

Abstract
AIMS
Human pregnane X receptor (PXR/NR1I2) is the master regulator of CYP3A4, which metabolizes >50% of drugs on the market. This study investigated the relationship between the two most frequent haplotypes [H1 (TCAGGGGCCACC) and H2 (CCGAAAACTAAT)] of PXR and basal and St John's wort (SJW)-induced CYP3A4 activity.
METHODS
Ten healthy subjects carrying H1 and H2 haplotypes (three subjects with H1/H1, four with H1/H2 and three with H2/H2) entered this study. The 10 subjects did not carry CYP3A4*4, *5 and *6. All subjects were administrated a 300-mg SJW tablet three times daily for 14 days, and CYP3A4 activity was measured using nifedipine (NIF) as a probe. The plasma concentrations of NIF and dehydronifedipine (DNIF) were determined by a validated liquid chromatography/mass spectrometry/mass spectrometry method.
RESULTS
After administration of SJW, the AUC0–∞ of NIF decreased significantly, and the AUC0–∞ of DNIF increased significantly (P < 0.05). For H1/H2, the AUC0–∞ of NIF decreased by 42.4%, and the AUC0–∞ of DNIF increased by 20.2%; for H2/H2, the AUC0–∞ of NIF decreased by 47.9%, and the AUC0–∞ of DNIF increased by 33.0%; for H1/H1, the AUC0–∞ of NIF decreased by 29.0%, yet the AUC0–∞ of DNIF increased by 106.7%. The increase of the AUC0–∞ of DNIF in H1/H1 was significantly different from the other two haplotype pairs (P < 0.05). Meanwhile, before administration of SJW, the ratio of AUC0–∞(DNIF)/AUC0–∞(NIF) was the lowest for H1/H1 (22.1%), compared with H1/H2 (58.7%) and H2/H2 (30.0%).
CONCLUSIONS
H1/H1 of the human PXR gene had weaker basal transcriptional activity but greater inducible transcriptional activity to CYP3A4 than H1/H2 and H2/H2.
Keywords: CYP3A4, haplotype, induction, PXR, St John's wort
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Human pregnane X receptor (PXR/NR1I2) is a key regulator of cytochrome P450 3A4.
To date, there are 198 reported SNPs for the human PXR/NR1I2 gene.
Some of these SNPs are found to affect the inducing ability of PXR to CYP3A4.
WHAT THIS STUDY ADDS
This study, for the first time, has investigated the effect of PXR haplotype on basal and St John's wort-induced CYP3A4 activity in humans.
H1/H1 of the PXR gene had weaker basal transcriptional activity but greater inducible transcriptional activity to CYP3A4 than H1/H2 and H2/H2.
Introduction
Interindividual variability in response to drugs remains an important challenge to optimal drug therapy. An important determinant of pharmacological response is the pathways governing drug-metabolizing enzymes and drug transporters. Drug-metabolizing enzymes facilitate the conversion of xenobiotics to hydrophilic molecules more suitable for excretion into urine or bile. The importance of metabolism to drug effects and toxicity has been widely appreciated and studied. The cytochrome P450 (CYP) superfamily of enzymes is responsible for the metabolism, detoxification and bioactivation of the majority of pharmaceuticals and xenobiotics. CYP3A4 is the most abundantly expressed CYP and has been implicated in the metabolism of >50% of prescribed pharmaceuticals [1]. Variability in CYP3A4 expression and/or function can have a dramatic effect on the clinical response to many drugs. The expression levels of CYP3A4 vary up to 100-fold between individuals [1]. Induction or inhibition of CYP3A4 is a common cause of adverse drug–drug interactions, which are a major public health problem worldwide [1]. Genetic polymorphisms including 304 single nucleotide polymorphisms (SNPs) in the CYP3A4 gene (http://www.ncbi.nlm.nih.gov/, access date 29 August 2008) can not explain such large interindividual variation.
