAEL (typically 10–100) may not provide sufficient safety margins in the FIH trial. Conversely, if the receptor concentration is higher and/or the affinity lower in humans compared with monkeys, the starting doses obtained by applying a nonmechanistic safety factor may be too low and result in the evaluation of multiple nonpharmacologically active doses in the FIH study.
Figure 2.

Schematic of a mechanism-based target-mediated drug disposition (TMD) model. Model accounts for antibody and receptor dynamics as well as complex formation and turnover. The broken arrows are processes that are assumed to be at equilibrium in the Kd-based receptor occupancy calculation
Figure 3.
Pharmacokinetic–pharmacodynamic model-based prediction of human dose (mg kg−1)–response (%RO) based on cynomolgus monkey dose–response using target-mediated drug disposition model
Example 2: calculation of human dose–RO response based on preclinical PK data
If preclinical target or mechanism biomarker data are not available, the in vivo PK profile can, in some cases, be used to provide information on the in vivo RO [22, 23]. The dose-dependant clearance of an anti-muC18 MAB in primates was described by a saturable component (Michaelis–Menten-type equation) representing TMD and a concentration-independent component. The TMD clearance parameters were assumed to represent the effective target levels and the in vivo target binding potency. These parameters were scaled up to human using the relative target concentrations and the binding affinities in humans and primates, and human simulations of dose–RO response were obtained. The starting dose was chosen to obtain 10% peak RO. Even though these scale-up assumptions may not be strictly valid under all circumstances, human predictions appeared to confirm their accuracy in this case. A more mechanistic PK–PD model may also be used to infer in vivo RO from PK data, as illustrated by Wang and Balthasar [35], who also showed that the structure of the PK model used to describe the data could also influence the estimated RO.
Example 3: characterizing the in vivo dose–RO response using in vitro binding data: comparison of PK–PD modelling vs. equilibrium-based approaches
Where low RO is targeted at starting doses, a simple formula, based on equilibrium drug–receptor interaction theory and on known PK of MABs, has been suggested in the Expert Study Group report [1]. Based on standard assumptions such as excess of drug compared with receptor and rapid kon–koff rates, the RO is calculated based on equilibrium binding affinity (Kd) and the derived formula suggests that at a dose (calculated in mg kg−1) of Kd/2000 (Kd expressed in nM), 10% peak RO is obtained. As indicated above, some of these pharmacological assumptions, typically applied for small molecules, may not apply to biologics. One such instance, where in vivo RO is limited by turnover rate of the receptor–MAB complex, is illustrated in this example [36].
Using the model parameters described previously (Example 1) for an anti-IgE MAB, the impact of increasing the affinity of antibody by slowing the off-rate from the receptor (kon = 1 nM−1 day−1, koff = 0.001−10 day−1) on the dose required for 10% peak RO is shown below (Figure 4) for the equilibrium formula and the PK–PD simulation. At high in vitro affinity [Koff (Kd) < 0.1 units], there is no further increase in in vivo potency because the turnover rate of the complex is faster than the off-rate from the receptor. Thus, only a maximum of twofold increase in in vivo potency over a 1 nM affinity MAB (omalizumab) is predicted for manifold increase in affinity. However, the equilibrium formula does not account for the complex turnover and hence predicts continuously increasing potency with binding affinity and hence lower starting doses at higher affinities. These predictions agree well with recent observations [24].
Figure 4.

Comparison of predicted receptor occupancy based on Kd-based calculation (Kd/2000) and pharmacokinetic–pharmacodynamic (PK–PD) method. Evaluation of the impact of koff on the dose required for 10% receptor occupancy. (•), PK–PD model-based prediction; (▪), equilibrium-based prediction (Kd/2000)
It can also be shown through simulations that the slower on-rate of many macromolecules at their receptor compared with small molecules [14], high turnover rates of many macromolecule targets (e.g. cytokines [37, 38]),) and the lower tissue concentrations of many MABs compared with plasma concentrations [18, 19, 35], factors which are not accounted for in the Kd-based RO calculation, could result in substantial overprediction of in vivo RO using that method. This overprediction could result in the selection of multiple low doses that are pharmacologically inactive. Therefore, a PK–PD model-based approach, which more accurately captures the competing dynamic interactions, is preferable for MABEL dose calculation.