The pregnane X receptor (PXR/NR1I2), also known as steroid and xenobiotic receptor and pregnane-activated receptor, is a member of the nuclear receptor (NR) family of ligand-dependent transcription factors [2–5]. PXR/NR1I2 has been identified as a key regulator for the expression of genes involved in all stages of drug metabolism and transport [4, 5]. Phase I drug-metabolizing enzymes regulated by PXR/NR1I2 include several CYPs (e.g. CYP3A4, 3A5, 2B6 and 2C8), carboxylesterases and dehydrogenases [2–5]. The most common clinical implication for the activation of PXR/NR1I2 is the occurrence of drug–drug interactions mediated by upregulated CYP3A4. Therefore, altered function or expression of the PXR gene due to SNPs is considered an important additional source of interindividual variation in the expression and activity of CYP3A4. To date, there are 373 reported SNPs for the human NR1I2 gene in the SNP database at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/, access date 29 August 2008). Multiple SNPs of PXR have functional effects on the expression of human PXR. Zhang et al.[6] found that the -25385C→T was associated with a marked higher CYP3A4 induction ability by rifampin as determined by the erythromycin breath test, a marker of CYP3A4 hepatic activity. Individuals with the -25385C→T genotype had twofold higher CYP3A4 activity after treatment with rifampin compared with subjects with the wild-type genotype. Out of nine SNPs reported in the 3′-UTR of PXR/NR1I2, four demonstrated association with the expression levels of target genes. Hustert et al.[7] found three variants (V140M, D163G and A370T) with significant functional defects in terms of CYP3A4 transcription. A Q158K mutation of PXR has been linked to decreased rifampin-mediated CYP3A4 induction. Koyano et al.[8] have investigated the three variants [443G→A (R148Q), 1141C→T (R381W), 1207G→A (I403V)] of PXR and found that their basal and rifampicin-induced transactivation of the CYP3A4 enhancer/promoter was significantly reduced compared with the wild-type PXR [9]. Our previous study showed that the activity of the recombinants with alleles containing the -24622A→T in the 5′-untranslated region or -24446C→A in exon 1 was 30–40% higher than that in the reference genotype [10].
Human PXR has broad substrate specificity and thus may be activated by a large number of chemically diverse compounds found in the diet as well as therapeutic agents. It is now known that many clinically relevant drug interactions involving upregulation of CYP3A4 are mediated through the actions of human PXR [11, 12]. The objective of this study was to explore the influence of the most frequent haplotypes of human PXR genetic variations on basal and St John's wort (SJW)-induced CYP3A4 activity. CYP3A4 activity was evaluated using the dihydropyridine calcium channel inhibitor nifedipine (NIF) as a probe substrate [13]. It is well known that in humans, NIF is predominantly metabolized by CYP3A4 to its primary pyridine metabolite, dehydronifedipine (DNIF) [14, 15].
Materials and methods
Drugs and reagents
SJW preparation (300-mg tablet) containing 0.3% hypericin and 5% hyperforin was obtained from Dr Willmar Schwabe Pharmaceuticals (Karlsruhe, Germany). NIF (purity 99.9%) and nitrendipine (internal standard, purity 97.8%) were synthesized and provided by Baiyunshan Pharmaceutical Inc. (Guangzhou, China). DNIF (purity 99.9%) was obtained from Sigma-Aldrich Chemical Co. (St Louis, MO, USA).
Subjects
We initially recruited 12 unrelated healthy adult subjects, from a total of 210 healthy Han Chinese volunteers who had been screened for human PXR genotype and haplotype for this study [10]. In our previous study, 42 inferred haplotypes were identified in Han Chinese [10]. In the haplotype analysis, only those SNPs with a mutant allele frequency over 5% were included. These included 12 SNPs, namely -25385C→T in the 5′-UTR, -24381A→C in exon 1, -24113G→A in intron 1, 252A→G in intron 2, 275A→G in intron 2, 4760G→A in intron 4, 7635G→A in intron 5, 7675C→T in intron 5, and 10483C→T, 10719G→A, 11156A→C and 11193T→C all in the 3′-UTR [10]. The most frequent haplotypes in Han Chinese are H1 (TCAGGGGCCACC) and H2 (CCGAAAACTAAT), with a frequency of 15.1% [10]. After having signed the informed consent, two subjects (one carrying H1/H1, one carrying H2/H2) withdrew for personal reasons. Only 10 subjects completed the study. All volunteers were subjected to a complete physical examination and a series of biochemical tests for evaluating their health status before entering the study. Volunteers were excluded if they were found to have diseases of the heart, kidney, liver, blood, or other organs/systems. Ethical approval of the study was obtained from the Ethics Committee of Sun Yat-sen University (Guangzhou, China), and written informed consent was obtained from all participants.