Example 4: calculation of starting dose based on mechanism-based PK–PD model of biomarker change for an ERA
ERAs are recombinant human analogues of endogenous erythropoietin. Blood haemoglobin level is a known mechanism biomarker with high linkage to both clinical efficacy and safety of this mechanism. Also, agonism of erythropoietin receptor is known to result in maximum pharmacological effect at submaximal RO [27]. Considering these factors, a dose targeting a minimal increase in haemoglobin levels was chosen as the starting dose in the FIH trial of a next-generation ERA, AMG114. A mechanism-based PK–PD model, developed for a previous ERA, darbepoetin alfa [39], was used to obtain the dose–response relationship of both darbepoetin alfa and AMG 114 in a preclinical model of anaemia. Human PK–PD model parameters for AMG114 were obtained based on prior experience with darbepoetin alfa in the clinic and the relative potencies of AMG114 and darbepoetin alfa in the preclinical model. The starting dose (15 µg) was selected to ensure adequate characterization of the PK and limited change in haemoglobin concentration in subjects [40].
Example 5: characterising the in vivo dose–RO response using in vitro biomarker data
In the absence of preclinical in vivo data, the in vitro data may be used to set the starting doses in FIH studies. An illustration of this methodology for a MAB acting through the ADCC mechanism has been presented by Pilaro [26]. In vitro binding and ADCC activity (percent specific killing) were measured for the MAB, which does not cross-react with any animal test species. The starting clinical dose was selected so that the predicted maximum plasma concentration (assuming initial distribution volume = plasma volume − 40 ml kg−1) was approximately 3–10-fold lower than the half-maximal binding and ADCC effect concentrations measured from in vitro data.
Discussion
The new EMEA guidance [2] emphasizes selecting the starting dose in FIH trials based on pharmacological considerations in addition to toxicology (Figure 1) for high-risk compounds such as some biologics. By considering in vitro safety testing in addition to in vivo toxicological testing, it offers a framework to arrive at starting doses even in cases where relevant animal species may not be available [26]. This methodology is especially valuable in the case of biologics such as MABs, which have high specificity to their target; hence toxicological effects are in many cases due to exaggerated pharmacology.
In general, for antagonist-type biologics, targeting a low RO at the starting dose may ensure minimum biological effect, while providing high confidence in the preclinical-clinical translation of response. A mechanism-based PK–PD model-driven approach to human RO prediction may be preferable to simpler equilibrium-based methods, as shown in Examples 1–3, but this approach may require substantial investment in complex biomarker assays to measure target expression levels, turnover rates, and impact of MAB binding in the FIH study population prior to the clinical study. However, as illustrated in Example 1, selecting starting clinical doses based on the NOAEL without consideration of the underlying pharmacological differences could result in too high or too low a starting dose. This could result in either unexpected exaggerated pharmacological response at the starting dose, or multiple nonpharmacological doses. In case of extrapolation from in vitro data, the PK–PD model-based approach is likely to be most successful for a well-studied class of biologics such as MABs, whose PK properties are, in general, uniform based on their isotype [41]. The extent of RO at the starting dose should consider the relationship between the RO and downstream biological effect, with potentially lower starting RO for an unprecedented agonist mechanism and higher for an antagonist. When targeting a low RO in FIH trial may not be appropriate, other mechanism biomarkers may be used to select a MABEL dose, but understanding the relationship between biomarker changes and the clinical safety/efficacy profile may be necessary to ensure safety.
While the scientific basis of the use of these models to predict clinical dose–RO response is sound, a more comprehensive evaluation of this extrapolation procedure using empirical data is required to identify its limitations. Also in cases where the candidate does not cross-react with relevant animal species, it may be possible to characterize the dose–exposure–response in preclinical studies using surrogate MABs [42] or using transgenic animals [43]. However, more data are required on the ability to predict human response using PK–PD data from these test platforms.
The role of PK–PD modelling and simulation in the rational selection of FIH doses for high-risk biologics has been illustrated. PK–PD models, in general, need to move to a more mechanistic basis, and the data analysis tools need to evolve to support these models. More publications from industry and regulatory agencies should highlight the use of these models to support clinical trial design, especially FIH dosing rationale. This would provide clarity around the interpretation of the EMEA guidance on this topic and help provide an impetus to PK–PD science in general and translational PK–PD in particular.