Study design and clinical protocol
The study was conducted before and after a 14-day treatment period with a SJW preparation at 300 mg three times daily. Two weeks before and during the experiment, the subjects have been forbidden to take any kind of food substances known to induce or inhibit CYP3A4, including grapefruit juice, red wine, alcoholic beverages, teas and herbs. CYP3A4 activity before and after administration of SJW was measured using NIF as a probe. After overnight fasting (10 h), volunteers were treated with a single oral dose of NIF (10-mg tablet) with 200 ml of warm water. The volunteers were monitored during the experimental period for the development of adverse effects commonly associated with SJW and NIF, such as dry mouth and dizziness. Regular standardized low-fat meals were provided until 4 h after dose administration; water intake was allowed after 2 h of drug administration. Following drug administration, venous blood samples (5 ml) were collected into heparinized brown tubes according to the following schedule: immediately before drug administration (0 min) and 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 5, 8, 12 and 24 h after dosing. Blood samples were centrifuged at 2000 g for 10 min, and plasma was separated and clearly labelled and stored at −30°C until analysis.
Analytical method
Plasma concentrations of NIF and its metabolite, DNIF, were quantified by a validated liquid chromatography/mass spectrometry/mass spectrometry (LC/MS/MS) method according to the method previously described [16]. A Waters 2695 separation module (alliance) (Avondale, CA, USA) was used for solvent and sample delivery. Chromatographic separation was achieved by using a C18 column (Hypersil BDS C18, i.d. 2.1 × 50 mm, 3 µm; Elite HPLC Inc., Dalian, China) at room temperature. The mobile phase consisted of methanol–water (containing 1% formic acid) (80:20, v/v), pumped at a flow rate of 200 µl min−1.
In brief, a mixture of 500 µl of a plasma aliquot and 20 µl of nitrendipine (internal standard) was extracted with 2 ml of extraction solvent (ether : n-hexane, 3:1, v/v). After centrifugation at 2500 g for 10 min, the organic layer was decanted and evaporated to dryness using nitrogen gas. The residues were dissolved in 100 µl methanol/water (50:50, v/v) and an aliquot (10 µl) of the reconstituent was injected onto the LC/MS/MS for analysis. The method had a chromatographic running time of approximately 2.5 min and the lower limit of quantification of the analytical method was 0.5 ng ml−1 for both analytes.
Pharmacokinetic analysis
Pharmacokinetics parameters were calculated by noncompartmental model using WinNonlin program version 1.0 (Scientific Consulting Inc., Cary, NC, USA). The AUC from time zero to the last quantifiable time point (AUC0–t) and from time zero to infinity AUC0–∞ were estimated using the log-linear trapezoidal rule. The terminal elimination rate constant (β) was determined by log linear regression, and the terminal elimination half-life (t1/2β) was determined by the following relationship: t1/2β= 0.693/β. Peak plasma concentration (Cmax) was determined by visual inspection. Mean residence time (MRT) was calculated by the total area under the first-moment curve divided by AUC0–t. F (%) was the relative bioavailability of NIF before and after oral administration of SJW, obtained by (AUC0–t after SJW/AUC0–t before SJW)× 100%.
Statistical analysis
Statistical analysis was performed in SPSS system for Windows version 11.0 (SPSS Inc, Chicago, IL, USA). Data were expressed as mean ± SD. The changes of pharmacokinetic parameters in 10 individuals after administration of SJW were analysed with Student's t-test for paired design. The differences of pharmacokinetic parameters among the three PXR haplotype groups were analysed using the Kruskal–Wallis H test. P < 0.05 was regarded as being of statistical significance.
Results and discussion
The clinical features and PXR haplotypes of the 10 healthy subjects are summarized in Table 1. The 10 healthy subjects involved in the pharmacokinetic study contained the two most frequent haplotypes {[H1(TCAGGGGCCACC) and H2(CCGAAAACTAAT)]}. Three individuals were identified to be with the haplotype pairs H1/H1; four were with the haplotype pairs H1/H2; and the other three were owned by the haplotype pairs H2/H2. After administration of SJW, the area under the plasma concentration–time profile (AUC) of NIF in all subjects decreased significantly, and the AUC of its major metabolite DNIF increased significantly (P < 0.05, see Table 2), indicating that SJW induced CYP3A4-catalysed metabolism of NIF to form DNIF. For the three haplotype pairs of PXR, there was a significant difference in the change of the AUC0–∞ of NIF and DNIF. For H1/H2, the AUC0–t and AUC0–∞ of NIF decreased by 46.4 and 42.4%, respectively, and the AUC0–t and AUC0–∞ of DNIF increased by 25 and 20.2%, respectively (Figure 1). For H2/H2, the AUC0–t and AUC0–∞ of NIF decreased by 44.7 and 47.9%, respectively, and the AUC0–t and AUC0–∞ of DNIF increased by 33.3 and 33.0%, respectively. For H1/H1, the AUC0–t and AUC0–∞ of NIF decreased by 34.5 and 29.0%, respectively, whereas the AUC0–t and AUC0–∞ of DNIF increased by 109.3 and 106.7%, respectively. Using Kruskal–Wallis H-test, the increases of the AUC0–t and AUC0–∞ of DNIF in H1/H1 were significantly different from the other two haplotype pairs (P < 0.05), with induced transcriptional activity of H1/H1 being stronger than that of the H1/H2 and H2/H2. These results indicate that in subjects with H1/H1, the inducers of PXR can lead to much higher inducible metabolic activity of CYP3A4 than in subjects with H1/H2 and H2/H2. Since the subgroup size of three to four is quite small, large-scale studies are needed to verify the findings and to investigate the functional impact of other haplotypes. None of the subjects carried the three common allelic variants of CYP3A4*4 (I118V), CYP3A4*5 (P218R) or CYP3A4*6 (17776A ins).
Table 1.
General data and haplotype of the 10 volunteers
| Subject | Gender | Age (years) | Height (cm) | Body weight (kg) | Heart rate (beat/min) | Blood pressure (mmHg) | Haplotype of PXR |
|---|---|---|---|---|---|---|---|
| 1 (H26) | Male | 23 | 171 | 61 | 75 | 120/80 | (1,2) |
| 2 (H19) | Female | 23 | 158 | 45 | 81 | 115/65 | (1,2) |
| 3 (H12) | Female | 21 | 164 | 54 | 86 | 112/73 | (1,2) |
| 4 (H92) | Female | 22 | 168 | 55 | 70 | 100/80 | (1,2) |
| 5 (H66) | Male | 22 | 172 | 63 | 82 | 105/68 | (2,2) |
| 6 (H115) | Male | 21 | 169 | 62 | 75 | 120/75 | (2,2) |
| 7 (H171) | Female | 24 | 158 | 50 | 88 | 112/65 | (2,2) |
| 8 (H36) | Male | 24 | 176 | 70 | 83 | 108/72 | (1,1) |
| 9 (H45) | Female | 21 | 166 | 58 | 70 | 115/76 | (1,1) |
| 10 (H156) | Male | 23 | 170 | 70 | 90 | 120/75 | (1,1) |
Table 2.
The pharmacokinetic parameters for nifedipine (NIF) and dehydronifedipine (DNIF) in the study population (n = 10)