Acknowledgments
The author is grateful to Dr Joe P. Balthasar (Professor, State University of Buffalo, New York, USA) and Dr Ellen Wang (Pfizer, Inc.) for their helpful comments on this manuscript.
REFERENCES
- 1.Duff G. Expert Scientific Group on Phase One Clinical Trials: Final Report. London: Department of Health, UK; 2006. [Google Scholar]
- 2.EMEA. Guideline on strategies to identify and mitigate risks for first-in-human clinical trials with investigational medicinal products. London: EMEA; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.FDA. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Rockville MD: FDA; 2005. [Google Scholar]
- 4.Reigner BG, Blesch KS. Estimating the starting dose for entry into humans: principles and practice. Eur J Clin Pharmacol. 2002;57:835–45. doi: 10.1007/s00228-001-0405-6. [DOI] [PubMed] [Google Scholar]
- 5.Vaidya AB, Vaidya RA. Initial human trials with an investigational new drug (phase 1 and 2): planning and management. J Postgrad Med. 1981;27:197–213. [PubMed] [Google Scholar]
- 6.Cohen A. Should we tolerate tolerability as an objective in early drug development? Br J Clin Pharmacol. 2007;64:249–52. doi: 10.1111/j.1365-2125.2007.03023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Contrera JF, Matthews EJ, Kruhlak NL, Benz RD. Estimating the safe starting dose in phase I clinical trials and no observed effect level based on QSAR modeling of the human maximum recommended daily dose. Regul Toxicol Pharmacol. 2004;40:185–206. doi: 10.1016/j.yrtph.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 8.Lowe PJ, Hijazi Y, Luttringer O, Yin H, Sarangapani R, Howard D. On the anticipation of the human dose in first-in-man trials from preclinical and prior clinical information in early drug development. Xenobiotica. 2007;37:1331–54. doi: 10.1080/00498250701648008. [DOI] [PubMed] [Google Scholar]
- 9.Agoram BM, Martin SW, van der Graaf PH. The role of mechanism-based pharmacokinetic–pharmacodynamic (PK–PD) modelling in translational research of biologics. Drug Discov Today. 2007;12:1018–24. doi: 10.1016/j.drudis.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Reichert J, Pavolu A. Monoclonal antibodies market. Nat Rev Drug Discov. 2004;3:383–4. doi: 10.1038/nrd1386. [DOI] [PubMed] [Google Scholar]
- 11.Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, Mistry Y, Dilger P, Liefooghe E, Cludts I, Fox B, Tarrant G, Robinson J, Meager T, Dolman C, Thorpe SJ, Bristow A, Wadhwa M, Thorpe R, Poole S. Cytokine storm’ in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol. 2007;179:3325–31. doi: 10.4049/jimmunol.179.5.3325. [DOI] [PubMed] [Google Scholar]
- 12.Mascelli MA, Zhou H, Sweet R, Getsy J, Davis HM, Graham M, Abernethy D. Molecular, biologic, and pharmacokinetic properties of monoclonal antibodies: impact of these parameters on early clinical development. J Clin Pharmacol. 2007;47:553–65. doi: 10.1177/0091270006298360. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi N, Tsukamoto Y, Sallas WM, Lowe PJ. A mechanism-based binding model for the population pharmacokinetics and pharmacodynamics of omalizumab. Br J Clin Pharmacol. 2007;63:548–61. doi: 10.1111/j.1365-2125.2006.02803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berzofsky JA. Fundamental Immunology. 5th edn. Philadelphia, PA: Lippincott, Williams and Wilkins; 1999. [Google Scholar]