| Tmax (h) | Cmax (µg l−1) | t1/2β (h) | MRT(h) | AUC0–t (µg l−1 h−1) | AUC0–∞ (µg l−1 h−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Haplotype | Before SJW | After SJW | Before SJW | After SJW | Before SJW | After SJW | Before SJW | After SJW | Before SJW | After SJW | Before SJW | After SJW | F (%) |
| NIF | |||||||||||||
| H1/H2 (n = 4) | 1.5 ± 0.7 | 1.3 ± 0.3 | 34.3 ± 13.0 | 22.7 ± 5.4* | 8.5 ± 4.4 | 7.4 ± 0.8 | 6.1 ± 0.4 | 5.0 ± 1.0 | 209.5 ± 32.6 | 112.2 ± 31.0* | 231.7 ± 47.7 | 133.4 ± 28.0* | 53.0 ± 8.0 |
| H2/H2 (n = 3) | 1.5 ± 0.0 | 1.5 ± 0.0 | 53.2 ± 6.3 | 30.8 ± 4.2* | 7.4 ± 2.7 | 4.8 ± 1.7 | 6.8 ± 1.0 | 5.8 ± 1.2 | 332.6 ± 76.4 | 183.9 ± 49.9* | 370.3 ± 102.5 | 193.0 ± 52.6* | 54.9 ± 3.1 |
| H1/H1 (n = 3) | 1.8 ± 0.6 | 1.8 ± 0.6 | 59.3 ± 13.2 | 33.2 ± 5.5* | 7.0 ± 1.7 | 9.5 ± 2.0 | 6.7 ± 0.9 | 7.6 ± 1.1 | 409.8 ± 176.2 | 268.4 ± 99.2* | 453.4 ± 221.1 | 322.1 ± 145.6* | 66.7 ± 4.4 |
| Total (n = 10) | 1.6 ± 0.5 | 1.5 ± 0.4 | 47.5 ± 15.4 | 28.3 ± 6.6* | 7.7 ± 3.0 | 7.2 ± 2.3 | 6.5 ± 0.8 | 6.0 ± 1.5 | 306.5 ± 128.5 | 180.6 ± 87.8* | 339.8 ± 154.2 | 207.9 ± 111.7* | 57.7 ± 8.2 |
| DNIF | |||||||||||||
| H1/H2 (n = 4) | 1.8 ± 0.5 | 1.6 ± 0.7 | 23.8 ± 6.8 | 39.5 ± 11.7* | 4.5 ± 1.3 | 4.3 ± 1.0 | 4.2 ± 0.5 | 4.0 ± 0.5 | 124.8 ± 36.6 | 155.7 ± 48.1* | 136.0 ± 35.0 | 163.6 ± 48.7* | – |
| H2/H2 (n = 3) | 1.8 ± 0.6 | 1.3 ± 0.3 | 21.0 ± 1.9 | 31.1 ± 2.4* | 4.2 ± 2.2 | 4.1 ± 1.7 | 5.1 ± 1.3 | 4.5 ± 0.7 | 105.1 ± 13.1 | 140.4 ± 16.0* | 111.2 ± 13.8 | 147.9 ± 22.3* | – |
| H1/H1 (n = 3) | 1.7 ± 0.8 | 1.8 ± 0.6 | 17.8 ± 5.8 | 36.4 ± 11.0* | 4.5 ± 0.9 | 5.1 ± 0.6 | 4.8 ± 1.6 | 5.2 ± 1.1 | 94.6 ± 59.5 | 197.9 ± 80.9* | 100.2 ± 62.1 | 207.1 ± 82.8* | – |
| Total (n = 10) | 1.8 ± 0.5 | 1.6 ± 0.6 | 21.2 ± 5.6 | 36.1 ± 9.3* | 4.4 ± 1.3 | 4.5 ± 1.1 | 4.7 ± 1.1 | 4.5 ± 0.9 | 109.8 ± 38.2* | 163.8 ± 53.7* | 117.8 ± 39.7 | 171.9 ± 55.3* | – |
P < 0.05, before vs. after SJW administration. Tmax, time to maximum plasma concentration; Cmax, peak concentration; t1/2β, elimination half-life; MRT, mean residence time; AUC0–t, area under concentration–time curve up to the last measured time point; AUC0–∞, AUC0–t extrapolated to infinity; F, relative bioavailability of NIF before and after oral administration of SJW, obtained by (AUC0–t after SJW/AUC0–t before SJW) × 100%; SJW, St John's wort.
Figure 1.
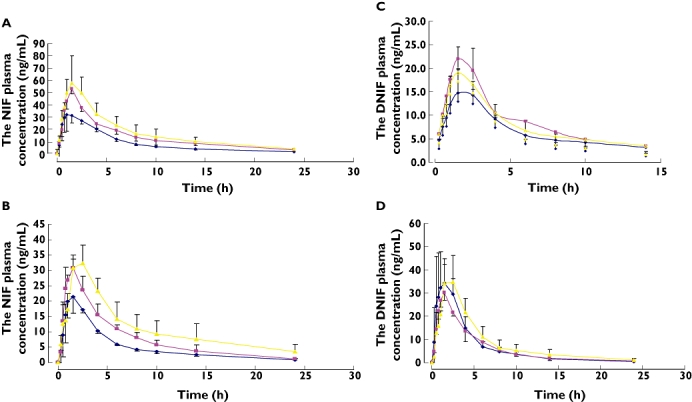
The plasma concentration–time profiles of nifedipine (NIF) (A,B) and dehydronifedipine (DNIF) (C,D) before (A,C) and after (B,D) administration of St John's wort (SJW) for 2 weeks in three different groups of PXR haplotype. Subjects were grouped into H1/H2, H2/H2 and H1/H1 according to the haplotype of the PXR gene. H1&2 ( ); H2&2 (
); H2&2 ( ); H1&1 (
); H1&1 ( )
)
Meanwhile, before administration of SJW, the ratio of AUCDNIF/AUCNIF was the lowest for H1/H1 (22.1%), presuming that the basal transcriptional activity was the weakest among the three haplotypes (H1/H2 of 58.7% and H2/H2 of 30.0%). However, for the three haplotype pairs, the elimination half-life (t1/2β) and MRT for both NIF and DNIF were not significantly different.
There is wide interindividual variation and population variation in the pharmacokinetics and clinical response of NIF. Reports on oral NIF pharmacokinetics have shown that peak plasma levels and AUC values are higher in Mexican and Japanese than in European and North American subjects [17, 18]. Following an oral 10-mg dose, the maximum concentrations (Cmax) of NIF range from 17 to 80 ng ml−1 with elimination half-lives (t1/2β) between 1 and 34 h [17, 18], and the Cmax values of DNIF vary from 8 to 37 ng ml−1 after an oral dose of 10 mg NIF [17, 18]. These significant differences were also observed in our study. The ratio of plasma level of DNIF to that of NIF is often used as a reliable indicator of CYP3A4 activity.
The frequencies of SNPs in PXR/NR1I2 genes vary in different ethnic groups, including White, African-American and Chinese populations [9, 19]. Since haplotype analysis (i.e. identification of SNP blocks) can often provide more useful information on the association of genes to a phenotype and give a better prediction of drug response (efficacy and toxicity) than an individual SNP genotype, studies are required to identify potential haplotypes in the PXR/NR1I2 gene that make a connection to a complex phenotype such as drug response.
Hyperforin present in SJW is the most potent inducer of CYP3A4 and P-glycoprotein [20]. SJW has been reported to decrease the systemic exposure of a number of drugs that are known substrates of CYP3A4 and/or P-glycoprotein [20]. Hyperforin is a high-affinity ligand that binds to PXR and consequently transactivates its target genes such as CYP3A4. Upon ligand binding, PXR forms a heterodimer with retinoid X receptor α (RXRα) and transactivates everted repeat with 6-bp spacer (ER6) elements upstream of the CYP genes [21]. RXRα serves as a common heterodimerization partner for many orphan nuclear receptors, including constitutive androstane receptor. The binding of PXR/RXRα to ER6 is followed by recruitment of coactivator proteins, e.g. steroid receptor coactivator-1, and transcriptional activation of the respective gene [22]. There is evidence for a second binding site for PXR in the ∼7800 bp upstream 5′-flanking region of the CYP3A4 gene having ER6-like binding sites [23]. PXR and RXRα are induced by glucocorticoid receptor [24]. Thus, the activation of glucocorticoid receptor by glucocorticoids, such as dexamethasone, leads to the induction of PXR/RXRα and to the increase of CYP3A4 induction by endogenous and exogenous compounds. PXR knockout mice showed no induction by typical mouse Cyp3a inducers, and loss of PXR did not alter the basal Cyp3a expression [25]. Transgenic mice containing human PXR have also been generated showing induction by human specific inducers, such as rifampicin [26]. In this study, we found that haplotype of PXR could affect its inducibility by SJW by unknown mechanisms. We speculate that different haplotypes of PXR would result in PXR variant proteins with distinct binding properties to ER6 of the CYP3A4 gene and thus have different inducing ability by SJW.
SNPs inducing changes in the function or expression of human PXR are considered to be potential sources for variation in CYP3A4 activity, since several SNPs of PXR have been found to alter basal and/or induced transactivation of the CYP3A gene [6–8]. In our study, we found that H1/H1 of the PXR gene had weaker basal transcriptional activity, but stronger induced transcriptional activity on CYP3A4 than H1/H2 and H2/H2, suggesting that different haplotypes of PXR can lead to different transactivation on CYP3A4, which provide further evidence for the great variation of the activity and expression of CYP3A4 enzyme.