- 15.Gross AW, Lodish HF. Cellular trafficking and degradation of erythropoietin and novel erythropoiesis stimulating protein (NESP) J Biol Chem. 2006;281:2024–32. doi: 10.1074/jbc.M510493200. [DOI] [PubMed] [Google Scholar]
- 16.Meno-Tetang GM, Lowe PJ. On the prediction of the human response: a recycled mechanistic pharmacokinetic/pharmacodynamic approach. Basic Clin Pharmacol Toxicol. 2005;96:182–92. doi: 10.1111/j.1742-7843.2005.pto960307.x. [DOI] [PubMed] [Google Scholar]
- 17.Ng CM, Stefanich E, Anand BS, Fielder PJ, Vaickus L. Pharmacokinetics/pharmacodynamics of nondepleting anti-CD4 monoclonal antibody (TRX1) in healthy human volunteers. Pharm Res. 2006;23:95–103. doi: 10.1007/s11095-005-8814-3. [DOI] [PubMed] [Google Scholar]
- 18.Agoram BM, Sutjandra L, Jang G, Molineux G, Elliot S. Tissue distribution and excretion of 125I-darbepoetin alfa in Sprague-Dawley rats following a single subcutaneous or intravenous administration. Nephrol Dial Transplant. 2006;21:iv304. [Google Scholar]
- 19.Baxter LT, Zhu H, Mackensen DG, Butler WF, Jain RK. Biodistribution of monoclonal antibodies: scale-up from mouse to human using a physiologically based pharmacokinetic model. Cancer Res. 1995;55:4611–22. [PubMed] [Google Scholar]
- 20.Ducrot T, Beliard R, Glacet A, Klein P, Harbonnier S, Benmostefa N, Bourel D. Use of the DAF assay to assess the functional properties of polyclonal and monoclonal RhD antibodies. Vox Sang. 1996;71:30–6. doi: 10.1046/j.1423-0410.1996.7110030.x. [DOI] [PubMed] [Google Scholar]
- 21.Martis L, Patel M, Giertych J, Mongoven J, Taminne M, Perrier MA, Mendoza O, Goud N, Costigan A, Denjoy N, Verger C, Owen WF., Jr Aseptic peritonitis due to peptidoglycan contamination of pharmacopoeia standard dialysis solution. Lancet. 2005;365:588–94. doi: 10.1016/S0140-6736(05)17908-2. [DOI] [PubMed] [Google Scholar]
- 22.Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28:507–32. doi: 10.1023/a:1014414520282. [DOI] [PubMed] [Google Scholar]
- 23.Tabrizi MA, Roskos LK. Preclinical and clinical safety of monoclonal antibodies. Drug Discov Today. 2007;12:540–7. doi: 10.1016/j.drudis.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 24.Putnam WS, Li J, Haggstrom J, Ng C, Kadkhodeyan-Fischer S, Cheu M, Deniz Y, Lowman H, Fielder PJ, Visich J, Joshi A, Jumbe S. Use of quantitative pharmacology in the development of HAE1, a high-affinity anti-IgE monoclonal antibody. AAPS J. 2008;10:425–30. doi: 10.1208/s12248-008-9045-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuideveld KP, Van der Graaf PH, Peletier LA, Danhof M. Allometric scaling of pharmacodynamic responses: application to 5-Ht1A receptor mediated responses from rat to man. Pharm Res. 2007;24:2031–9. doi: 10.1007/s11095-007-9336-y. [DOI] [PubMed] [Google Scholar]
- 26.Pilaro A. Case studies in toxicokinetics of monoclonal antibodies. 2007. AAPS National biotechnology Conference, San Diego.