Acknowledgments
The authors appreciate the financial supports provided by the National Natural Science Foundations of China (No. 30873124; No. 30873125; No. 30572231) and the Excellent Young Teachers Program of the national ‘985’ Project.
Competing interests
None declared.
REFERENCES
- 1.Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome p450 3a4. Curr Drug Metab. 2008;9:310–22. doi: 10.2174/138920008784220664. [DOI] [PubMed] [Google Scholar]
- 2.Moore DD, Kato S, Xie W, Mangelsdorf DJ, Schmidt DR, Xiao R, Kliewer SA. International Union of Pharmacology. LXII. The NR1H and NR1I receptors: constitutive androstane receptor, pregnene X receptor, farnesoid X receptor alpha, farnesoid X receptor beta, liver X receptor alpha, liver X receptor beta, and vitamin D receptor. Pharmacol Rev. 2006;58:742–59. doi: 10.1124/pr.58.4.6. [DOI] [PubMed] [Google Scholar]
- 3.Stanley LA, Horsburgh BC, Ross J, Scheer N, Wolf CR. PXR and CAR: nuclear receptors which play a pivotal role in drug disposition and chemical toxicity. Drug Metab Rev. 2006;38:515–97. doi: 10.1080/03602530600786232. [DOI] [PubMed] [Google Scholar]
- 4.Synold TW, Dussault I, Forman BM. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med. 2001;7:584–90. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- 5.Matic M, Mahns A, Tsoli M, Corradin A, Polly P, Robertson GR. Pregnane X receptor: promiscuous regulator of detoxification pathways. Int J Biochem Cell Biol. 2007;39:478–83. doi: 10.1016/j.biocel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Kuehl P, Green ED, Touchman JW, Watkins PB, Daly A, Hall SD, Maurel P, Relling M, Brimer C, Yasuda K, Wrighton SA, Hancock M, Kim RB, Strom S, Thummel K, Russell CG, Hudson JR., Jr Schuetz EG, Boguski MS. The human pregnane X receptor: genomic structure and identification and functional characterization of natural allelic variants. Pharmacogenetics. 2001;11:555–72. doi: 10.1097/00008571-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Hustert E, Zibat A, Presecan-Siedel E, Eiselt R, Mueller R, Fuss C, Brehm I, Brinkmann U, Eichelbaum M, Wojnowski L, Burk O. Natural protein variants of pregnane X receptor with altered transactivation activity toward CYP3A4. Drug Metab Dispos. 2001;29:1454–9. [PubMed] [Google Scholar]
- 8.Koyano S, Kurose K, Saito Y, Ozawa S, Hasegawa R, Komamura K, Ueno K, Kamakura S, Kitakaze M, Nakajima T, Matsumoto K, Akasawa A, Saito H, Sawada J. Functional characterization of four naturally occurring variants of human pregnane X receptor (PXR): one variant causes dramatic loss of both DNA binding activity and the transactivation of the CYP3A4 promoter/enhancer region. Drug Metab Dispos. 2004;32:149–54. doi: 10.1124/dmd.32.1.149. [DOI] [PubMed] [Google Scholar]
- 9.Lim YP, Liu CH, Shyu LJ, Huang JD. Functional characterization of a novel polymorphism of pregnane X receptor, Q158K, in Chinese subjects. Pharmacogenet Genomics. 2005;15:337–41. doi: 10.1097/01213011-200505000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Wang XD, Li JL, Su QB, Deng XY, Lu Y, Chen J, Zhang JX, Zhao LZ, Zuo Z, Chan E, Chen X, Chowbay B, Xue CC, Huang M, Zhou SF. A pharmacogenetic study of pregnane X receptor (NR1I2) in Han Chinese. Curr Drug Metab. 2007;8:778–86. doi: 10.2174/138920007782798199. [DOI] [PubMed] [Google Scholar]
- 11.Lamba JK, Lin YS, Schuetz EG, Thummel KE. Genetic contribution to variable human CYP3A-mediated metabolism. Adv Drug Deliv Rev. 2002;54:1271–94. doi: 10.1016/s0169-409x(02)00066-2. [DOI] [PubMed] [Google Scholar]
- 12.Mannel M. Drug interactions with St John's wort: mechanisms and clinical implications. Drug Saf. 2004;27:773–97. doi: 10.2165/00002018-200427110-00003. [DOI] [PubMed] [Google Scholar]