- 27.Macdougall IC. Optimizing the use of erythropoietic agents – pharmacokinetic and pharmacodynamic considerations. Nephrol Dial Transpl. 2002;17(Suppl. 5):66–70. doi: 10.1093/ndt/17.suppl_5.66. [DOI] [PubMed] [Google Scholar]
- 28.Bleeker WK, Lammerts van Bueren JJ, van Ojik HH, Gerritsen AF, Pluyter M, Houtkamp M, Halk E, Goldstein J, Schuurman J, van Dijk MA, van de Winkel JG, Parren PW. Dual mode of action of a human anti-epidermal growth factor receptor monoclonal antibody for cancer therapy. J Immunol. 2004;173:4699–707. doi: 10.4049/jimmunol.173.7.4699. [DOI] [PubMed] [Google Scholar]
- 29.Ricart AD, Tolcher AW. Technology insight: cytotoxic drug immunoconjugates for cancer therapy. Nat Clin Pract Oncol. 2007;4:245–55. doi: 10.1038/ncponc0774. [DOI] [PubMed] [Google Scholar]
- 30.Johnson M. Molecular mechanisms of beta(2)-adrenergic receptor function, response, and regulation. J Allergy Clin Immunol. 2006;117:18–24. doi: 10.1016/j.jaci.2005.11.012. quiz 25. [DOI] [PubMed] [Google Scholar]
- 31.Chang TW, Wu PC, Hsu CL, Hung AF. Anti-IgE antibodies for the treatment of IgE-mediated allergic diseases. Adv Immunol. 2007;93:63–119. doi: 10.1016/S0065-2776(06)93002-8. [DOI] [PubMed] [Google Scholar]
- 32.Fatkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, Saag MS, Goebel FD, Rockstroh JK, Dezube BJ, Jenkins TM, Medhurst C, Sullivan JF, Ridgway C, Abel S, James IT, Youle M, van der Ryst E. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med. 2005;11:1170–2. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- 33.DeGeorge JJ, Ahn CH, Andrews PA, Brower ME, Giorgio DW, Goheer MA, Lee-Ham DY, McGuinn WD, Schmidt W, Sun CJ, Tripathi SC. Regulatory considerations for preclinical development of anticancer drugs. Cancer Chemother Pharmacol. 1998;41:173–85. doi: 10.1007/s002800050726. [DOI] [PubMed] [Google Scholar]
- 34.Fick RB, Jr, Fox JA, Jardieu PM. Immunotherapy approach to allergic disease. Immunopharmacology. 2000;48:307–10. doi: 10.1016/s0162-3109(00)00229-0. [DOI] [PubMed] [Google Scholar]
- 35.Wang EQ, Balthasar JP. American Association of Pharmaceutical Scientists. Toronto, Canada: AAPS: 2008. Evaluation of PK/PD Models for Antibodies Exhibiting Target Mediated Disposition. National Biotechnology Conference. [Google Scholar]
- 36.Agoram B, Martin SW, Davis JD. Use of PKPD modelling for starting dose selection in first-in-human trials with high-risk monoclonal antibodies. Clin Pharmacol Ther. 2008;83(Suppl. 1):pS62. [Google Scholar]
- 37.Charles P, Elliott MJ, Davis D, Potter A, Kalden JR, Antoni C, Breedveld FC, Smolen JS, Eberl G, deWoody K, Feldmann M, Maini RN. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J Immunol. 1999;163:1521–8. [PubMed] [Google Scholar]
- 38.Lu ZY, Brailly H, Wijdenes J, Bataille R, Rossi JF, Klein B. Measurement of whole body interleukin-6 (IL-6) production: prediction of the efficacy of anti-IL-6 treatments. Blood. 1995;86:3123–31. [PubMed] [Google Scholar]
- 39.Agoram B, Heatherington AC, Gastonguay MR. Development and evaluation of a population pharmacokinetic–pharmacodynamic model of darbepoetin alfa in patients with nonmyeloid malignancies undergoing multicycle chemotherapy. AAPS J. 2006;8:E552–63. doi: 10.1208/aapsj080364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Osterborg AC, De Boer R, Clemens M, Renczes G, Kotasek D, Prausova J, Marschner N, Hedenus M, Hendricks L, Amado R. A novel erythropoiesis-stimulating agent (AMG114) with 131-hour half-life effectively treats chemotherapy-induced anemia when administered as 200 mcg every 3 weeks. J Clin Oncol. 2006;24:8626. [Google Scholar]
- 41.Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93:2645–68. doi: 10.1002/jps.20178. [DOI] [PubMed] [Google Scholar]
- 42.Wu B, Joshi A, Ren S, Ng C. The application of mechanism-based PK/PD modeling in pharmacodynamic-based dose selection of muM17, a surrogate monoclonal antibody for efalizumab. J Pharm Sci. 2006;95:1258–68. doi: 10.1002/jps.20475. [DOI] [PubMed] [Google Scholar]
- 43.Alifrangis L, André P, Overgaard RV, Sola C, Tisserant A, Wagtmann N, Romagne F, Ingwersen SH. Population Analysis Group in Europe 16, Copenhagen. Denmark: 2007. Setting a Safe Starting Dose for a First-in-Man trial of a Monoclonal Antibody Based on Population PK–PD Predictions. [Google Scholar]