- 13.Ball SE, Scatina J, Kao J, Ferron GM, Fruncillo R, Mayer P, Weinryb I, Guida M, Hopkins PJ, Warner N, Hall J. Population distribution and effects on drug metabolism of a genetic variant in the 5′ promoter region of CYP3A4. Clin Pharmacol Ther. 1999;66:288–94. doi: 10.1016/S0009-9236(99)70037-8. [DOI] [PubMed] [Google Scholar]
- 14.Funaki T, Soons PA, Guengerich FP, Breimer DD. In vivo oxidative cleavage of a pyridine-carboxylic acid ester metabolite of nifedipine. Biochem Pharmacol. 1989;38:4213–6. doi: 10.1016/0006-2952(89)90517-0. [DOI] [PubMed] [Google Scholar]
- 15.Watkins PB. Noninvasive tests of CYP3A enzymes. Pharmacogenetics. 1994;4:171–84. doi: 10.1097/00008571-199408000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Wang XD, Li JL, Lu Y, Chen X, Huang M, Chowbay B, Zhou SF. Rapid and simultaneous determination of nifedipine and dehydronifedipine in human plasma by liquid chromatography-tandem mass spectrometry: application to a clinical herb–drug interaction study. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;852:534–44. doi: 10.1016/j.jchromb.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 17.Balogh A, Gessinger S, Svarovsky U, Hippius M, Mellinger U, Klinger G, Hoffmann A, Oettel M. Can oral contraceptive steroids influence the elimination of nifedipine and its primary pyridine metabolite in humans? Eur J Clin Pharmacol. 1998;54:729–34. doi: 10.1007/s002280050543. [DOI] [PubMed] [Google Scholar]
- 18.Castaneda-Hernandez G, Hoyo-Vadillo C, Palma-Aguirre JA, Flores-Murrieta FJ. Pharmacokinetics of oral nifedipine in different populations. J Clin Pharmacol. 1993;33:140–5. doi: 10.1002/j.1552-4604.1993.tb03934.x. [DOI] [PubMed] [Google Scholar]
- 19.King CR, Xiao M, Yu J, Minton MR, Addleman NJ, Van Booven DJ, Kwok PY, McLeod HL, Marsh S. Identification of NR1I2 genetic variation using resequencing. Eur J Clin Pharmacol. 2007;63:547–54. doi: 10.1007/s00228-007-0295-3. [DOI] [PubMed] [Google Scholar]
- 20.Zhou SF, Lai X. An update on clinical drug interactions with the herbal antidepressant St. John's wort. Curr Drug Metab. 2008;9:394–409. doi: 10.2174/138920008784746391. [DOI] [PubMed] [Google Scholar]
- 21.Waxman DJ. P450 gene induction by structurally diverse xenochemicals: central role of nuclear receptors CAR, PXR, and PPAR. Arch Biochem Biophys. 1999;369:11–23. doi: 10.1006/abbi.1999.1351. [DOI] [PubMed] [Google Scholar]
- 22.Lanz RB, McKenna NJ, Onate SA, Albrecht U, Wong J, Tsai SY, Tsai MJ, O'Malley BW. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell. 1999;97:17–27. doi: 10.1016/s0092-8674(00)80711-4. [DOI] [PubMed] [Google Scholar]
- 23.Goodwin B, Hodgson E, Liddle C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol Pharmacol. 1999;56:1329–39. doi: 10.1124/mol.56.6.1329. [DOI] [PubMed] [Google Scholar]
- 24.Pascussi JM, Gerbal-Chaloin S, Duret C, Daujat-Chavanieu M, Vilarem MJ, Maurel P. The tangle of nuclear receptors that controls xenobiotic metabolism and transport: crosstalk and consequences. Annu Rev Pharmacol Toxicol. 2008;48:1–32. doi: 10.1146/annurev.pharmtox.47.120505.105349. [DOI] [PubMed] [Google Scholar]
- 25.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, Waxman DJ, Evans RM. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci USA. 2001;98:3375–80. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, Nelson MC, Neuschwander-Tetri BA, Brunt EM, Guzelian PS, Evans RM. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature. 2000;406:435–9. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]